

Abstract
The Wilms tumor 1 (WT1) oncoprotein is an intracellular, oncogenic transcription factor that is overexpressed in a wide range of leukemias and solid cancers. RMFPNAPYL (RMF), a WT1-derived CD8+ T cell human leukocyte antigen (HLA)–A0201 epitope, is a validated target for T cell–based immunotherapy. Using phage display technology, we discovered a fully human “T cell receptor–like” monoclonal antibody (mAb), ESK1, specific for the WT1 RMF peptide/HLA-A0201 complex. ESK1 bound to several leukemia and solid tumor cell lines and primary leukemia cells, in a WT1-and HLA-A0201–restricted manner, with high avidity [dissociation constant (Kd) = 0.1 nM]. ESK1 mediated antibody-dependent human effector cell cytotoxicity in vitro. Low doses of naked ESK1 antibody cleared established, disseminated, human acute lymphocytic leukemia and Philadelphia chromosome–positive leukemia in nonobese diabetic/severe combined immunodeficient γc−/− (NSG) mouse models. At therapeutic doses, no toxicity was seen in HLA-A0201 transgenic mice. ESK1 is a potential therapeutic agent for a wide range of cancers overexpressing the WT1 oncoprotein. This finding also provides preclinical validation for the strategy of developing therapeutic mAbs targeting intracellular oncogenic proteins.
Introduction
Leukemias are difficult-to-treat neoplasms that are largely incurable in adults. Marketed therapeutic anticancer monoclonal antibodies (mAbs) recognize extracellular or cell surface proteins, which constitute only a small fraction of the cellular proteins and are not tumor-specific (1–3). In contrast, mutated or oncogenic tumor-associated proteins are typically nuclear or cytoplasmic (4–6). Intracellular proteins can be degraded in the proteasome, processed, and presented on the cell surface by major histocompatibility complex (MHC) class I molecules as T cell epitopes that are recognized by T cell receptors (TCRs) (7, 8). Therefore, generating therapeutic “TCR-like” mAbs that recognize intracellular tumor antigen–derived peptide/MHC complexes on the cell surface widens possible cancer target selection, enhances therapeutic potency, and provides the selectivity of T cell–like recognition.
Several TCR-like Fab or ScFv antibodies specific for cancer antigens have been successfully selected from mice or from phage display libraries (9–14). TCR-like Fab or ScFv specific for the melanoma antigens NY-ESO-1 or telomerase catalytic subunit–derived peptide, presented by human leukocyte antigen (HLA)–A01 or HLA-A02, among others, has been described (9–12) and is an excellent tool for studying antigen processing and presentation. Fab-toxin proteins, generated by fusing TCR-like Fab antibodies specific for melanoma antigens MART-1 26–35/A2 or gp100 280–288/A2 to a truncated form of Pseudomonas endotoxin, were shown to inhibit human melanoma xenografts in vivo (13).
Wilms tumor 1 (WT1) oncoprotein is a zinc finger transcription factor whose expression in normal adult tissue is rare but is overexpressed in leukemias of multiple lineages and a wide range of solid tumors, particularly in mesothelioma and ovarian cancer (15–19). WT1 expression is a biomarker and a prognostic indicator (20, 21). RNA interference knockdown studies of WT1 suggest that it has oncogenic potential (22) and it appears to be expressed in leukemia stem cell populations (23). A National Institutes of Health–convened panel recently ranked WT1 as the top cancer target for immunotherapy (24). WT1 is a nuclear protein, inaccessible to classical antibody therapy, but vaccine approaches are under way to generate WT1-specific cytotoxic T cell (CTL) responses that recognize peptides presented on the cell surface by MHC class I molecules (25–29). We and others have extensively studied the 9-mer WT1-derived peptide 126–134, RMFPNAPYL (“RMF”), that has been shown to be processed and presented by HLAA0201 molecules. This peptide induces cytotoxic CD8 T cells capable of killing WT1+ tumor cells in vitro and in human T cell–based and vaccine trials (30–33), thus providing a strong rationale for therapeutic targeting of the RMF epitopes with mAbs.
We report here the discovery of a fully human immunoglobulin G1 (IgG1) mAb, named “ESK1,” that is specific for the WT1 RMF peptide/HLA-A0201 complex (RMF/A2) found on many human cancers. The mAb mediated antibody-dependent cell-mediated cytotoxicity (ADCC) in a WT1-and HLA-A0201–restricted manner in vitro. In nonobese diabetic/severe combined immunodeficient (NOD/SCID) γc−/−(NSG) mice, ESK1 as a naked mAb showed potent antitumor efficacy against established disseminated human leukemia xenografts.
Results
Selection of ScFv specific for RMF/A2 complex and engineering of full-length human mAb
Single phage clones selective for the RMF/A2 complex were picked by a positive screen on A0201/RMF monomers and a negative screen on A0201/RHAMM-R3 control peptide monomers. Therefore, any phage that reacted with HLA-A02 and an irrelevant peptide would have been taken out of the system at the first step. Clones that had unique DNA coding sequences were characterized in secondary screens by binding to a transporter associated with antigen processing (TAP)-deficient, human HLA-A0201+ cell line (T2) alone or pulsed with RMF peptide or control peptides. Fifteen of 35 clones screened showed specific binding to T2 cells pulsed with RMF peptide. Those clones that showed binding to T2 cells without the RMF peptide were discarded. The phage clones with the strongest binding were further characterized for specificity and avidity against T2 pulsed with various peptides(Fig. 1A). Five of the 15 ScFv+ phage clones were engineered into full-length human monoclonal IgG1 for additional analyses.
Fig. 1.

Binding of the phage clones and mAb to WT1 RMFp/HLA-A0201 complexes on live cells measured by flow cytometry. (a) Phage clone of ESK1 binds to T2 cells pulsed with RMF peptide (green), but not to T2 cells alone (pink), T2 cells pulsed with control EW peptide (light blue), or mutant heteroclitic peptide WT1-A1 YMFPNAPLY (orange), compared to secondary goat anti-human-FITC only (dark blue). (b–d) Binding of: isotype control human IgG1 (brown), full-length IgG1 mAbs ESK1 (orange), ESK3 (dark blue), ESK5 (green), ESK15 (light blue) and ESK23 (pink) at 1 ug/ml, followed by secondary PE-goat anti-hum an, on T2 cells pulsed with 50ug/ml RMF peptide (b), without peptide (c), or 50ug/ml irrelevant RHAMM-R3 peptide (d). (e–f) T2 cells pulsed with 50 ug/ml RMF peptide were stained with full-length ESK1 (e) or isotype control human IgG1 (d) at: 0.01 ug/ml (pink), 0.1 ug/ml (light blue), 1ug/ml (orange) or 10ug/ml (green), or secondary alone (red). (g–h) Binding of ESK1, ESK3 and ESK5 at 1ug/mL on T2 cells pulsed with 1.6–50 ug/mL RMF peptide (g), or irrelevant RHAMM-R3 peptide (h).
Specificity of the IgG1 mAb
Human T2 cells, pulsed with or without RMF or control RHAMM-R3 peptides, initially were used to determine the binding specificity. Five human IgG1 (hIgG1) showed strong binding to the RMF-pulsed T2 cells (Fig. 1B). The binding strength of the mAbs was 50-to 100-fold enhanced compared to their parental ScFv phage clones. Two mAbs among the five showed binding to T2 cells alone or pulsed with the control peptide RHAMM-R3, suggesting that these two mAbs also cross-reacted with HLA-A2 molecule epitopes and therefore were excluded from further investigation (Fig. 1, C and D). A comparison of the binding avidity of the three remaining mAbs specific for the RMF/A2 complex first was investigated by titration of the mAbs and of the RMF peptide. mAb ESK1 showed the strongest binding, down to a mAb concentration of 0.01 μg/ml (Fig. 1, E and F). ESK1 could detect the RMF/A2 complex in a peptide concentration–dependent manner at concentrations as low as 1.6 μg/ml, with higher fluorescence intensity than ESK5 and ESK3 mAbs (Fig. 1, G and H).
Specificity was further confirmed through binding of ESK1 to T2 cells pulsed with analog RMF peptides (table S1). Analog RMF peptides were synthesized with amino acid substitutions, loaded onto T2 cells, and tested for ESK1 binding. Positions 2 and 9 of the RMF were left intact because these are the anchor residues for RMF peptide binding to the HLA-A0201 molecule. Alanine or tyrosine substitution at position 1 (WT1-A1 or WT1-B) reduced the binding of ESK1 the most compared to the native WT1-A RMF peptide (fig. S1A). The loss of the binding was not due to the reduction of the peptide binding affinity to the HLA-A2 molecule, because both these peptides showed strong binding in T2 stabilization assays (fig. S1B). These results showed that nearly identical peptides were not adequate replacements for the ESK1 epitope and that binding to the HLA-A0201 epitopes alone on the cell surface was not sufficient.
ESK1 was able to recognize the naturally processed WT1 epitope RMF presented by HLA-A0201 molecules on the cell surface in a panel of cell lines. ESK1 bound to six of seven human mesothelioma cell lines that are positive for both HLA-A0201 and WT1 mRNA, but not to cells that were either HLA-A0201− or WT1− (Table 1 and Fig. 2, A and B). Similarly, among 11 human hematopoietic cell lines tested, ESK1 bound to cell lines BV173, BA25, Set2, AML14, and ALL-3 (Table 1 and Fig. 2, C to F), which are positive for both WT1 mRNA and HLA-A0201. ESK1 did not bind to the HLA-A2− HL-60 or WT1− cell lines B-Jab and SKLY-16. Cell lines that were genotypically HLA-A02+ but expressed little HLA-A02 on their surface (such as K562) were also negative for binding to ESK1 (Fig. 2D). A panel of 28 fresh healthy donor cells (fig. S2 and Table 1) and fresh leukemia cells (Fig. 2 and Table 1) was examined by direct flow cytometry for binding to ESK1 and costained with various lineage markers. ESK1 bound to fresh acute myeloid leukemia (AML) CD34+/CD33+ leukemia cells in an HLA-A2– and WT1-restricted manner (Fig. 2, G and H), but not to normal CD33+ peripheral blood mononuclear cells (PBMCs) (fig. S2). Healthy donor cells were gated for CD3+ T cells, CD19+ B cells, and CD33+ monocytic/myeloid cells and examined separately. For 15 samples of HLA-A0201–cells, no cell populations were positive. Among 13 HLAA0201+ donors, normal T cells and monocytic/myeloid cells were also negative (fig. S2 and Table 1), whereas samples of leukemias were positive (Fig. 2 and Table 1). There were two A02+ leukemia samples that were only weakly ESK1+, but the expression of WT1 in these samples was not known, so these could be either false negatives or true negatives. Cells in two of the B cell populations from healthy A02+ donors were weakly positive as well. It is not known whether the cross-reactivity was due to WT1 overexpression in a subset of cells or another cross-reacting epitope. Lymphoid cells from one sample from a patient with myelodysplastic syndrome were also weakly positive, suggesting that it was part of the malignant clone, but lymphoid cells from the other patients were negative. However, the sum of this large amount of data on numerous samples of fresh cells shows that there is no general cross-reactivity of ESK1 with HLA-A02+/WT1− cells in typical healthy or leukemic cell populations.
Table 1.
Expression of WT1 RNA, HLA-A0201 expression and ESK1 binding*
| Cancer cell | HLA-A2 expression | WT1 mRNA or protein** | ESK1 binding | Binding Ratio: BB7.2 to isotype IgG |
|---|---|---|---|---|
| Solid tumor cell lines | ||||
| JMN | Positive | + | Positive | 248 |
| Meso 37 | Positive | + | Positive | 68 |
| Meso 47 | Positive | + | Positive | 17 |
| H2452 | Positive | + | Positive | 20 |
| Meso34 | Positive | + | Positive | 37 |
| Meso-56 | Positive | + | Positive | 23 |
| H2373 | Positive | + | Negative | 1.6 |
| MSTO | Negative | + | Negative | 1.4 |
| VAMT | Negative | + | Negative | NT |
| SKOV3 | Positive | +** | Positive | 14.7 |
| OVCAR3 | Positive | +** | Positive | > 20 |
| CC-228 | Positive | +** | Positive | > 20 |
| RMS-13 | Positive | +** | Positive | > 20 |
| Leukemias and Hematopoietic cells | ||||
| BV173 | Positive | + | Positive | 196 |
| BA25 | Positive | + | Positive | 118 |
| ALL-3 | Positive | + | Positive | 60 |
| U266 | Positive | + | Negative | 1.8 |
| 697 | Positive | + | Negative | 4.1 |
| LAMA | Positive | + | Negative | 6 |
| SKLY-16 | Positive | − | Negative | 1.9 |
| HL-60 | Negative | + | Negative | 0.4 |
| K562 | Negative | + | Negative | 1.5 |
| B-Jab | Positive | − | Negative | 11 |
| T2 | Positive | − | Negative | >20 |
| Fresh AML cells (n=4) | Positive | NT | 2 positive; 2 weakly positive | N/A |
| Fresh AML cells (n=8) | Negative | NT | Negative | N/A |
| Healthy CD3+ cells (n=9) | Positive | NT | Negative | N/A |
| Healthy CD33+ cells (n=9) | Positive | NT | Negative | N/A |
| Healthy CD19+ cells (n=9) | Positive | NT | 2 weakly positive | N/A |
| Healthy CD3+ cells (n=7) | Negative | NT | Negative | 1–3 |
| Healthy CD33+ cells (n=7) | Negative | NT | Negative | 1–3 |
| Healthy CD19+ cells (n=7) | Negative | NT | Negative | 1–3 |
HLA-A2 is by genotype. WT1 mRNA expression level was estimated according to our previous study (29), in which WT1 levels are shown as fold increase in WT1 expression level relative to that expressed in K562 cells.
In four lines Western blot was done instead of PCR. ESK1 binding and BB7.2 (cell surface HLA-A02) were measured by flow cytometry.
NT= not tested. N/A= not applicable.
Fig. 2.

Recognition of the naturally presented RMF/A2 complex on the cell surface by ESK1 in a HLA-A0201 and WT1 restricted manner. Binding of ESK1 to human mesothelioma cell lines JMN (a) and MSTO (b), to human leukemia cell lines BV173 (c). K562 (d), BA25 (e), and SET2 (f) with 10 ug/ml ESK1. Binding of ESK1 to AML blasts was measured on gated CD34+/CD33+ cells from HLA-A2 positive (g) or HLA-A2 negative (h) patients. Isotype human IgG1 was used as negative control. HLA and WT1 phenotype is shown for each cell type. Indirect flow cytometry was used in a–e and direct flow cytometry in f–h.
As expected, intensity of binding also appeared to be directly associated with the expression level of HLA-A0201 molecule. Although H2373, 697, LAMA, and U266 cell lines were positive for both WT1 transcripts and HLA-A02, the expression level of the HLA-A02 was low (Table 1) and the mAb did not bind. Notably, ESK1 did not bind to T2 cells alone or pulsed with other HLA-A0201–binding peptides such as a RHAMM-R3, Ewing sarcoma– derived peptide (EW), or the mutated heteroclitic RMF peptide WT1-A1 (fig. S1). The latter two peptides have been shown to have higher affinity than RMF for the HLA-A0201 molecule in T2 stabilization assays (28). These results confirmed ESK1 recognition that is specific for the epitopes jointly composed of the MHC peptide complex. As a further control on specificity for RMF/A0201, we tested whether ESK1 would bind to fresh cells from the spleens of HLAA0201 transgenic B6 mice. These mice overexpress human HLA-A0201 especially in their lymphoid organs. Although HLA-A0201 was detected on these cells, no binding of ESK1 was seen (fig. S3).
Binding avidity of the ESK1 mAb
A radioimmunoassay (RIA) using 125I-labeled ESK1 was used to determine an avidity constant on JMN cells [dissociation constant (Kd) = ~0.1 nM] (Fig. 3, A and B). In addition to confirming specificity, the RIA was also used to determine the number of antibody binding sites on a panel of cell lines and fresh cells, because this number is of great interest to understanding the effector mechanisms of the mAb and the biochemical features of RMF presentation (Fig. 3C). The background for detecting binding in this assay was in the range of 200 to 400 sites per cell on cell lines and about 100 sites on fresh cells, because that was the calculated signal on HLA-A02− or WT1− cells. Therefore, determination of site numbers at or below this range was not possible. ESK1 bound to the JMN mesothelioma and the BA25, BV173, and SET2 leukemias, which are positive for both HLA-A0201 and WT1 mRNA. ESK did not bind to HLA-A0201− cells (HL60 or MSTO) or WT1 mRNA –negative cells (SKLY-16).
Fig. 3.
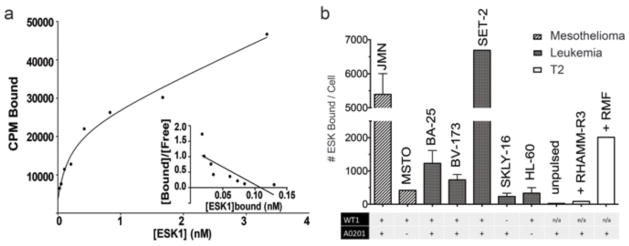
Radioimmunoassay was used to measure number of antibody bound per cell. (a) ESK1 was labeled with I-125 using chloramine-T to a specific activity of 4.3 mCi per mg and used for Scatchard analysis. (b) ESK1 binding to mesothelioma and leukemia cell lines, with HLA-A2 and WT1 phenotype indicated. Because we cannot determine whether the bivalent mAb is binding to 1 or 2 epitopes on the surface, total epitopes per cell could be as high as twice the number of mAb binding sites.
ESK1 also did not bind to 697 cells, which are both HLA-A0201+ and WT1+, but express low levels of HLA-A0201 (Table 1), confirming that a certain level of total MHC molecules is needed to present sufficient WT1 peptide for ESK1 binding. T2 pulsed with RMF bound ~2000 mAbs per cell and JMN cells bound ~6000 ESK1 mAbs per cell (Fig. 3B). RIA showed variable binding of ESK1 to AML blasts that were HLA-A2+ and WT1 mRNA–positive (0 to 1300 mAb sites over that of background binding to PBMCs that were A02−). WT1− patients bound 0 to 175 mAbs per cell over background. The RIA results in Fig. 3 confirmed the flow cytometry analysis in Fig. 2 and Table 1. These RIA data showed that the level of RMF/A2 on the surface of many examples of cancer and leukemia cells is adequate to allow reactivity with ESK1 and that the levels of epitope on WT1− healthy cells are low or absent.
ESK1 effector mechanisms
There are four general mechanisms by which mAbs affect cytotoxicity: ADCC, complement-mediated cytotoxicity (CMC), antibody-dependent cellular phagocytosis (ADCP), and direct killing via apoptosis. ADCC is considered to be one of the major effector mechanisms of therapeutic mAbs in humans. ESK1 was active in ADCC assays against multiple cell types (Fig. 4). In the presence of human PBMCs, ESK1 mediated dose-dependent ADCC against T2 cells loaded with RMF peptide (Fig. 4A) and against the RMF epitope naturally presented by HLA-A0201 molecules on JMN mesothelioma (Fig. 4B), BV173 leukemia (Fig. 4C), SKOV ovarian carcinoma (Fig. 4E), CC228 colon carcinoma cell lines (Fig. 4E), and fresh AML cells (Fig. 4F). Control T2 cells with no peptide or pulsed with the irrelevant A0201-binding peptide RHAMM-R3 (Fig. 4A) as well as A02− fresh AML blast cells (Fig. 4F), HL60 (Fig. 4D), or MSTO (Fig. 4B) were not killed. These results demonstrated that ESK1 mediated ADCC against cells that naturally express RMF/A2 complex at physiologic levels and with easily achievable mAb levels.
Fig. 4.

ESK1 mediates ADCC with human PBMC effectors at indicated concentrations. (a) T2 cells alone, or pulsed with RMF or irrelevant RHAMM-R3 peptides at 50 ug/ml, incubated with human PBMC effectors at E:T of 35:1 with cytotoxicity measured by LDH assay. (b) Similarly, JMN and MSTO cells at E:T of 30:1. (c) ADCC against BV173 was measured by 4hr-51Cr-release assay at E:T of 50:1, and HL-60 cells by LDH assay at E:T of 12.5:1. (d) ADCC against SKOV and CC228 cells was measured by 4hr-51Cr-release assay at E:T of 60:1. (e) ADCC against primary AML blasts from an HLA-A2 positive patient and an HLA-A2 negative patient, measured by LDH assay at E:T of 15:1. The data are representative of 1–3 experiments each. Killing was consistently observed at 1ug/ml ESK1 with multiple donors. Each data point was the average of triplicates.
We could not demonstrate evidence of cytotoxicity of the ESK1 mAb via CMC (table S2). No direct killing in vitro was seen either (fig. S4). The antibody alone or cross-linked with soluble goat anti-hIg, soluble or dried on plates, at concentrations from 0.03 to 30 μg/ml showed no effect on cell viability or proliferation for up to 96 hours. There were no consistent reproducible trends in killing or a clear dose response relationship.
Multiple attempts also were made to demonstrate ADCP in vitro and in vivo in NSG mice. These experiments were done with ESK1 antibody in the presence of macrophages drawn from naïve mice or with macrophages from mice that had been injected first with the mAb and target cells to mimic the in vivo situation. In no case could we observe, by flow cytometry or microscopic immunofluorescence methods, in vitro or in tissues, opsonization or phagocytosis mediated by ESK1 (fig. S5).
ESK1 therapy of human BV173 leukemia cells in NSG mice
We tested the efficacy of ESK1 in vivo in NSG mice xenografted intravenously 6 days previously with BV173 bcr/abl+ acute lymphoblastic leukemia (ALL) cells. At the time of treatment, mice had disseminated leukemia visible in their liver, spleen, and bone marrow. NSG mice lack mature B, T, and natural killer (NK) cells, and we hypothesized that introducing human effector cells (CD3−/CD34− PBMCs) along with ESK1 treatment would recapitulate in vivo the ADCC-mediated antitumor effects observed in vitro. Indeed, injection of effectors along with two 100-μg doses of ESK1 nearly ablated the leukemia in comparison to controls treated with IgG/effectors or effectors only (Fig. 5A). Although only two doses of mAb were given, this effect was durable over the course of the experiment (Fig. 5B). Early on in the trials, human effector cells, alone or combined with control hIgG1, appeared to promote more rapid growth of leukemia relative to untreated mice with leukemia (Fig. 5A). This suggests that the antitumor effect was clearly not related to the human effectors by themselves; perhaps, these infused cells enriched the murine microenvironment for the growth of the human leukemia. Sixty percent of the control mice given effectors (with or without control mAb) died early in the experiment with massive infiltration of the BV173, whereas all mice given the combination of human effectors plus the ESK1 had prolonged and sometimes leukemia-free survival (Fig. 5, A and B). At the time of termination of this trial on day 70, most mice in the ESK1 plus effector treatment group had little leukemia, and one of five mice had still not relapsed. Surprisingly, ESK1 mAb alone without infusion of human effectors markedly reduced tumor burden as well as ESK1 combined with human effectors (Fig. 5A); however, the leukemia eventually relapsed more quickly in the ESK1 mAb alone group (Fig. 5B). Because there were residual leukemia cells in these mice after the limited dosing schedule of mAb, this relapse was expected. Two doses of 100, 50, or 25 μg of ESK1 alone on days 6 and 10 cleared leukemia at all doses tested (Fig. 5C) and significantly improved survival at all dose levels (Fig. 5D). The leukemia relapsed slowly in a dose-dependent manner after antibody therapy alone (no effectors) was stopped.
Fig 5.

ESK1 effectively treats human BV173 leukemia in NSG mice. Tumor burden was calculated by summing the luminescent signal of each mouse in four positions, and average signal for each group (n=5) is plotted. Where noted, human effectors were given intravenously on day 6, and antibody (ESK1 or human IgG1 isotype control) was administered intravenously on days 6 and 10. (a). 100μg ESK1 alone or in combination with human effectors significantly reduces tumor burden at early times. (b). After one month, mice treated with ESK1 alone begin to relapse, while ESK1 with human effectors had prolonged durability. 1 of 5 mice was alive without leukemia signal at 70 days. (c) ESK1 significantly reduced tumor burden in a dose-dependent manner. One mouse died of an anesthesia accident in the 100ug group. (d) Survival of mice with disseminated BV173 leukemia treated with three different doses (25, 50, or 100 ug twice) of ESK1 alone on days 6 and 10 after leukemia engrafting. All treatment showed significantly prolonged survival as compared to untreated control animals or animals treated with isotype control hIgG (p< 0.01 by Log rank Mantel Cox test).
Therapy of other leukemias in mice: Mechanistic insights
To confirm that the marked antileukemic effects were not confined to the bcr/abl+ leukemias, we also studied ESK1 in a second disseminated leukemia NSG model, BA25 acute lymphocytic leukemia. Two doses of 100 μg of ESK1 alone were again able to effectively suppress the growth of the disseminated leukemia in comparison to control IgG (Fig. 6, A and B). We also attempted to treat the luciferase-marked ALL-3 Daudi (A02−/WT1−) cell line with the same schedule and dose as for BA25 ALL. There was no therapeutic effect(Fig. 6C). To ensure there was no cross-reactivity with HLA-A02+ cells in vivo in a therapeutic setting, we also conducted a therapeutic trial using the same conditions as for the BV173 leukemia on a luciferase-marked T2 leukemia, which is strongly HLA-A02+. No therapeutic activity was seen with ESK1 compared to the control IgG (Fig. 6D). These two control trials with epitope-negative leukemias showed that the presence of the RMF/A2 epitope was necessary for therapeutic effects.
Fig. 6. ESK1 specifically treats a second A2+ leukemia, but not A2− or WT1-tumors.

(a–b) Three million BA25 human acute lymphocytic leukemia cells (luciferase positive) were injected IV into NSG mice. On day 3, tumor engraftment was confirmed by firefly luciferase imaging in all mice that were to be treated; mice were then randomly divided into different treatment groups. On day 4 and day 8, 100ug mAb ESK1 or the isotype control mAb were injected IV. Tumor growth was assessed by luminescence imaging once to twice a week, and clinical measurements were assessed daily. Significant reductions in the growth of the ALL cells by the ESK1 mAb was seen relative to animals given the isotype control (a) by quantitation of luminescence from the supine animals (p < 0.04 and p < 0.02 on days 13 and 17, respectively.) These data are represented in a day 17 image (b). Treatment with 100ug mAb on days 6 and 10 after injection of 5 million Daudi ALL3. Leukemia cells (c) or 2 million T2 leukemia cells was followed and quantitated by bioluminescent imaging.
The mechanism of ESK1 action in vivo was further clarified by therapeutic injection of a F(ab′)2 form of ESK1 on the same dose and schedule as intact ESK1. This antibody was tested in the in vivo BV173 leukemia model. The F(ab′)2 maintained the same affinity for the RMF/A2 epitope as determined by interferometry, and was thus able to bind and cross-link the epitope equally well, but had no therapeutic effects (fig. S6). These data confirm that the Fc portion of the hIgG1 was necessary, and binding to or cross-linking of the epitopes alone was not effective.
To gain further insight into the mechanism, we engineered a ESK1 antibody with modified glycosylation Fc, called ESKM. The Fc glycol modification was done to give the mAb a higher affinity for human Fcg receptor IIIa (FcgRIIIa) (and mouse FcgRIV), but reduced affinity binding to human FcgRIIb (and mouse FcgRIIb), than antibody produced in wild-type Chinese hamsterovary (CHO). ESKM had improved ADCC activity (fig. S7A). In vitro, ESKM was 5-to 10-fold more potent at killing target cells than the original antibody, ESK1. Finally, we compared the modified ESKM mAb containing the Fc region with better ADCC to the original ESK1 antibody in the BV173 mouse therapy model. Treatment was done at very low doses (10 μg twice) to distinguish the effects of the two antibodies against the target cells. The modified antibody was more effective therapeutically in the leukemia model (fig. S7B), therefore showing that improved ADCC resulted in better therapeutic effects.
Toxicity studies
Because the peptide/MHC epitope is not present in wild-type mice or the NSG mice, we used a human HLA-A0201 transgenic mouse as an appropriate model to assess whether ESK1 would cause toxicity in vivo where all the tissues can express human HLA-A0201 and some tissues could express the WT1 RMF/A2 epitope as well, such as might be expected in people. These mice express the same RMF sequence of WT1 as humans. HLA-A0201 surface expression was confirmed in these mice by flow cytometry of spleen cells (fig. S3B). At the same dose used to achieve effective therapy (two doses of 100 μg, intravenously), no evidence of toxicity was seen clinically, by animal weight loss, spleen weight loss, or pathological microscopic evaluation of the major target tissues. (Spleen weight was analyzed because the tissue expresses HLA-A0201.) No inflammation or infiltrates nor losses of cells were observed (fig. S8 and tables S3 and S4).
Discussion
We hypothesized that the WT1 RMF peptide/HLAA2 complex would be a rational and critical target to test the concept for developing a potent new cancer therapeutic mAb with high selectivity and wide applicability. We describe here a potently therapeutically active, cancer-selective, TCR-like hIgG1 mAb, that is, an antibody with features that make it immediately promising as a new cancer drug.
Because tumor-associated proteins may be degraded, processed, and presented by MHC class I molecules and recognized by TCR of CTLs, active immunization with peptide vaccines or adoptive transfer of antigen-specific T cells has been widely used as an experimental approach to the specific immunologic treatment of human cancers. However, these strategies have been impeded by low TCR affinities, limited in vivo potent cytotoxic responses against high tumor burdens, the lack of effector cell persistence, and the generation of tolerance to the differentially expressed tumor “self-antigens.” To date, there have been only a limited number of examples of consistent and durable regressions induced by tumor-specific T cells. There are no U.S. Food and Drug Administration–approved cancer vaccines or CTL therapies.
Human mAb therapies alone, as cytotoxic conjugates, or in combination with other agents have proven to be potent, controllable, and highly effective treatment modalities for several cancers. The targets of marketed therapeutic mAb have been limited to soluble or cell surface proteins, however, owing to the difficulty of targeting intracellular antigens. Unfortunately, in cancer cells, many of the most interesting, important, and tumor-specific proteins are found intracellularly and are thus currently inaccessible to mAb therapy. Recently, others have demonstrated cytotoxic and therapeutic mAb to intracellular antigens, although the mechanisms of targeting and action differ from those described here (34–36). A TCR-like mouse mAb specific for the PR1/HLA-A0201 demonstrated inhibition of AML in vitro (14), and a mouse mAb specific for an epitope derived from human chorionic gonadotropin (hCG)-b bound to HLA-A02 was shown to be active against breast cancer in mice (37). However, no full-length human mAbs for intracellular antigens presented on the cell surface have been reported as therapeutic agents.
Use of phage display technology allowed us to prepare an agent of high specificity and avidity. The RMF peptide has been validated as a TCR epitope in multiple previous studies. However, when compared to mAbs, T cells are capable of effectively recognizing peptide/MHC complexes in smaller numbers on the cell surface. We chose this epitope because of its previous validation (16, 19, 22, 24–33) as a target on human tumor cells recognizable by HLA-A2–restricted, peptide specific T cells both in vitro and in vivo. Other intracellular protein epitopes/mAb systems might not display similar high expression or immunogenicity, and the RMF epitope may not be adequately expressed on the surface in all target cancer cells that express WT1 protein. Similar high surface expression has been observed for the PR1 peptide (14). We found that the detection of the antigen was strongly dependent on the avidity of the mAb. For example, two other mAbs with apparent lower avidity showed far less binding to target cells at usable concentrations. ESK1’s subnanomolar Kd was sufficiently low to be therapeutically useful in mice and also to be administered at feasible doses in humans. Other TCR-like mAbs to peptides have shown variable avidities, often depending on whether the Ig was monovalent or bivalent, such as NY-ESO-1 (47 nM), gp100 (294 nM), and PR1 (9.9 nM) (14). For cells that express too few peptide/MHC epitopes to achieve adequate binding of the mAb, it may be possible to upregulate MHC by use of interferon or other cytokine treatments (38).
The evidence in vitro and in vivo in this study implicates ADCC as the dominant mechanism for ESK1 killing leukemia cells; however, it is not possible to exclude with certainty other mechanisms in vivo. ADCC will be limited by the low numbers of NK cells in NSG mice, the low density of target sites (1000 to 6000 RMF/A2 per cell on the cancer lines), and the lower affinity of the hIgG1 for the mouse FcR. Because NSG mice have macrophages, neutrophils, and monocytes, particularly in the organs where the leukemias reside, ADCC should still be active at the starting concentrations of mAb in vivo (0.3 to 0.4 μM). Other limitations to the use of TCR-like mAbs include the possibility of low levels of expression of the complex, owing to either downregulation of the MHC or reduced processing of the peptide. To potentially circumvent this issue for future translation, we chose RMF, which has been shown in many preclinical and clinical studies to be present on several cancer cell types (19, 24–26, 29). This suggested that the biochemistry of this epitope was particularly favorable for a TCR like mAb approach. A second issue for use of this mAb is its HLA restriction; however, the widespread presence of HLA-A0201 among 40% of Caucasians and to a lesser extent in other groups, will make this agent widely usable. In addition, other HLA-A02 haplotypes bearing RMF may cross-react with ESK1 because the amino acid sequences of the subtypes of HLA-A02 differ minimally. The addition of high-avidity TCR-like mAb to WT1 epitopes presented by three to four other prevalent HLAs could allow coverage for up to 85% of patients in the United States (39).
ESK1 TCR-like mAb detects the target epitope complex on the surface of tumor cells; therefore, it may be possible to use the mAb as a companion diagnostic tool to discriminate which cancers in patients will be good targets for ongoing treatments with adoptive transfer of WT1-specific T cells, WT1 vaccines, or mAb therapy, and possibly to predict therapeutic activity. This is in contrast to reverse transcription–PCR or Western blotting for WT1, which are not measures of the amount of RMF/MHC epitope presented on the cell surface.
ESK1 showed fast and potent therapeutic activity alone in both a disseminated human bcr/abl+ acute leukemia xenograft model and a disseminated pre-B ALL model. The addition of CD3−/CD34− human effector cells further improved long-term outcomes and prolonged survival. Leukemia returned more quickly in the mAb alone group compared to ESK1 combined with effectors, showing that ESK1-mediated human ADCC likely plays an important role in eliminating tumor cells long term. Judging from efficacy in these models alone, predicting the depth and duration of effects that will be seen in humans is difficult. However, other mAbs that are effective at ADCC, such as rituximab and trastuzumab, have been effective agents in humans (1, 3).
With the data presented here showing efficacy in two mouse models and the additional planned rigorous toxicity testing, this TCR-like, fully human IgG mAb may be taken into human trials. Other mAbs (native hIgG1 and radioconjugates) developed by us to this stage proceeded directly to human trials in acute leukemia (40, 41). This ESK1 mAb is specific for a widely expressed oncogenic product for which there is currently no way to construct a drug. More than 1 million patients worldwide may have a WT1+ tumor or leukemia and up to one-third of these may have an HLA-A02 haplotype. Hence, an effective antibody therapeutic agent could have large clinical impact.
Materials and Methods
Cell samples, cell lines, and antibodies
After informed consent on Memorial Sloan-Kettering Cancer Center (MSKCC) Institutional Review Board–approved protocols, PBMCs from HLA-typed healthy donors and patients were obtained by Ficoll density centrifugation. A listing of the hematopoietic and solid tumor cell lines can be found in the Supplementary Materials and Methods. The sources for obtaining human mesothelioma cell lines are described previously (29).
Monoclonal antibodies against human HLA-A2 (clone BB7.2) conjugated to fluorescein isothiocyanate (FITC) or allophycocyanin (APC), and its isotype control mouse IgG2b/FITC or APC, to human or mouse CD3, CD19, CD56, CD33, CD34 (BD Biosciences), goat F(ab′)2 anti-hIgG conjugated with phycoerythrin (PE) or FITC, and goat F(ab′)2 anti-mouse Ig conjugated to FITC were purchased from Invitrogen. Mouse mAb to HLA class I (W6/32) was obtained from the MSKCC Monoclonal Antibody Core Facility. Human isotype control hIgG1 antibody was provided by Eureka Therapeutics (catalog number ET901)
Peptides
All peptides were purchased and synthesized by Genemed Synthesis Inc. Peptides were >90% pure (table S1). The peptides were dissolved in dimethyl sulfoxide and diluted in saline at 5 mg/ml and frozen at −80°C. Biotinylated single-chain WT1 peptide/HLA-A0201 and RHAMMR3/HLA-A0201 complexes were synthesized by refolding the peptides with recombinant HLA-A2 and β2-microglobulin at the Tetramer facility at MSKCC.
Animals
Eight-to 10-week-old male NOD.Cg-Prkdc SCID IL2rgtm1Wjl/SzJ mice, known as NSG, were purchased from The Jackson Laboratory or obtained from the MSKCC animal breeding facility. C57BL/6 and C57BL/-Tg (HLA-A2.1) 1 Enge/J (6 to 8 weeks old, male) were also purchased from The Jackson Laboratory.
Flow cytometry analysis
For cell surface staining, cells were incubated with appropriate mAbs for 30 min on ice, washed, and incubated with secondary antibody reagents when necessary. Flow cytometry data were collected on a FACSCalibur (Becton-Dickinson) and analyzed with FlowJo V8.7.1 and 9.4.8 software.
ScFv clones specific for WT1 peptide/HLA-A0201 complexes
A human ScFv antibody phage display library (7 × 1010 clones) was used for the selection of mAb clones. Selection and characterization of the ScFv, as well as engineering of full-length ESK1 and the other mAbs using the selected ScFv fragments, are described in the Supplementary Materials and Methods.
Characterization of the full-length hIgG1 for the RMF/A2 complex
Initially, the specificities of the fully human IgG1mAbs for the RMF/A2 complex were determined by staining T2 cells pulsed with or without RMF or RHAMM-R3 control peptides, followed by secondary goat F(ab′)2 anti-hIgG mAb conjugated to PE or F ITC. The fluorescence intensity was measured by flow cytometry. The same method was used to determine the binding of the mAb to fresh tumor cells and cell lines. ESKM, a form of ESK1 that has altered sugar structures and hence 10-fold improved ADCC, was made in an engineered CHO line (42).
Antibody-dependent cellular cytotoxicity
Target cells used for ADCC were T2 cells pulsed with or without WT1 or RHAMM-R3 peptides, and cancer cell lines without peptide pulsing and fresh cells without peptide pulsing (see list in the Cell samples, cell lines, and antibodies section). ESK1 or its isotype control hIgG1 at various concentrations was incubated with target cells and fresh PBMCs at different effector/target ratios for 16 hours. The supernatant was harvested, and the cytotoxicity was measured by L DH release assay with CytoTox 96 Non-Radioreactive kit from Promega following their instruction. Cytotoxicity was also measured by standard 4-hour 51Cr release assay.
Other mechanisms of cytotoxicity
Methods used to assess CMC, ADCP (2, 43, 44), and the ability of the mAb to directly reduce proliferation of the cells or kill them without effector cells are described in the Supplementary Materials.
Transduction and selection of luciferase/green fluorescent protein–positive cells
BV173 and JMN cells were engineered to express high level of green fluorescent protein (GFP)–luciferase fusion protein with retroviral vectors containing a plasmid encoding luc/GFP. Cells showing high level GFP expression were selected by flow cytometry analysis and were used for the animal studies.
Therapeutic trials of ESK1 in a human leukemia xenograft NSG model
Two million BV173 human leukemia cells were injected intravenously into NSG mice. On day 5, tumor engraftment was confirmed by firefly luciferase imaging in all mice that were to be treated; mice were then randomly divided into different treatment groups. On days 6 and 10, mAb ESK1 or the isotype control mAbs were injected intravenously. In animals that also received human effector cells with or without mAb, cells (CD34-and CD3-depleted PBMCs from healthy donor) were injected intravenously into mice (107 cells per mouse) on day 6. Alternatively, 3 million BA25 human acute lymphocytic leukemia cells (luciferase-positive) were injected intravenously into NSG mice, and treatments were on days 4 and 8, without effectors. Tumor growth was assessed by luminescence imaging once to twice a week, and clinical measurements were assessed daily.
Luciferase-infected Daudi ALL3 cells (5 × 106 cells per animal) and luciferase-infected T2 cells (2 × 106 cells per animal) were also used as negative control cell lines in vivo.
Toxicity studies
ESK1 or isotype control mAb (100 μg) was injected into human HLAA0201 transgenic mice (The Jackson Laboratory) (n = 5 per mAb) on days 0 and 4 to mimic the maximum dose and therapeutic schedule used in the therapy experiments. Clinical parameters and weights were recorded on days 0, 6, and 14. On day 6 (for early effects), two mice were sacrificed. On day 14 (for late effects or recovery), three mice were sacrificed. Histopathologic examination was of the major possible WT1+ target organs (spleen, bone and bone marrow, liver, thymus, and kidney on days 6 and 14) as well as heart, lung, and ileum (on day 6 only).
Statistical analysis
Values reported represent means ± SEM (or ±SD, where noted). P values were calculated with GraphPad Prism, with P< 0.05 considered significant. Experiments were done three to five times except where noted, and the particular statistical analyses used in the experiments are noted in the figure captions. Statistics were performed to illustrate significance between groups where n ≥ 3. ADCC assays were not statistically comparable between experiments because the sources of human cells changed; in individual experiments, means of triplicate results were normalized. Linear and nonlinear regression analyses were used to fit curves. Statistical analysis was not performed in some animal experiments because death of animals led to data censoring (Fig. 5, A and B), because data were graphed ± SEM and the differences were not overlapping, or because the data were descriptive.
Supplementary Material
Acknowledgments
We thank Ingrid. Leiner for synthesizing biotinylated peptide/HLA-A0201 complexes; Dr. Annamalai Selvakumar for transducing JMN cells with luciferase/GFP; A lice. Yeh for HLA genotyping; and Drs. Dimiter Tassev, Leo Dubrovnik, KathyHsu, and Mr. Elliot Brea for helpful discussions.
Funding: This work is supported by grants awarded to D.A.S. from the Leukemia and Lymphoma Society, P01 CA23766 (also to R.J.O.), R01 CA55349, the SKI Technology Development Fund, the Tudor and Glades Foundations, the Merker Fund, and the Lymphoma Foundation and to T.D. from Mesothelioma Applied Research Foundation.
Footnotes
Author contributions: T.D., D.A.S., and N.V. designed and analyzed most of the immunology and animal experiments, and wrote the manuscript. D.P., N.V., E.D., and R.J.O. designed and analyzed mouse experiments. J.W. did pathology. S.Y., L.Z., T.T., C.L., and H.L. did the mAb engineering. T.D., A.S., V.Z., and T.K. did in vitro immunology assays. A.S., E.C., N.V., T.D., and T.K. did cytometry. M.C. and N.V. did RIA.
Competing interests: Sloan-Kettering and Eureka Inc. have filed for patent protection for the mAb. The authors have no other competing interests.
Data and materials availability: The ESK1 mAb is proprietary and will need to be obtained under a Materials Transfer Agreement from either Sloan-Kettering(contact Sharon Seiler) or Eureka Therapeutics (contact Elisa Pan).
References
- 1.Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. 2012;148:1081–1084. doi: 10.1016/j.cell.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R. Anti-CD47 antibody synergizes with rituximabto promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 4.Houghton AN, Guevara-Patiño JA. Immune recognition of self in immunity against cancer. J Clin Invest. 2004;11:468–471. doi: 10.1172/JCI22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sensi M, Anichini MA. Unique tumor antigens: Evidence for immune control of genome integrity and immunogenic targets for T cell–mediated patient-specific immunotherapy. Clin Cancer Res. 2006;12:5023–5032. doi: 10.1158/1078-0432.CCR-05-2682. [DOI] [PubMed] [Google Scholar]
- 6.Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia. 2007;9:1859–1874. doi: 10.1038/sj.leu.2404787. [DOI] [PubMed] [Google Scholar]
- 7.Morris E, Hart D, Gao L, Tsallios A, Xue S, Stauss H. Generation of tumor-specific T-cell therapies. Blood Rev. 2006;20:61–69. doi: 10.1016/j.blre.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 8.König R. Interactions between MHC molecules and co-receptors of the TCR. Curr Opin Immunol. 2002;14:75–83. doi: 10.1016/s0952-7915(01)00300-4. [DOI] [PubMed] [Google Scholar]
- 9.Noy R, Eppel M, Haus-Cohen M, Klechevsky E, Mekler O, Michaeli Y, Denkberg G, Reiter Y. T-cell receptor-like antibodies: Novel reagents for clinical cancer immunology and immunotherapy. Expert Rev Anticancer Ther. 2005;5:523–536. doi: 10.1586/14737140.5.3.523. [DOI] [PubMed] [Google Scholar]
- 10.Chames P, Hufton SE, Coulie PG, Uchanska-Ziegler B, Hoogenboom HR. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1–MAGE-A1 from a nonimmunized phage-Fab library. Proc Natl Acad Sci USA. 2000;97:7969–7974. doi: 10.1073/pnas.97.14.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Held G, Matsuo M, Epel M, Gnjatic S, Ritter G, Lee SY, Tai TY, Cohen CJ, Old LJ, Pfreundschuh M, Reiter Y, Hoogenboom HR, Renner C. Dissecting cytotoxic T cell responses towards the NY-ESO-1 protein by peptide/MHC-specific antibody fragments. Eur J Immunol. 2004;34:2919–2929. doi: 10.1002/eji.200425297. [DOI] [PubMed] [Google Scholar]
- 12.Lev A, Denkberg G, Cohen CJ, Tzukerman M, Skorecki K, Chames P, Hoogenboom HR, Reiter Y. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic subunit. Cancer Res. 2002;62:3184–3194. [PubMed] [Google Scholar]
- 13.Klechevsky E, Gallegos M, Denkberg G, Palucka K, Banchereau J, Cohen C, Reiter Y. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res. 2008;68:6360–6367. doi: 10.1158/0008-5472.CAN-08-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, McIntyre BW, Ma Q, Li D, St John L, Clise-Dwyer K, Molldrem JJ. An anti–PR1/HLA-A2 T-cell receptor–like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood. 2011;117:4262–4272. doi: 10.1182/blood-2010-07-299248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development. 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- 16.Keilholz U, Menssen HD, Gaiger A, Menke A, Oji Y, Oka Y, Scheinbenbogen C, Stauss H, Thiel E, Sugiyama H. Wilms’ tumor gene 1 (WT 1) in human neoplasia. Leukemia. 2005;19:1318–1323. doi: 10.1038/sj.leu.2403817. [DOI] [PubMed] [Google Scholar]
- 17.Takigawa N, Kiura K, Kishimoto T. Medical treatment of mesothelioma: Anything new? Curr Oncol Rep. 2011;4:265–271. doi: 10.1007/s11912-011-0172-1. [DOI] [PubMed] [Google Scholar]
- 18.Raja S, Murthy SC, Mason DP. Malignant pleural mesothelioma. Curr Oncol Rep. 2011;4:259–264. doi: 10.1007/s11912-011-0177-9. [DOI] [PubMed] [Google Scholar]
- 19.Oka Y, Tsuboi A, Elisseeva OA, Nakajima H, Fujiki F, Kawakami M, Shirakata T, Nishida S, Hosen N, Oji Y, Kawase I, Sugiyama H. WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. Scientific World Journal. 2007;7:649–665. doi: 10.1100/tsw.2007.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H, Kita K, Hiraoka A, Masaoka T, Nasu K. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood. 1994;84:3071–3079. [PubMed] [Google Scholar]
- 21.Ogawa H, Tamaki H, Ikegame K, Soma T, Kawakami M, Tsuboi A, Kim EH, Hosen N, Murakami M, Fujioka T, Masuda T, Taniguchi Y, Nishida S, Oji Y, Oka Y, Sugiyama H. The usefulness of monitoring WT1 gene transcripts for the prediction and management of relapse following allogeneic stem cell transplantation in acute type leukemia. Blood. 2003;101:1698–1704. doi: 10.1182/blood-2002-06-1831. [DOI] [PubMed] [Google Scholar]
- 22.Yamagarni T, Sugiyama H, Inoue K, Ogawa H, Tategawa T, Hirata M, Kudoh T, Akiyama T, Murakami A, Maekawa T. Growth inhibition of human leukemic cells by WT1 (Wilms tumorgene) antisense oligodeoxynucleotides: Implications for the involvement of WT1 in leukemogenesis. Blood. 1996;87:2878–2884. [PubMed] [Google Scholar]
- 23.Gerber JM, Qin L, Kowalski J, Smith D, Griffin CA, Vala MS, Collector MI, Perkins B, Zahurak M, Matsui W, Gocke CD, Sharkis S, Levitsky H, Jones RJ. Characterization of chronic myeloid leukemia stem cells. Am J Hematol. 2011;86:31–37. doi: 10.1002/ajh.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, Matrisian LM. The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellantuono I, Gao L, Parry S, Marley S, Dazzi F, Apperley J, Goldman JM, Stauss HJ. Two distinct HLA-A0201–presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood. 2002;100:3835–3837. doi: 10.1182/blood.V100.10.3835. [DOI] [PubMed] [Google Scholar]
- 26.Van Driessche A, Berneman ZN, Van Tendeloo VF. Active specific immunotherapy targeting the Wilms’ tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: Lessons from early clinical trials. Oncologist. 2012;17:250–259. doi: 10.1634/theoncologist.2011-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezvani K, Yong AS, Savani BN, Mielke S, Keyvanfar K, Gostick E, Price DA, Douek DC, Barrett AJ. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood. 2007;110:1924–1932. doi: 10.1182/blood-2007-03-076844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia. 2006;20:2025–2033. doi: 10.1038/sj.leu.2404380. [DOI] [PubMed] [Google Scholar]
- 29.May RJ, Dao T, Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH, Maslak P, Scheinberg DA. Peptide epitopes from the Wilms’ tumor 1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin Cancer Res. 2007;13:4547–4555. doi: 10.1158/1078-0432.CCR-07-0708. [DOI] [PubMed] [Google Scholar]
- 30.Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, Hofmann WK, Uharek L, Thiel E, Scheibenbogen C. A clinical and immunologic phase 2 trial of Wilms tumor gene product (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113:6541–6548. doi: 10.1182/blood-2009-02-202598. [DOI] [PubMed] [Google Scholar]
- 31.Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, Jafarpour B, Boss C, Barrett AJ. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111:236–242. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maslak P, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, Zhang R, Wolchok JD, Yuan J, Pinilla-Ibarz J, Berman E, Weiss M, Jurcic J, Frattini MG, Scheinberg DA. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses inpatients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi: 10.1182/blood-2009-10-250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krug LM, Dao T, Brown A, Maslak P, Travis W, Bekele S, Korontsvit T, Zakhaleva V, Wolchok J, Yuan JD, Li H, Tyson L, Scheinberg DA. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunol Immunother. 2010;59:1467–1479. doi: 10.1007/s00262-010-0871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo K, Li J, Tang JP, Tan CP, Hong CW, Al-Aidaroos AQ, Varghese L, Huang C, Zeng Q. Targeting intracellular oncoproteins with antibody therapy or vaccination. Sci Transl Med. 2011;3:99ra85. doi: 10.1126/scitranslmed.3002296. [DOI] [PubMed] [Google Scholar]
- 35.Ferrone S. Hidden immunotherapy targets challenge dogma. Sci Transl Med. 2011;3:99ps38. doi: 10.1126/scitranslmed.3002821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong CW, Zeng Q. Awaiting a new era of cancer immunotherapy. Cancer Res. 2012;72:3715–3719. doi: 10.1158/0008-5472.CAN-12-0063. [DOI] [PubMed] [Google Scholar]
- 37.Verma B, Neethling FA, Caseltine S, Fabrizio G, Largo S, Duty JA, Tabaczewski P, Weidanz JA. TCR mimic monoclonal antibody targets a specific peptide/HLA class I complex and significantly impedes tumor growth in vivo using breast cancer models. J Immunol. 2010;184:2156–2165. doi: 10.4049/jimmunol.0902414. [DOI] [PubMed] [Google Scholar]
- 38.Bao L, Dunham K, Lucas K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol Immunother. 2011;60:1299–1307. doi: 10.1007/s00262-011-1037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsh SGE, Parham P, Barber LD. The HLA Facts Book. Academic Press; New York: 2000. [Google Scholar]
- 40.Rosenblat TL, McDevitt MR, Mulford DA, Pandit-Taskar N, Divgi CR, Panageas KS, Heaney ML, Chanel S, Morgenstern A, Sgouros G, Larson SM, Scheinberg DA, Jurcic JG. Sequential cytarabine and a-particle immunotherapy with bismuth-213–lintuzumab (HuM195)for acute myeloid leukemia. Clin Cancer Res. 2010;16:5303–5311. doi: 10.1158/1078-0432.CCR-10-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jurcic JG, Larson SM, Sgouros G, McDevitt MR, Finn RD, Divgi CR, Ballangrud AM, Hamacher KA, Ma D, Humm JL, Brechbiel MW, Moline R, Scheinberg DA. Targeting a particle immunotherapy for myeloid leukemia. Blood. 2002;100:1233–1239. [PubMed] [Google Scholar]
- 42.Cheng L, Xiang JY, Yan S, Wang P, Tai WC. U.S. Patent 2010/0081195. Modified host cells and uses thereof. 2010
- 43.Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, Park CY, Weissman IL, Majeti R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71:1374–1384. doi: 10.1158/0008-5472.CAN-10-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jung ST, Kelton W, Kang TH, Ng DT, Andersen JT, Sandlie I, Sarkar CA, Georgiou G. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcgRIIa affinity and selectivity. ACS Chem Biol. 2013;8:368–375. doi: 10.1021/cb300455f. [DOI] [PubMed] [Google Scholar]
- 45.Persic L, Roberts A, Wilton J, Cattaneo A, Bradbury A, Hoogenboom HR. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene. 1997;187:9–18. doi: 10.1016/s0378-1119(96)00628-2. [DOI] [PubMed] [Google Scholar]
- 46.Tomimatsu K, Matsumoto S, Yamashita M, Teruya K, Katakura Y, Kabayama S, Shirahata S. Production of human monoclonal antibodies against FceRIa by a method combining in vitro immunization with phage display. Biosci Biotechnol Biochem. 2009;73:1465–1469. doi: 10.1271/bbb.80640. [DOI] [PubMed] [Google Scholar]
- 47.Caron PC, Co MS, Bull MK, Avdalovic NM, Queen C, Scheinberg DA. Biological and immunological features of humanized M195 (anti-CD33) monoclonal antibodies. Cancer Res. 1992;52:6761–6767. [PubMed] [Google Scholar]
- 48.McDevitt MR, Chattopadhyay D, Kappel BJ, Jaggi JS, Schiffman SR, Antczak C, Njardarson JT, Brentjens R, Scheinberg DA. Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes. J Nucl Med. 2007;48:1180–1189. doi: 10.2967/jnumed.106.039131. [DOI] [PubMed] [Google Scholar]
- 49.Greiner J, Schmitt A, Giannopoulos K, Rojewski MT, Götz M, Funk I, Ringhoffer M, Bunjes D, Hofmann S, Ritter G, Döhner H, Schmitt M. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica. 2010;95:1191–1197. doi: 10.3324/haematol.2009.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dao T, Korontsvit T, Zakhaleva V, Haro K, Packin J, Scheinberg DA. Identification of a human cyclin D1-derived peptide that induces human cytotoxic CD4 T cells. PLoS One. 2009;4:e6730. doi: 10.1371/journal.pone.0006730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.