

Abstract
Objective
Split-hand/foot malformation type 1 is an autosomal dominant condition with reduced penetrance and variable expression. We report three individuals from two families with split-hand/split-foot malformation (SHFM) in whom next generation sequencing was performed to investigate the cause of their phenotype.
Methods and results
The first proband has a de novo balanced translocation t(2;7)(p25.1;q22) identified by karyotyping. Whole genome sequencing showed that the chromosome 7 breakpoint is situated within the SHFM1 locus on chromosome 7q21.3. This separates the DYNC1I1 exons recently identified as limb enhancers in mouse studies from their target genes, DLX5 and DLX6. In the second family, X-linked recessive inheritance was suspected and exome sequencing was performed to search for a mutation in the affected proband and his uncle. No coding mutation was found within the SHFM2 locus at Xq26 or elsewhere in the exome, but a 106 kb deletion within the SHFM1 locus was detected through copy number analysis. Genome sequencing of the deletion breakpoints showed that the DLX5 and DLX6 genes are disomic but the putative DYNC1I1 exon 15 and 17 enhancers are deleted.
Conclusions
Exome sequencing identified a 106 kb deletion that narrows the SHFM1 critical region from 0.9 to 0.1 Mb and confirms a key role of DYNC1I1 exonic enhancers in normal limb formation in humans.
Keywords: Clinical Genetics, Copy-Number, Genetics, Chromosomal
Split-hand/split-foot malformation is a congenital limb abnormality characterised by the absence of one or more median rays or digits that results in cone-shaped clefts of hands and/or feet. It is often accompanied by other limb anomalies including monodactyly, syndactyly, and aplasia or hypoplasia of the phalanges, metacarpals, and metatarsals. The malformation can be isolated or syndromic (intellectual disability in 33%, craniofacial malformations in >35%, and deafness in 35%; split-hand/split-foot malformation 1D (SHFM1D), OMIM #220600), and the severity can be variable between patients as well as between different limbs of the same individual. The condition is genetically heterogeneous with six loci defined, including the SHFM1 locus 7q21 (OMIM #183600) and SHFM2 locus Xq26 (OMIM #313350). The 7q21 locus contains the candidate SHFM genes distal-less homeobox 5 and 6 (DLX5/6); double-knockout mice exhibit severe skeletal abnormalities and die shortly after birth,1 while a homozygous missense mutation in DLX5 has recently been reported to cause split-hand/foot malformation and hearing loss in a consanguineous family.2 No other known DLX5/6 coding mutations leading to SHFM1 have been reported to date. Several chromosomal abnormalities have been reported in this region that lead to SHFM1; some do not affect the DLX5/6 genes and are thought to disrupt one or more regulatory elements, or result in a physical separation of such elements from the most likely target genes, DLX5/6. The smallest reported deletion is of 880 kb encompassing SLC25A13 and part of DYNC1I1, but leaving DLX5 and DLX6 intact.3 In the developing limb, both Dlx5 and Dlx6 are thought to be regulated by the transcription factor tumour protein p634 and TP63 mutations cause split-hand/split-foot malformation 4 (SHFM4; OMIM #605289). An enhancer element with a p63 binding site has been reported within the SHFM1 locus 300 kb proximal to Dlx5/6.3
Recent studies in mouse and zebrafish models identified novel tissue-specific enhancers that correlate with limb, craniofacial and hearing phenotypes observed in individuals with chromosomal rearrangements.5 6 Two of these enhancers are located within the coding exons 15 and 17 of Dync1i1 (dynein, cytoplasmic 1, intermediate chain 1), a gene that is not expressed during limb development. DYNC1I1 eExon (exonic enhancer) 15 is marked by an enhancer chromatin signature and physically interacts with the Dlx5/6 promoter regions 900 kb distal to DYNC1I1, specifically in the limb.5 An additional multi-tissue enhancer was identified in an intron of Slc25a13 (solute carrier family 25 member 13), driving the expression of Dlx5/6 in otic vesicle, forebrain, branchial arch and limb—a pattern that correlates with some but not all SHFM1 phenotypes observed in humans.6
We studied three patients with SHFM from two families. Informed consent was obtained from all participants or their parents. Proband 1 had split-hand/split-foot malformation (figure 1A) affecting three limbs, and sensorineural deafness. Array comparative genomic hybridisation (CGH) analysis indicated normal dosage, but subsequent karyotyping showed a de novo balanced translocation (2;7)(p25.1;q22). Paired-end whole genome sequencing was performed on a single lane of Illumina HiSeq 2000 to map the translocation breakpoints. We obtained average coverage of eight reads per base, and looked for reads where one of the pair mapped at or near the chr2 region, and the other at or near the chr7 region. We identified seven such split pair reads, and a further six reads mapping directly over the translocation breakpoints (see online supplementary figure 1A) which showed that the chromosome 7 breakpoint is within 7q21.3, not 7q22. The precise breakpoints were confirmed by Sanger sequencing of PCR products spanning the breakpoints (see online supplementary figure 1B), and the translocation was defined as t(2;7)(p25.1;q21.3)(oNC_000007.13:g.96229309::CTCTGC::NC_000002.11:g.10585833;NC_000007.13:g.96229314::G::oNC_000002.11:g.10585828). The chromosome 2 breakpoint is located within intron 1 of the ornithine decarboxylase 1 (ODC1) gene encoding the rate-limiting enzyme of the polyamine biosynthesis pathway. A polymorphism in this gene has been associated with colon cancer risk,7 but there are no reported disease-causing mutations. The chromosome 7 breakpoint is situated within the 7q21.3 SHFM1 locus, in the intergenic region 370 kb upstream of DLX5/6 genes and 500 kb downstream of the DYNC1I1 exons 15 and 17 identified as exonic enhancers in mice and zebrafish (figure 2).5
Figure 1.
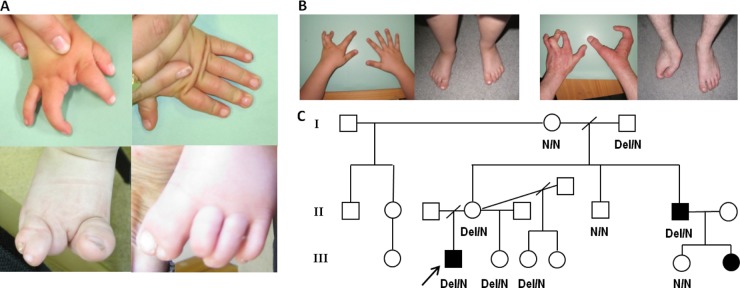
Variable malformation severity and incomplete penetrance in the two split-hand/split-foot malformation 1 (SHFM1) families. (A) Hands and feet of the family 1 patient with a de novo translocation. The right hand has a deficient central ray (upper left image). The left hand is normal (upper right image). The right foot shows deficiency of the second and third rays, partial syndactyly of toes 4–5, and a large great toe (lower left). The left foot shows syndactyly of toes 1–2, and deficiency of the terminal phalanx of the third toe with anonychia (lower right). (B) Hands and feet of the family 2 patients with the deletion. In the proband, both feet and the right hand are normal. In the left hand, the central ray is deficient, and in the ring finger there is camptodactyly at the proximal interphalangeal joint (left image). The proband's maternal uncle is severely affected, with the exception of his left foot, which is normal. His left hand is similar to that of his affected nephew, with deficiency of the central ray, camptodactyly of the ring finger, and some shortening and stiffness of the second (index) finger. In the right hand the second and third rays are absent and there is fusion/syndactyly of rays 4–5 with a single digit. The right foot is also severely affected, with absence of the second and third rays, and fusion of rays 4–5 with a single digit (right image). The uncle's affected daughter (no images) has deficiency of the central ray of the right hand and right foot, with the left hand and left foot unaffected. The right hand closely resembles the left hands of her father and proband. (C) Family 2 pedigree and the results of junction fragment PCR sequencing of both affected (black shapes) and unaffected (white shapes) family members, showing incomplete penetrance of the mutation. The two individuals in whom we performed exome sequencing are the proband (black arrow) and his affected uncle. After the deletion was identified, a third individual (the first female affected in this family) was diagnosed with suspected split-hand/split-foot malformation on routine antenatal ultrasound. She has missing central rays (third digit) of the right hand and right foot only.
In family 2 the proband and his affected uncle have isolated split-hand/split-foot malformation affecting one or three limbs (figure 1B). The uncle has complained of mild hearing difficulty for about 5 years and an audiogram revealed unilateral high tone loss; however, he used to work in a machine tool environment, which may have been the cause. Array CGH analysis using an Agilent 44K oligo array was normal and the mode of inheritance was thought most likely to be X-linked recessive. We performed exome sequencing of both affected individuals to look for shared mutations within the putative SHFM2 region at Xq26.3, but failed to find a likely causal mutation at Xq26 or elsewhere within the exome. We then carried out copy number analysis of the entire chromosome 7 using the method published by Nord et al8 and identified a single shared ∼100 kb deletion on 7q21, partially encompassing exons 11–18 of the SLC25A13 gene and exons 14–17 of the DYNC1I1 gene (see online supplementary figure 1C). Biallelic mutations in SLC25A13 cause citrin deficiency, an adult- or neonatal-onset metabolic disorder, while homozygous Slc25a13 knock-out mice show no skeletal defects9; hence a heterozygous partial deletion of this gene is unlikely to be associated with SHFM1 phenotype. The DYNC1I1 gene encodes the intermediate chain 1 subunit of the cytoplasmic dynein motor protein complex, the primary motor protein responsible for retrograde axonal transport in neurons. Since this gene is not expressed in the developing limb a partial gene deletion is not predicted to cause split-hand/foot malformation. However, the deleted DYNC1I1 exons include the enhancers identified by Birnbaum et al.5 Visual inspection of the reads indicated that the breakpoints were not located within the exons, so in order to accurately map the deletion we performed paired-end whole genome sequencing of the proband. We looked for read pairs with large (>100 kb) insert sizes within the introns on either side of the last deleted exon, and identified four such read pairs, as well as two reads mapping directly across the breakpoints (see online supplementary figure 1D). Sanger sequencing of a PCR product spanning the breakpoints confirmed a 105 935bp deletion; NC_000007.13:g.95704812_95810747del (see online supplementary figure 1E). The presence of the junction fragment was confirmed in the two affected individuals and also in four non-penetrant unaffected family members (figure 1C). Incomplete penetrance of rearrangements affecting the SHFM1 locus has been reported previously.10–13 After the deletion was identified, a third affected individual was diagnosed during routine antenatal scanning (figure 1C). At birth, this baby girl was noted to have a normal left hand and left foot. However, the right hand and right foot were affected to a similar extent, with absence of the central ray and, in the hand, camptodactyly affecting the ring finger, as well as the second (index) finger to a lesser extent.
The 106 kb deletion identified in both affected members of the second family is the smallest deletion identified within the SHFM1 region. It results in haploinsufficiency for most 3′ DYNC1I1 exons 14–17. This result suggests that DYNC1I1 exons 15 and 17 act as tissue-specific enhancers of DLX5/6 expression in humans as well as in mouse and zebrafish. DLX5 and DLX6 are transcription factors essential for epidermal morphogenesis and limb development, and targeted inactivation of the Dlx5/6 gene pair in mice results in severe limb, craniofacial, and axial skeletal defects.1 However, only a single human mutation in DLX5/6 has been identified to date,2 and the reported chromosomal aberrations leading to SHFM1 encompass but the breakpoints do not directly disrupt either of the two genes, suggesting that DLX5/6 cis-regulatory elements play a crucial role in human limb development. Interestingly, the deletion in family 2 does not extend to the p63 binding site in the enhancer identified by Kouwenhoven et al.3
In addition to DYNC1I1 eExons 15 and 17, several other putative mouse and zebrafish enhancers have been identified within the minimal SHFM1 region that extends between the DYNC1I1 and DLX5/6 genes.6 The conserved sequences were shown to drive Dlx5/6 expression in the developing limb, fin, brain, ear, branchial arch (gill and jaw), and genital tissues, and the position of these functional enhancers compared to SHFM1-associated chromosome rearrangements suggests that their disruption might, in some cases, explain the additional clinical phenotypes such as hearing loss and craniofacial defects.6 Within intron 14 of SLC25A13 is an enhancer with activity in the otic vesicle (figure 2) that might explain the sensorineural deafness in proband 1 through the physical separation of this enhancer from the DLX5/6 genes. Although the deletion in family 2 also includes SLC25A13 intron 14, only one of the six individuals with the deletion is reported to have mild hearing difficulties, likely to be coincidental, and no other additional clinical features were noted. It is possible that these enhancers either act in a slightly different way in humans compared to mouse and zebrafish, or there may be some redundancy given that several enhancers have been identified for the same tissue, and single enhancers have been observed in multiple tissues.6
Figure 2.
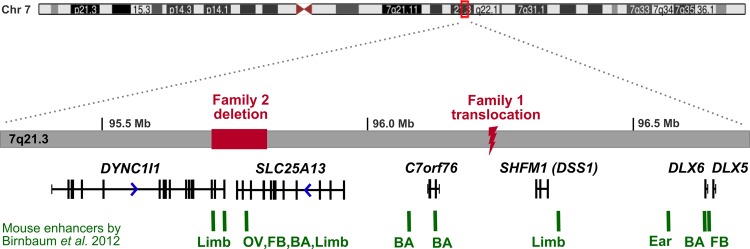
The full SHFM1 locus showing the locations of the two mutations identified in this study relative to the previously published mouse (E11.5) tissue-specific enhancers. The deletion and translocation breakpoints physically separate tissue-specific enhancers from DLX5/6 target genes. BA, Branchial arch, FB, Forebrain; OV, Otic vesicle.
Our whole genome sequencing demonstrates that, even at a relatively low mean coverage (8 reads per base per proband), it is possible to accurately resolve translocation or deletion breakpoints. For proband 1 the genome sequencing was more accurate than metaphase chromosome analysis and resulted in a reassignment of the 7q breakpoint to the neighbouring chromosome band. The application of next generation sequencing for precise characterisation of breakpoints is likely to find clinical utility as it can show whether the coding region of a candidate disease gene is disrupted or, as in this study, identify the spatial relationship between regulatory elements and their target genes.
The use of paired-end whole genome sequencing to map rearrangement breakpoints to the exact nucleotide has previously been described for just seven patients.14–16 In all cases the approximate location of the rearrangement, or at least the chromosomes involved, was already known to the analysts, and one of the next challenges is to be able to identify such rearrangements without the need for prior cytogenetic testing. We used the method developed by Nord et al8 to identify the causative deletion in family 2. A number of different tools have been described for detecting copy number variants (CNVs) from exome data. Some have been successfully applied to discovering CNVs in the general population17 18 or in multifactorial disease cohorts,19 20 but there are very few examples of novel pathogenic deletions causing monogenic disorders detected through normalised read depth analysis of exome sequence data.21
In summary, by using exome sequencing copy number analysis and whole genome sequencing to map deletion and translocation breakpoints, our study demonstrates that exonic enhancers recently discovered through mouse and zebrafish models are also critical for limb development in humans.
Supplementary Material
Acknowledgments
Karen Stals provided technical assistance. SE is a Wellcome Trust Senior Investigator and a member of the Exeter NIHR Clinical Research Facility staff. MNW is supported by Wellcome Trust Biomedical Informatics Hub funding. We thank Konrad Paszkiewicz and colleagues at the University of Exeter Sequencing Service for their assistance with the genome sequencing. The assistance of Dr Peter Taschner and Professor Johan den Dunnen (Leiden University) with the mutation descriptions is much appreciated.
Footnotes
Contributors: SE designed the study. HLA, WX and XX performed the bioinformatic analyses. RC performed the Next Generation and Sanger sequencing experiments. CW did the array CGH testing. PDT and CLST analysed the clinical data. HLA, MNW and SE prepared the draft manuscript. All authors contributed to discussion of the results and manuscript preparation.
Funding: HLA, MNW and SE are Wellcome Trust funded.
Competing interests: None.
Patient consent: Obtained.
Ethics approval: No approval from an ethics committee or institutional review board was obtained since the patients or their parents gave informed consent for genetic testing to determine the cause of their disease.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Robledo RF, Rajan L, Li X, Lufkin T. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev 2002;16:1089–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shamseldin HE, Faden MA, Alashram W, Alkuraya FS. Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. J Med Genet 2012;49:16–20 [DOI] [PubMed] [Google Scholar]
- 3.Kouwenhoven EN, van Heeringen SJ, Tena JJ, Oti M, Dutilh BE, Alonso ME, de la Calle-Mustienes E, Smeenk L, Rinne T, Parsaulian L, Bolat E, Jurgelenaite R, Huynen MA, Hoischen A, Veltman JA, Brunner HG, Roscioli T, Oates E, Wilson M, Manzanares M, Gomez-Skarmeta JL, Stunnenberg HG, Lohrum M, van Bokhoven H, Zhou H. Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet 2010;6:e1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo Iacono N, Mantero S, Chiarelli A, Garcia E, Mills AA, Morasso MI, Costanzo A, Levi G, Guerrini L, Merlo GR. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 2008;135:1377–88 [DOI] [PubMed] [Google Scholar]
- 5.Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, Gurrieri F, Everman DB, Schwartz CE, Birk OS, Bejerano G, Lomvardas S, Ahituv N. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res 2012;22:1059–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birnbaum RY, Everman DB, Murphy KK, Gurrieri F, Schwartz CE, Ahituv N. Functional characterization of tissue-specific enhancers in the DLX5/6 locus. Hum Mol Genet 2012;21:4930–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez ME, O'Brien TG, Fultz KE, Babbar N, Yerushalmi H, Qu N, Guo Y, Boorman D, Einspahr J, Alberts DS, Gerner EW. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proc Natl Acad Sci 2003;100:7859–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nord A, Lee M, King M-C, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics 2011;12:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinasac DS, Moriyama M, Jalil MA, Begum L, Li MX, Iijima M, Horiuchi M, Robinson BH, Kobayashi K, Saheki T, Tsui L-C. Slc25a13-knockout mice harbor metabolic deficits but fail to display hallmarks of adult-onset type II citrullinemia. Mol Cell Biol 2004;24:527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genuardi M, Pomponi MG, Sammito V, Bellussi A, Zollino M, Neri G. Split hand/split foot anomaly in a family segregating a balanced translocation with breakpoint on 7q22.1. Am J Med Genet 1993;47:823–31 [DOI] [PubMed] [Google Scholar]
- 11.Spranger M, Schapera J. Anomalous inheritance in a kindred with split hand, split foot malformation. Eur J Pediatr 1988;147:202–5 [DOI] [PubMed] [Google Scholar]
- 12. Temtamy SA, McKusick VA. The genetics of hand malformations. Birth Defects, Original article series New York, USA. Alan R. Liss Inc, 1978;14:3:619. [PubMed] [Google Scholar]
- 13.Zlotogora J. On the inheritance of the split hand/split foot malformation. Am J Med Genet 1994;53:29–32 [DOI] [PubMed] [Google Scholar]
- 14.Schluth-Bolard C, Labalme A, Cordier M-P, Till M, Nadeau Gl, Tevissen Hln, Lesca Gt, Boutry-Kryza N, Rossignol S, Rocas D, Dubruc E, Edery P, Sanlaville D. Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet 2013;50:144–50 [DOI] [PubMed] [Google Scholar]
- 15.Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, Kirby A, Liu S, Muddukrishna B, Ohsumi TK, Shen Y, Borowsky M, Daly MJ, Morton CC, Gusella JF. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 2011;88:469–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talkowski ME, Ordulu Z, Pillalamarri V, Benson CB, Blumenthal I, Connolly S, Hanscom C, Hussain N, Pereira S, Picker J, Rosenfeld JA, Shaffer LG, Wilkins-Haug LE, Gusella JF, Morton CC. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med 2012;367:2226–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krumm N, Sudmant PH, Ko A, O'Roak BJ, Malig M, Coe BP, Project NES, Quinlan AR, Nickerson DA, Eichler EE. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Grzeda K, Stewart C, Grubert F, Urban A, Snyder M, Marth G. Copy number variation detection from 1000 Genomes project exon capture sequencing data. BMC Bioinform 2012;13:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fromer M, Moran Jennifer L, Chambert K, Banks E, Bergen Sarah E, Ruderfer Douglas M, Handsaker Robert E, McCarroll Steven A, O'Donovan Michael C, Owen Michael J, Kirov G, Sullivan Patrick F, Hultman Christina M, Sklar P, Purcell Shaun M. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 2012;91:597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coin LJM, Cao D, Ren J, Zuo X, Sun L, Yang S, Zhang X, Cui Y, Li Y, Jin X, Wang J. An exome sequencing pipeline for identifying and genotyping common CNVs associated with disease with application to psoriasis. Bioinformatics 2012;28:i370–i4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO, Thrasher AJ, Kumararatne D, Doffinger R, Nejentsev S. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012; 28:2747–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.