

The time is ripe to assess whether pharmacogenomics research—the study of the genetic basis for variation in drug response—has provided important insights into a personalized approach to prescribing and dosing medications. Here, we describe the status of the field and approaches for addressing some of the open questions in pharmacogenomics research and use of genetic testing in guiding drug therapy.
With the sequencing of the human genome more than a decade ago, promises were made for safer and more effective medications that could be personalized on the basis of a patient’s particular genetic makeup. Ten years later, it is timely to reflect on those promises. Are personalized drugs being developed for individuals who harbor particular genetic variants? Has sufficient research been carried out in pharmacogenomics, the study of the genetic bases for variations in drug response? Is genetic information being used to prescribe or dose medications? Last, how should scientists attempt to define and fill the gaps in translation of pharmacogenomics research? In this Perspective, we address these questions and offer recommendations for future research in the field.
GENETIC VARIANTS, PERSONALIZED DRUGS?
Pharmacogenomic studies are performed to identify genetic risk factors for nonresponse to medications and adverse drug reactions. The vast majority of published articles in the pharmacogenomics field has focused on approved prescription drugs. For example, studies of cholesterol-lowering statin drugs have revealed single-nucleotide polymorphisms (SNPs) that are associated with the statin-driven muscle toxicities observed in some patients (1). However, with the exception of the recently approved drug ivacafor, which targets a specific defective allele in cystic fibrosis (2), few new drugs have been developed on the basis of individual hereditary genetic information (as opposed to the various nongermline or somatic mutations that occur in tumors). Because the development of a new drug requires, on average, greater than 10 years, a wide range of approved genotype-targeted drugs would not yet be expected; however, to our knowledge genotype-targeted drugs are not in clinical trials or under development.
In fact, in several cases the opposite trend has occurred: New drugs have been developed that circumvent genetic testing. For example, a genetic variant in the CYP2C19 gene, which encodes a drug-metabolizing enzyme that is required for metabolic activation of the antiplatelet drug clopidogrel, is associated with reduced response to the drug (3). A new drug, prasugrel, is not activated by CYP2C19 and thus does not require genetic testing for CYP2C19 variants before prescribing. But prasugrel likely will be much more expensive than generic clopidogrel, which will enter the market soon and create a much cheaper alternative for the majority of patients who have adequate CYP2C19 activity.
In a similar vein, although the findings are controversial the antiestrogen receptor drug tamoxifen has been found to be less active in preventing recurrence of breast cancer in women who carry CYP2D6 variants such as CYP2D6*10 and CYP2D6*4 that result in reduced enzyme activity (4). To avoid this risk, endoxifen—the active metabolite of tamoxifen—is now in clinical trials (http://clinicaltrials.gov/ct2/show/NCT01273168). Therefore, although it has informed the commercial sector, genetic information mostly has been used to continue the old drug-development paradigm of one-size-fits-all. Commercial benefits for developing new, more effective drugs are strong, whereas the commercial potential for developing individual pharmacogenomic tests is relatively limited. In particular, genetic tests would not have to be repeated, in contrast to many other clinical laboratory tests such as those that measure blood lipid concentrations or hemoglobin A1c.
ENOUGH ALREADY?
For approved drugs, candidate gene studies have dominated the pharmacogenomic literature, with many studies centered on a handful of variants in a few genes, especially those that encode polymorphic drug-metabolizing enzymes or drug transporters. Newer methodologies such as genome-wide association (GWA) or whole-exome sequencing studies—methods that take an agnostic approach to variant discovery—have only recently been investigated for pharmacogenomic phenotypes. For example, the U.S. National Human Genome Research Institute (NHGRI) has cataloged only 102 GWA studies of pharmacogenomic phenotypes, in comparison with more than 1000 GWA studies centered on disease risk and complex traits. The NHGRI-cataloged pharmacogenomics GWA studies are generally smaller than traditional molecular epidemiology studies, having approximately 1/10 the number of samples (mean of ~670 samples/study in the 104 studies) compared with GWA studies of disease and complex traits (mean of ~6500 samples/study in 1212 studies) (5). A survey of 15 published GWA pharmacogenomic studies (1, 6–19) reveals similar results, with the number of cases (that is, patients who experienced an adverse event or did not respond to a drug) ranging between 14 and 293.
The reasons for the markedly fewer studies and lower sample sizes of pharmacogenomic GWA studies compared with GWA studies of disease risk are related to the complexity of conducting pharmacogenomic studies and an expectation of larger effect sizes of a significant SNP on the phenotypic trait. If a SNP has a large effect on a trait, fewer samples are required to observe a significant result. For example, for studies of common human disease or complex traits ascertainment of cases and controls is generally straightforward. Data from electronic medical records are used to identify individuals with particular diseases (for example, type 2 diabetes), and for many common diseases and traits, genotypes obtained from geographically matched populations can be used as controls. For pharmacogenomic studies, however, control samples must be collected from individuals who are taking the same drug but do not experience the phenotype (for example, the adverse drug reaction). Thus, simple population controls cannot be used. Furthermore, medical records do not routinely convey information about adverse drug reactions or nonresponse to medications, and thus such data must be collected specifically for each study. The common practice of polypharmacy—the use of several medications simultaneously in a single patient—can confound the interpretation of the phenotype. For example, because serotonin-specific reuptake inhibitor (SSRI) antidepressants such as fluoxetine inhibit CYP2D6, a patient with normal CYP2D6 alleles who is taking fluoxetine will metabolize a substrate of CYP2D6, such as tamoxifen, at a rate similar to that of a patient with a reduced-function CYP2D6 allele; thus, use of SSRIs in a pharmacogenetic study of tamoxifen will confound the interpretation of the data. Further, some deleterious adverse drug reactions may be rare and thus difficult to study. Last, randomized clinical trials—ideal settings for pharmacogenomic studies of drug response phenotypes—are costly, take years to complete, and are rarely repeated using the exact protocols, making replication of GWA signals identified in such trials a challenge.
Despite the relatively low number of samples and few studies, pharmacogenomic GWA studies have been particularly fruitful. For example, only 105 SNPs with large effect sizes (odds ratios > 3 and P < 1.0 × 10−5) were identified in the 1215 NHGRI-cataloged GWA studies (as of 19 July 2012) of disease and complex traits, whereas 64 SNPs with large effect sizes (odds ratios > 3 and P < 1.0 × 10−5) were reported in the 102 pharmacogenomic GWA studies (as of 19 July 2012). Thus, the yield of high-effect–size SNPs is ~7 times more frequent in studies of drug response as compared with those of human disease risk or complex traits. Furthermore, substantially more SNPs with large effect sizes were associated with adverse drug reactions compared with therapeutic response, suggesting that studies of adverse drug reactions may be particularly informative.
We suspect that there are at least two major reasons for the large effect sizes of SNPs identified in pharmacogenomic studies. First, until recently humans had not been exposed to synthetic drugs—modern drugs with the longest histories have been available only for a little over 100 years—and so there has been little negative evolutionary pressure for drug use, allowing polymorphisms to become relatively common in human populations. Second, there might be a large interplay between the gene and the drug effect. For example, individuals with a genetic variant in the human leukocyte antigen (HLA) locus are highly susceptible to carbamazepine-induced very severe skin-adverse reaction (20). Because the phenotypes in question (such as skin-adverse reaction) manifest only if an individual takes a certain drug (such as carbamazepine), the sizes of the subject population required to obtain significantly different allele frequencies in individuals with the phenotype versus those without might be expected to be relatively small.
Irrespective of the mechanisms that specify the observed large-effect sizes for SNPs that are associated with pharmacogenomic traits such as adverse drug reactions, the data to date suggest that pharmacogenomic GWA studies that focus on adverse drug reactions likely will identify genetic factors that increase a patient’s risk of suffering an adverse reaction to a given drug. As with GWA studies of disease risk, few pharmacogenomic studies have been performed in individuals who are not of European or Asian background (Fig. 1), a gap that must be filled if we are to detect ethnicity-specific adverse drug reactions. Several ongoing pharmacogenomic studies such as those of the Indian Society of Agricultural Engineers consortium include patients of non-European ancestries (21, 22).
Fig. 1. Ethnic representation.
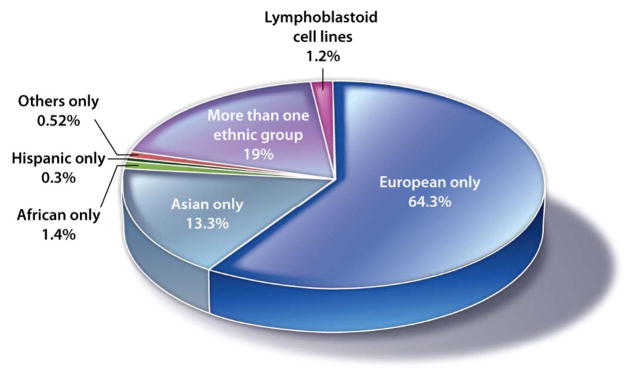
Shown are participants in pharmacogenomics GWA studies categorized by ethnicity. Data were obtained from www.genome.gov/admin/gwascatalog.txt (accessed 19 July 2012). At this time, 102 pharmacogenomics research GWA studies were included. Blue, European only (51 studies, 52,784 participants, 64.3%); turquoise, Asian only (23 studies, 10,957 participants, 13.3%); green, African only (2 studies, 1119 participants, 1.4%); black, Hispanic only (1 study, 229 participants, 0.3%); red, Others only (1 study, 429 participants, 0.52%); purple, More than one ethnic group (20 studies, 15,604 participants, 19.0%); magenta, Lymphoblastoid cell lines (4 studies, 995 participants, 1.2%).
PERSONALIZED PRESCRIPTIONS
Regulation
Because genetic variants with large effect sizes have strong predictive values, the U.S. Food and Drug Administration (FDA) has begun to change the labels of prescription drugs to include pharmacogenetic information (Table 1). These label changes have centered largely on issues of safety, which is consistent with FDA’s mandate to protect the public. Notable examples of adverse drug effect–associated genomic variations detected in post-market pharmacogenomic studies include the HLA genotype HLA-*1502, which is associated with a life-threatening skin hypersensitivity reaction to carbamazepine (a drug to treat epilepsy and bipolar disorder) and common variants in the UGT1A1 gene that are associated with an increased risk of neutropenia after treatment with irinotecan (an anticancer agent). Labels of some new drugs include genetic information and recommendation for genetic testing. For example, the label of tetrabenazine, a new drug used in the treatment of chorea associated with Huntington’s disease, recommends CYP2D6 genotyping before administration of large doses.
Table 1. FDA drug labels that include genetic and genomic prescribing information.
Shown are selected drugs that contain pharmacogenomic information in label sections Boxed Warning, Warnings and Precautions, Indications and Usage, or Dosage and Administration. A full list of pharmacogenomic biomarkers in drug labels is available at the FDA Web site (www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm). Three different types of pharmacogenomics study approaches were used to identify genetic variations associated with drug-response phenotypes in study populations: (i) GWA studies are based on an agnostic approach in which more than 100,000 genetic variants that scan the entire genome are each tested for a significant association with a phenotypic trait such as an adverse drug reaction in a study cohort; (ii) candidate gene studies are based on a priori knowledge that is used to select a limited number of genetic variants and associate them with a phenotypic trait such as drug response in a study cohort; and (iii) in prospective clinical trials, patients are randomly assigned to one of several treatment groups; the treatments are initiated, and the results from the different treatments groups are compared at a predetermined end point.
| Drugs | Label sections | Genetic test (germline variants; recommendation in drug label) | Types of reactions | Types of pharmacogenomics research (GWA studies/candidate gene studies/clinical trial) | Clinical guidelines |
|---|---|---|---|---|---|
| Abacavir | Boxed Warning, Warnings and Precautions | HLA-B*5701; screening for the HLA-B*5701 allele is recommended. | HLA-B-*5701 allele; high risk of hypersensitivity to abacavir. | Prospective clinical trial (43) | FDA-approved drug label (33) |
| Aripiprazole | Dosage and Administration | CYP2D6 activity; the Dosage and Administration section of the label says that “dosing for CYP2D6 poor-metabolizers should initially be reduced to one-half of the usual dose and then adjusted to achieve a favorable clinical response.” | CYP2D6 poor-metabolizers have 80% increase blood aripiprazole concentrations relative to controls. Elderly poor-metabolizers have increased risk of death. Patients with major depressive disorders may experience worsening of their depression or suicidal ideation and behavior. | None of the above | (33); variants in CYP2D6 could be found in (44, 45). |
| Azathioprine 6-mercaptopurinethioguanine | Dosage and Administration, Warnings and Precautions | Thiopurine methyltransferase (TPMT) genomic variants (TPMT*2, TPMT*3A, and TPMT*3C) and TPMT activity; genetic and phenotypic testing of TPMT can be used to identify patients with absent or reduced TPMT activity but cannot replace complete blood count monitoring in patients. | Myelosuppression (low activity of the bone marrow resulting in low red blood cells, white blood cells, and platelets) in patients with an inherited deficiency of TPMT. Lower dose is recommended in patients with reduced TPMT activity. | Candidate gene study (46–48) | (32, 33) |
| Carbamezepine | Carbamezepine Boxed Warning, Warnings and Precautions | HLA-B*1502; FDA recommends that patients of Chinese ancestry be genotyped for this variant prior to initiating treatment. | HLA-B*1502 allele; risk of developing Stevens-Johnson syndrome (inflammatory eruption of the skin and mucous membranes) or toxic epidermal necrolysis. | Candidate gene study (20) | FDA-approved drug label |
| Clopidogrel | Boxed Warning, Warnings and Precautions, Dosage and Administration | CYP2C19 variants; Boxed Warning drug label section says that “a test is available to identify the CYP2C19 genotype and can be used as an aid in determining therapeutic strategy.” | CYP2C19*2 and CYP2C19*3 alleles yield poor-metabolizers who display lower active metabolite exposure, lower inhibition of platelet aggregation, and higher cardiovascular event rates after acute coronary syndrome or percutaneous coronary intervention, relative to normal metabolizers. | Candidate gene and GWA studies (3, 49) | (33, 50) |
| Irinotecan | Irinotecan Dosage and Administration, Warnings and Precautions | UGT1A1*28 allele; Warnings and Precautions section of the drug label section says that patients with this allele are at greater risk for neutropenia. | UGT1A1*28 allele results in reduced UGT1A1 enzyme activity and increased risk for neutropenia. | Candidate gene study (51) | (33) |
| Ivacaftor | Indications and Usage | cystic fibrosis transmembrane conductance regulator gene (CFTR); this drug is approved for cystic fibrosis patients (>6 years old) with the G551D mutation in CFTR (occurs in 4 to 5% of patients with cystic fibrosis) (2, 52). | Patients who are homozygous for the CFTR F508del mutation will not respond to this drug. | Prospective clinical trial (2) | FDA-approved drug label |
| Tetrabenazine | Tetrabenazine Dosage and Administration, Warnings and Precautions | CYP2D6 activity; the Dosage and Administration section of the label says that “before prescribing a daily dose of tetrabenazine that is greater than 50 mg per day, it is necessary to determine whether patients are poor, extensive, or intermediate metabolizers.” | May increase the risk of depression and suicidal thoughts and behavior. Other Warnings and Precautions are QTc prolongation, sedation, fatigue, and akathisia. | None of the above | FDA-approved drug label; CYP2D6 variants can be found in (44, 45). |
| Warfarin | Dosage and Administration, Warnings and Precautions | CYP2C9 (CYP2C9*2 or CYP2C9*3) and VKORC1 variants (−1639G>A); FDA recommends that genotype information can assist in selection of the starting dose | CYP2C9*2 or CYP2C9*3 alleles are associated with increased risk of bleeding. The VKORC (−1639G>A) variant is associated with lower warfarin dose requirements | Candidate gene and GWA studies (23, 29, 53) | (24); warfarin algorithm* |
Warfarin algorithms: 1, www.warfarindosing.org/Source/Home.aspx; 2, (25); and 3, (23).
However, although a variety of prescription drug labels have been modified to include genetic information, many products for which genetic information is reasonably well documented in the scientific literature do not yet contain this information in their labels. For example, information about genotypes that increase one’s risk for (i) high-dose simvastatin-induced myopathy, (ii) aromatase inhibitor–induced musculoskeletal adverse events, and (iii) liver toxicity caused by various drugs is not included in the relevant drug labels. Clearly, vigilant survey with careful attention on the quality of the evidence is needed by regulatory agencies in order to update product labels to include well documented genetic information.
Doctor’s orders
Another bottleneck to clinical use of pharmacogenomics data occurs in the doctor’s office owing to a lack of familiarity with the field of pharmacogenomics on the part of the physician. Although FDA has changed the labels of many drugs to include relevant genomic information, few clinicians make use of these data when prescribing medications or selecting drug doses for treatment. FDA label changes are neither necessary nor sufficient to change medical practice. Drugs are widely prescribed off-label, and many in vitro diagnostic tests (IVDs) are marketed (and reimbursed) without FDA approval as laboratory-developed tests (LDTs). Conversely, changes to labels that were made to include genomic polymorphism data are considered to be largely informational rather than mandates for changes in prescribing practices—unless the new information is elevated to a boxed warning (for example, in the case of abacavir). Similarly, the absence of an FDA label change should not constitute a barrier to the use of genetic or genomic test data to inform drug therapy, particularly if the clinical evidence is substantive.
Additional barriers to the use of genomic information in patient care are the limited availability and current costs of pharmacogenomic testing, uncertainty regarding reimbursement, reluctance to delay prescribing until a test result is obtained, and physician apprehension regarding the interpretation of results. These larger issues will not be readily addressed or given appropriate priority by researchers, insurers, or policy-makers if the relative importance of pharmacogenomic testing is judged in the context of IVDs. As noted above, because pharmacogenomic testing for a particular genetic variant needs to only be performed once, the commercial potential of individual pharmacogenomic tests is limited, this is in contrast to often-repeated IVDs such as those for viral load (for example, HIV) or hemoglobin A1C (to monitor diabetic patients).
Success stories
Despite these substantial barriers to the routine implementation of pharmacogenomic testing data in patient care, there is at least one notable success. Much of the progress in bringing abacavir-related pharmacogenomics analysis into clinical practice can be attributed to a partnership between academic investigators and the drug’s manufacturer, GlaxoSmithKline (GSK). Specifically, GSK funded a large randomized clinical trial (1956 patients) that compared standard-of-care prescribing of abacavir with prospective screening for the HLA-B*5701 variants in patients before abacavir treatment decisions were made. In the screening arm of the trial, if individuals had the HLA-B*5701 variant, abacavir was not prescribed, whereas the drug was used in individuals that did not carry the genetic variant. The standard-of-care arm involved no genotyping. The trial results demonstrated that immunologically confirmed hypersensitivity reactions to abacavir could be eliminated. It was noted that the HLA-B*5701 had a 100% negative predictive power—that is, individuals without the allele did not experience the adverse drug event. Further, the positive predictive power was also high (50%). Reduction of severe adverse drug reactions by 50% should greatly improve the quality of life of patients at risk and contribute to reduced medical costs. The high positive and negative predictive power of the test may have contributed to the wide use of this test in prescribing abacavir. The FDA issued a black box warning in 2008 recommending genetic testing before prescribing abacavir. Genetic testing for HLA-*1502 is also being done before prescribing carbamazepine. Other examples of FDA recommendations are presented on at the FDA Web site (www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm).
Failure to launch
As one of the most disappointing translational failures, the anticoagulant drug warfarin widely used to prevent strokes and pulmonary embolism (Fig. 2) is the subject of many academic studies (23– 25), most of which support genotype-based prescribing. Unfortunately, this research has resulted in comparatively little clinical uptake (26–28). It has long been known that the plasma levels of S-warfarin (the active enantiomer) are increased in CYP2C9 poor-metabolizers, which can be tested through genotyping of CYP2C9 (Fig. 2). More recently, individuals with variants of VKORC1 (vitamin K epoxide reductase complex subunit 1)— the target molecule of warfarin, which is involved in vitamin K recycling—have been shown to have increased sensitivity to the effects of warfarin (29). These studies imply that such patients with variants in both genes, VKORC1 and CYP2C9, would require lower doses of warfarin (Fig. 2) (23). Numerous investigators have studied the association of warfarin sensitivity with genetic variations in the CYP2C9 and VKORC1 genes by assessing the weekly doses required to observe an appropriate therapeutic effect on blood clotting.
Fig. 2. Personalized medicine.

Shown are the effects of the CYP2C9 and VKORC1 genomic variants on warfarin pharmacokinetics and pharmacodynamics in human subjects.
On the basis of these findings, specific treatment guidelines have been published (24, 25). However, the vast majority (more than 80%) of prescribing physicians do not perform genetic testing before prescribing warfarin, and concerns have been raised regarding the clinical utility and cost-effectiveness of genomic testing–based prescribing of warfarin (28). In particular, the opponents of genetic testing claim that because blood-clotting measures (for example, INR) are routinely monitored to guide warfarin dosing, performance of genetic testing, which is not directly related to blood clotting, is unnecessary. Proponents claim that genetic testing would aid in selection of the first dose, which is currently based on age, gender, and ethnicity, and therefore reduce the number of life-threatening bleeding events that occur before INR measurements have been made. In addition, dabigatran and ximelagatran direct inhibitors of thrombin—a coagulation factor that catalyzes the conversion of fibrinogen to the clotting protein fibrin—recently were demonstrated to be superior to warfarin for some indications such as strokes and embolism (30). Expansion of dabigatran prescribing in patient populations likely will stifle additional studies on genomic testing– based prescribing of warfarin.
At the heart of the warfarin controversy and indeed the controversy about use of pharmacogenomic testing in clinical trials is the issue of level of evidence. In particular, there is disagreement about the level of evidence that should be required for implementation of a genetic test in clinical practice and reimbursement by third parties. It is argued that before a genetic test is used in medical practice, a clinical trial must be conducted to show that the test improves outcome and is cost-effective. Because clinical trials are often lengthy and costly, and there is little motivation for industry to support the trial, such trials are often not performed. Accordingly, Altman has proposed that a standard of “noninferiority” be adopted for implementation of genetic testing in clinical practice (31). Noninferiority implies that the genetic testing before drug selection is no worse than the current prescribing practice, which does not involve genetic testing. The argument for noninferiority is based on low costs of genetic tests and low risk to the patients. We agree with Altman and support the idea of immediate implementation of genetic testing for many drugs in which a compelling body of literature exists to support the use of genetic testing.
TRANSLATIONAL PARTNERSHIPS
The relative success of genomic testing–based prescribing of abacavir versus warfarin illustrates the importance of commercial support, both financially and administratively. Because use of abacavir was important to GSK, the company invested in a large randomized trial that was successfully completed. For drugs without an interested sponsor, randomized trials would need to be supported by public or foundation funding, which is another barrier to implementation. More importantly, it is not feasible from a public and societal perspective to perform prospective randomized trials for every drug-gene interaction.
Large-scale implementation of pharmacogenomic testing in patient care will require a quantum leap from the current model of genotyping before prescribing a particular drug of interest. Economic barriers will need to be eliminated, allowing a focus on utility rather than cost-effectiveness. A transformation of this magnitude most likely will involve the application of preemptive genotyping or whole-genome sequencing and incorporation of genetic information into the patients’ electronic medical records, which can then be used for future genomic information–based medical decisions. The U.S. National Institutes of Health (NIH) is now funding new initiatives to facilitate the introduction of whole-genome information into electronic medical records, and the commercial sector is gearing up to develop software to aid in analyses of these data. The NIH Pharmacogenomics Research Network (PGRN) is driving a large effort to draft evidence-based guidelines that outline how clinicians should use genetic information to inform the selection and dosing of medications (32). Furthermore, simple algorithms are presented for prescribing medications on the basis of genetic and demographic information and for selecting appropriate drug doses that yield optimal therapy. Guidelines have been published for dosing of warfarin, clopidogrel, thiopurines, and other commonly prescribed drugs in a series of papers that includes references to the primary research (Table 1) (32, 33). These guidelines will need to be encoded into “user-friendly” decision support tools for physicians and coupled to genomic information in electronic medical records to inform clinicians about dosing and drug selection. The guidelines will need to be updated as new information is obtained. Pharmacists who have education in pharmacogenomic testing may be of great assistance in selecting drugs and doses on the basis of genetic information.
Public education, which has played an important role in preventative health care such as mammography and HIV testing, may play an equally important role in the successful use of genetic testing to guide treatment. With increased awareness about pharmacogenomics-based testing, patients may provide genetic information to their physicians to help inform drug product selection. In particular, direct-to-consumer companies have emerged that provide genotyping services for disease-risk and drug-response genes (34, 35). The information provided by these companies may allow consumers to require more personalized medical treatments from their physicians. Unfortunately, the routine clinical application of this information would present substantial practical challenges in most medical centers because guidelines describing how to use the information to inform drug or dose choice are not routinely available. As with all laboratory tests, genetic tests may be prone to errors (36). A recent controversy surrounding genotyping for CYP2D6 polymorphisms in prescribing tamoxifen underscores problems in genetic testing (37) and in particular genotyping from tumor DNA, which is known to harbor gene deletions, amplifications, loss of heterozygosity, and others. Because use of genetic tests for patient care and not research purposes requires very high accuracy, the test should be conducted in Clinical Laboratory Improvement Amendments–certified laboratories, which will help to reduce genotyping errors as much as possible.
It has been suggested that pharmacogenomics will be among the first broad clinical applications of genomics research. But for pharmacogenomics to fulfill its destiny, researchers must expand the small numbers of pharmacogenomic studies that have taken advantage of genome-wide methods. Consortia such as the Global Alliance in Pharmacogenomics formed between the NIH PGRN and the Center for Genomic Medicine, RIKEN, are needed to increase the numbers of such studies and to enhance sample collection, replication, and population diversity. It important to acknowledge, however, that sample sizes for pharmacogenomic studies will never be as large as those for studies of disease risk, and clinical trial replication presents a particular problem. Randomized clinical trials take years to complete, are expensive, and at least in the pharmacogenomic–drug interaction realm, are rarely replicated precisely; yet, such trials are the best source of samples for the study of drug-response phenotypes.
However, pharmacogenomic studies also have considerable strengths. As noted, SNPs with high odds ratios are more likely to be found in pharmacogenomic GWA studies than in those of disease risk. Further, a large body of knowledge exists with respect to drug metabolism and mechanisms of action in humans, making drugs powerful molecular probes of human biology. For example, small-molecule inhibitors of specific drug-metabolizing enzymes and transporters are approved drugs and can be used to probe the function of enzymes and transporters in studies of human biology. Therefore, even though every effort must be made to identify appropriate pharmacogenomic replication studies, researchers should also look to functional validation and mechanistic insights provided by our knowledge of drug effects as additional ways to enhance our confidence in signals identified during genome-wide pharmacogenomic studies. It would be unwise to delay investigations into the mechanistic bases of these signals until one has identified the inevitable false positives because the purpose of genome-wide pharmacogenomic studies is to advance our understanding of mechanisms of human physiology and pathophysiology. The emerging field of systems pharmacology will inform gene-by-drug interaction studies and expand our understanding of pharmacological pathways such as those posted in the Pharmacogenetics and Pharmacogenomics Knowledgebase (www.pharmGKB.org). Just as one-size-fits-all is not the ideal approach for the selection of a drug or drug dose— especially when we consider the enormous genetic variation among individuals—such a strategy also is not ideal for the pursuit of all genome-wide pharmacogenomic signals, especially if functional validation and mechanistic pursuit provide biological insights or biomarkers for drug response (15, 38, 39). Other supporting studies may include use of medical informatics to harvest data from the electronic medical records or chemoinformatics to identify drugs with similar properties, which may be expected to cause similar adverse effects (40–42).
For the pharmacogenomics field to advance, it is crucial that manuscript reviewers and journal editors recognize that replication of findings from pharmacogenomic GWA studies is intrinsically difficult and, in some cases, not feasible. Because of the large effect sizes of SNPs identified in GWA studies of pharmacogenomic phenotypes, the need for replication may be less fundamental than it is in GWA studies of disease.
Pharmacogenomics research and development has the potential both to fine-tune and transform medical care, potentially in the not-too-distant future. As our understanding of drug-genome interactions grows, regulatory agencies will enhance their requirements for the inclusion of genomic information in product labels. Use of electronic medical records that contain genomic information will expand. User-friendly physician-decision support systems to interface between medical practitioners and the electronic medical records will be used to guide clinicians in using genomic information in the selection of drugs and doses.
Acknowledgments
Funding: We acknowledge funding from the U.S. National Institutes of Health and the U.S. National Institute of General Medical Sciences (GM61390, GM061393, GM61388). Competing interests: K. M. G. and S. W. Y. declare that they have no competing financial interests. M. J. R. is a co-inventor on multiple pending and issued patents related to pharmacogenetic diagnostics and receives royalties related to UGT1A1 genotyping.
REFERENCES AND NOTES
- 1.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. SEARCH Collaborative Group, SLCO1B1 variants and statin-induced myopathy— A genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 2.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS. VX08-770-102 Study Group, A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB, Damcott CM, Pakyz R, Tantry US, Gibson Q, Pollin TI, Post W, Parsa A, Mitchell BD, Faraday N, Herzog W, Gurbel PA. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302:849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, Fritz P, Simon W, Suman VJ, Ames MM, Safgren SL, Kuffel MJ, Ulmer HU, Boländer J, Strick R, Beckmann MW, Koelbl H, Weinshilboum RM, Ingle JN, Eichelbaum M, Schwab M, Brauch H. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA. 2009;302:1429–1436. doi: 10.1001/jama.2009.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe’er I, Floratos A, Daly MJ, Goldstein DB, John S, Nelson MR, Graham J, Park BK, Dillon JF, Bernal W, Cordell HJ, Pirmohamed M, Aithal GP, Day CP. DILIGEN Study, International SAE Consortium, HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, Ito Y, Mita E, Tanaka E, Mochida S, Murawaki Y, Honda M, Sakai A, Hiasa Y, Nishiguchi S, Koike A, Sakaida I, Imamura M, Ito K, Yano K, Masaki N, Sugauchi F, Izumi N, Tokunaga K, Mizokami M. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 8.Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, Wright TM, Meyer J, Paulding CA. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42:711–714. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 9.Ozeki T, Mushiroda T, Yowang A, Takahashi A, Kubo M, Shirakata Y, Ikezawa Z, Iijima M, Shiohara T, Hashimoto K, Kamatani N, Nakamura Y. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20:1034–1041. doi: 10.1093/hmg/ddq537. [DOI] [PubMed] [Google Scholar]
- 10.McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, Carrington M, Sills GJ, Marson T, Jia X, de Bakker PI, Chinthapalli K, Molokhia M, Johnson MR, O’Connor GD, Chaila E, Alhusaini S, Shianna KV, Radtke RA, Heinzen EL, Walley N, Pandolfo M, Pichler W, Park BK, Depondt C, Sisodiya SM, Goldstein DB, Deloukas P, Delanty N, Cavalleri GL, Pirmohamed M. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364:1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fellay J, Thompson AJ, Ge D, Gumbs CE, Urban TJ, Shianna KV, Little LD, Qiu P, Bertelsen AH, Watson M, Warner A, Muir AJ, Brass C, Albrecht J, Sulkowski M, McHutchison JG, Goldstein DB. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010;464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka Y, Kurosaki M, Nishida N, Sugiyama M, Matsuura K, Sakamoto N, Enomoto N, Yatsuhashi H, Nishiguchi S, Hino K, Hige S, Itoh Y, Tanaka E, Mochida S, Honda M, Hiasa Y, Koike A, Sugauchi F, Kaneko S, Izumi N, Tokunaga K, Mizokami M. Genome-wide association study identified ITPA/DDRGK1 variants reflecting thrombocytopenia in pegylated interferon and ribavirin therapy for chronic hepatitis C. Hum Mol Genet. 2011;20:3507–3516. doi: 10.1093/hmg/ddr249. [DOI] [PubMed] [Google Scholar]
- 13.Tohkin M, Kaniwa N, Saito Y, Sugiyama E, Kurose K, Nishikawa J, Hasegawa R, Aihara M, Matsunaga K, Abe M, Furuya H, Takahashi Y, Ikeda H, Muramatsu M, Ueta M, Sotozono C, Kinoshita S, Ikezawa Z. A whole-genome association study of major determinants for allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J. 2011 doi: 10.1038/tpj.2011.41. [DOI] [PubMed] [Google Scholar]
- 14.Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, Day CP, Ruiz-Cabello F, Donaldson PT, Stephens C, Pirmohamed M, Romero-Gomez M, Navarro JM, Fontana RJ, Miller M, Groome M, Bondon-Guitton E, Conforti A, Stricker BH, Carvajal A, Ibanez L, Yue QY, Eichelbaum M, Floratos A, Pe’er I, Daly MJ, Goldstein DB, Dillon JF, Nelson MR, Watkins PB, Daly AK. Spanish DILI Registry; EUDRAGENE; DILIN; DILIGEN; International SAEC, Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011;141:338–347. doi: 10.1053/j.gastro.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingle JN, Schaid DJ, Goss PE, Liu M, Mushiroda T, Chapman JA, Kubo M, Jenkins GD, Batzler A, Shepherd L, Pater J, Wang L, Ellis MJ, Stearns V, Rohrer DC, Goetz MP, Pritchard KI, Flockhart DA, Nakamura Y, Weinshilboum RM. Genome-wide associations and functional genomic studies of musculoskeletal adverse events in women receiving aromatase inhibitors. J Clin Oncol. 2010;28:4674–4682. doi: 10.1200/JCO.2010.28.5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uher R, Perroud N, Ng MY, Hauser J, Henigsberg N, Maier W, Mors O, Placentino A, Rietschel M, Souery D, Za-gar T, Czerski PM, Jerman B, Larsen ER, Schulze TG, Zobel A, Cohen-Woods S, Pirlo K, Butler AW, Muglia P, Barnes MR, Lathrop M, Farmer A, Breen G, Aitchison KJ, Craig I, Lewis CM, McGuffin P. Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am J Psychiatry. 2010;167:555–564. doi: 10.1176/appi.ajp.2009.09070932. [DOI] [PubMed] [Google Scholar]
- 17.Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA, Li X, Wilke RA, Rieder MJ, Williams PT, Ridker PM, Chatterjee A, Rotter JI, Nickerson DA, Stephens M, Krauss RM. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS ONE. 2010;5:e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aberg K, Adkins DE, Bukszár J, Webb BT, Caroff SN, Miller DD, Sebat J, Stroup S, Fanous AH, Vladimirov VI, McClay JL, Lieberman JA, Sullivan PF, van den Oord EJ. Genomewide association study of movement-related adverse antipsychotic effects. Biol Psychiatry. 2010;67:279–282. doi: 10.1016/j.biopsych.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, Burch L, Carr F, Strange A, Freeman C, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Craddock N, Deloukas P, Dronov S, Duncanson A, Edkins S, Gray E, Hunt S, Jankowski J, Langford C, Markus HS, Mathew CG, Plomin R, Rautanen A, Sawcer SJ, Samani NJ, Trembath R, Viswanathan AC, Wood NW, Harries LW, Hattersley AT, Doney AS, Colhoun H, Morris AD, Sutherland C, Hardie DG, Peltonen L, McCarthy MI, Holman RR, Palmer CN, Donnelly P, Pearson ER. GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group; Wellcome Trust Case Control Consortium 2; MAGIC investigators, Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43:117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, Chen YT. Medical genetics: A marker for Stevens-Johnson syndrome. Nature. 2004;428:486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 21.Pirmohamed M, Aithal GP, Behr E, Daly A, Roden D. The phenotype standardization project: Improving pharmacogenetic studies of serious adverse drug reactions. Clin Pharmacol Ther. 2011;89:784–785. doi: 10.1038/clpt.2011.30. [DOI] [PubMed] [Google Scholar]
- 22.Burchard EG, Ziv E, Coyle N, Gomez SL, Tang H, Karter AJ, Mountain JL, Pérez-Stable EJ, Sheppard D, Risch N. The importance of race and ethnic background in biomedical research and clinical practice. N Engl J Med. 2003;348:1170–1175. doi: 10.1056/NEJMsb025007. [DOI] [PubMed] [Google Scholar]
- 23.Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, Limdi NA, Page D, Roden DM, Wagner MJ, Caldwell MD, Johnson JA. International Warfarin Pharmacogenetics Consortium, Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009;360:753–764. doi: 10.1056/NEJMoa0809329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JA, Gong L, Whirl-Carrillo M, Gage BF, Scott SA, Stein CM, Anderson JL, Kimmel SE, Lee MT, Pirmohamed M, Wadelius M, Klein TE, Altman RB. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90:625–629. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gage BF, Eby C, Johnson JA, Deych E, Rieder MJ, Ridker PM, Milligan PE, Grice G, Lenzini P, Rettie AE, Aquilante CL, Grosso L, Marsh S, Langaee T, Farnett LE, Voora D, Veenstra DL, Glynn RJ, Barrett A, McLeod HL. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84:326–331. doi: 10.1038/clpt.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kangelaris KN, Bent S, Nussbaum RL, Garcia DA, Tice JA. Genetic testing before anticoagulation? A systematic review of pharmacogenetic dosing of warfarin. J Gen Intern Med. 2009;24:656–664. doi: 10.1007/s11606-009-0949-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flockhart DA, O’Kane D, Williams MS, Watson MS, Flockhart DA, Gage B, Gandolfi R, King R, Lyon E, Nussbaum R, O’Kane D, Schulman K, Veenstra D, Williams MS, Watson MS. ACMG Working Group on Pharmacogenetic Testing of CYP2C9, VKORC1 Alleles for Warfarin Use, Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med. 2008;10:139–150. doi: 10.1097/GIM.0b013e318163c35f. [DOI] [PubMed] [Google Scholar]
- 28.Kadafour M, Haugh R, Posin M, Kayser SR, Shin J. Survey on warfarin pharmacogenetic testing among anticoagulation providers. Pharmacogenomics. 2009;10:1853–1860. doi: 10.2217/pgs.09.117. [DOI] [PubMed] [Google Scholar]
- 29.Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, Blough DK, Thummel KE, Veenstra DL, Rettie AE. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005;352:2285–2293. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal S, Hachamovitch R, Menon V. Current trial associated outcomes with warfarin in prevention of stroke in patients with nonvalvular atrial fibrillation: A meta-analysis. Arch Intern Med. 2012;172:623–631. doi: 10.1001/archinternmed.2012.121. discussion 631–633. [DOI] [PubMed] [Google Scholar]
- 31.Altman RB. Pharmacogenomics: “Noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther. 2011;89:348–350. doi: 10.1038/clpt.2010.310. [DOI] [PubMed] [Google Scholar]
- 32.Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, Stein CM, Carrillo M, Evans WE, Klein T48E. Clinical Pharmacogenetics Implementation Consortium, Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swen JJ, Wilting I, de Goede AL, Grandia L, Mulder H, Touw DJ, de Boer A, Conemans JM, Egberts TC, Klungel OH, Koopmans R, van der Weide J, Wilffert B, Guchelaar HJ, Deneer VH. Pharmacogenetics: From bench to byte. Clin Pharmacol Ther. 2008;83:781–787. doi: 10.1038/sj.clpt.6100507. [DOI] [PubMed] [Google Scholar]
- 34.Prainsack B, Wolinsky H. Direct-to-consumer genome testing: Opportunities for pharmacogenomics research? Pharmacogenomics. 2010;11:651–655. doi: 10.2217/pgs.10.33. [DOI] [PubMed] [Google Scholar]
- 35.Kaufman DJ, Bollinger JM, Dvoskin RL, Scott JA. Risky business: Risk perception and the use of medical services among customers of DTC personal genetic testing. J Genet Couns. 2012;21:413–422. doi: 10.1007/s10897-012-9483-0. [DOI] [PubMed] [Google Scholar]
- 36.Kaiser J. Clinical medicine. Biomarker tests need closer scrutiny, IOM concludes. Science. 2012;335:1554. doi: 10.1126/science.335.6076.1554. [DOI] [PubMed] [Google Scholar]
- 37.Regan MM, Leyland-Jones B, Bouzyk M, Pagani O, Tang W, Kammler R, Dell’orto P, Biasi MO, Thürlimann B, Lyng MB, Ditzel HJ, Neven P, Debled M, Maibach R, Price KN, Gelber RD, Coates AS, Goldhirsch A, Rae JM, Viale G. Breast International Group (BIG) 1-98 Collaborative Group, CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: The breast international group 1-98 trial. J Natl Cancer Inst. 2012;104:441–451. doi: 10.1093/jnci/djs125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L. FKBP51 regulation of AKT/protein kinase B phosphorylation. Curr Opin Pharmacol. 2011;11:360–364. doi: 10.1016/j.coph.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wheeler HE, Dolan ME. Lymphoblastoid cell lines in pharmacogenomic discovery and clinical translation. Pharmacogenomics. 2012;13:55–70. doi: 10.2217/pgs.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tatonetti NP, Ye PP, Daneshjou R, Altman RB. Data-driven prediction of drug effects and interactions. Sci Transl Med. 2012;4:125ra31. doi: 10.1126/scitranslmed.3003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu T, Altman RB. Using multiple microenvironments to find similar ligand-binding sites: Application to kinase inhibitor binding. PLOS Comput Biol. 2011;7:e1002326. doi: 10.1371/journal.pcbi.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatonetti NP, Denny JC, Murphy SN, Fernald GH, Krishnan G, Castro V, Yue P, Tsau PS, Kohane I, Roden DM, Altman RB. Detecting drug interactions from adverse-event reports: Interaction between paroxetine and pravastatin increases blood glucose levels. Clin Pharmacol Ther. 2011;90:133–142. doi: 10.1038/clpt.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, Jägel-Guedes E, Rugina S, Kozyrev O, Cid JF, Hay P, Nolan D, Hughes S, Hughes A, Ryan S, Fitch N, Thorborn D, Benbow A. PREDICT-1 Study Team, HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 44.Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. doi: 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- 45.Crews KR, Gaedigk A, Dunnenberger HM, Klein TE, Shen DD, Callaghan JT, Kharasch ED, Skaar TC. Clinical Pharmacogenetics Implementation Consortium, Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther. 2012;91:321–326. doi: 10.1038/clpt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, Pui CH, Evans WE. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 47.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 48.Lennard L, Van Loon JA, Weinshilboum RM. Pharmacogenetics of acute azathioprine toxicity: Relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther. 1989;46:149–154. doi: 10.1038/clpt.1989.119. [DOI] [PubMed] [Google Scholar]
- 49.Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias W, Braunwald E, Sabatine MS. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 50.Scott SA, Sangkuhl K, Gardner EE, Stein CM, Hulot JS, Johnson JA, Roden DM, Klein TE, Shuldiner AR. Clinical Pharmacogenetics Implementation Consortium, Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther. 2011;90:328–332. doi: 10.1038/clpt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 52.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ash-lock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cooper GM, Johnson JA, Langaee TY, Feng H, Stanaway IB, Schwarz UI, Ritchie MD, Stein CM, Roden DM, Smith JD, Veenstra DL, Rettie AE, Rieder MJ. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112:1022–1027. doi: 10.1182/blood-2008-01-134247. [DOI] [PMC free article] [PubMed] [Google Scholar]