

Abstract
Abstract: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a progressive inherited heart disease characterized by ventricular arrhythmias and sudden cardiac death especially in the young. ARVC has been traditionally associated with the Mediterranean basin, as many seminal studies on the disease have originated from research groups of this region. Today, however, numerous ARVC registries from all over the world emphasize that the disease does not have a specific racial or geographical predilection. This work provides a review on the global perspective of ARVC.
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inheritable cardiac muscle disease characterized pathologically by fibrofatty replacement –usually but not exclusively- of the right ventricular (RV) myocardium. The estimated prevalence of ARVC is 1:2500 to 1:5000 with male predominance [1]. The disease shows a familial pattern in 40–70% of the cases and molecular genetic studies demonstrated that ARVC is a desmosomal disease resulting from genetically defective cell adhesion proteins such as plakoglobin, desmoplakin, plaokophillin-2, desmoglein-2, and desmocollin [2,3]. Clinical manifestations are related to electrical instability, including either ventricular tachycardia of RV origin or ventricular fibrillation, which may lead to sudden cardiac death (SCD), mostly in young people and athletes [4–6]. In rare cases, the progressive loss of ventricular myocardium may lead to right ventricular or biventricular heart failure [4].
ARVC had been traditionally associated with the Mediterranean region, as many seminal studies had originated from research groups in France, Greece, and Italy. Today, however, numerous ARVC registries from all over the world emphasize that the disease does not have a specific racial or geographical predilection. This work aims to provide a review on the global perspective of ARVC.
Historical review
Giovanni Maria Lancisi (1654–1720) was an anatomist, clinician, epidemiologist, advisor to Pope Clement XI and one of the greatest Italian physicians of the first half of the 18th century (Figure 1). In his book, De Motu Cordis et Aneurysmatibus, Lancisi reported a four generation family with sudden deaths and dilatation and aneurysms of the right ventricle [7]. Two hundred and fifty years later, in 1982, Dr. Frank Marcus (University of Arizona, United States) collaborating with a French group of researchers described the disease of right ventricular dysplasia in a series of 24 patients and referred to La Salpetrière Hospital and Jean Restand Hospital in Paris from 1973 to 1980. The group characterized some of the fundamental clinical elements of the disease including the T wave inversion in the right precordial leads on the surface electrocardiogram (ECG), the left bundle branch block pattern of the QRS complex during spontaneous ventricular tachycardia, and the echocardiographic features of a normal left ventricle with an enlarged and aneurismal right ventricle [8]. In 1986, Protonotarios et al. studied nine cases of palmoplantar keratosis in the Greek island of Naxos, of which seven showed symptoms and signs of heart disease. Three cases had episodes of ventricular tachycardia (VT) and a fourth patient died suddenly. All patients with cardiac signs and symptoms showed echocardiographic enlargement of the right ventricle and a right ventricular band. Moreover, the left ventricle was also affected in three patients [9]. The described cardiocutaneous syndrome, coined as “Naxos disease”, is an autosomal recessive form of ARVC. The current concept of the clinical and pathological picture of ARVC was established in Padua, Italy. Familial occurrence of ARVC with an autosomal dominant pattern and incomplete penetrance was proven by Nava et al. in 1988 [10]. Furthermore, Thiene et al. associated the pathology of ARVC with a series of SCD in the young ( ≤ 35 years), mostly during effort. All the cases had T wave inversion on the right precordial surface ECG at rest and left bundle branch block QRS pattern during ventricular tachycardia [11]. This was the first time ARVC was acknowledged as a major cause of death in the young.
Figure 1. .

Giovanni Maria Lancisi (1654–1720), an Italian physician, epidemiologist and anatomist. He observed marked neck venous distension with tricuspid valve insufficiency (later to be known as Lancisi's sign) and associated aortic arch aneurysms with syphilis in his masterpiece in cardiac pathology: De Motu Cordis et Aneurysmatibus (On the motion of the heart and on aneurysms), published posthumously in 1738. In his book, he also reported the first known family of sudden cardiac death and right ventricular aneurysms [7].
In 1994, Rampazzo et al. from the University of Padua, described the first gene locus associated with ARVC (14q23–24). To locate the disease gene, several polymorphic DNA markers were evaluated. Since some forms of hypertrophic cardiomyopathy are due to mutations of the β-myosin gene, chromosome 14 was selected as a first candidate for analysis, on the assumption that functionally related genes are often located on the same chromosome. The investigation was also extended to chromosomes 1, 2, 6, 12, and 17 that were linked to other hypertrophy cardiomyopathy gene mutations [12]. However, in 2000, the discovery that plakoglobin and desmoplakin gene mutations result in the autosomal recessive cardiocutaneous Naxos and Carvajal syndromes, respectively, led to the association between ARVC and desmosome gene mutation [13,14]. Desmoplakin was the first defective gene to be associated with autosomal dominant ARVC in 2002 [15]. Today, mutations in desmoplakin [15,16], plakoglobin [17], plakophilin-2 [18], desmoglein-2 [19], desmocollin-2 [20] and most recently desmin [21], have been found in dominant forms of ARVC with double and compound heterozygosity commonly reported [22,23]. This consistent protein alteration coined the concept of ARVC as a desmosomal disease in contrast to hypertrophic cardiomyopathy as a sarcomeric disease, and dilated cardiomyopathy as a cytoskeletal disease.
ARVC around the world
The current views concerning ARVC have been shaped based on results from vanguard national and international registries. The last three decades have witnessed an exponential increase in the scientific contribution to ARVC, not only in the number of published data on ARVC, but also on the number of centers involved in the research. Today, centers from all corners of the world add significant contribution to the global understanding of the pathology, molecular, genetics, epidemiology, clinical, electrophysiology, imaging, risk stratification and management aspects of ARVC.
In Europe, influential works have been published from France, Germany, Greece, Italy, the Netherlands, Spain and the United Kingdom. Hulot et al. published a study in 2004 on the clinical course of French patients with ARVC. Data were collected from 130 patients with confirmed ARVC from a tertiary center between 1977 and 2000. The study population characteristics showed male predominance (100 men) and a mean age at onset of symptoms of 31.8 ± 14.4 years. After a mean follow-up of 8.1 ± 7.8 years, there were 21 deaths with a cardiovascular origin (progressive heart failure in 14 patients and sudden death for the remaining 7 patients). All patients who died had a history of ventricular tachycardia. This shows that the main cause of death in this cohort was not arrhythmic death but rather congestive HF, highly attributable to the adequate implantable cardioverter-defibrillator (ICD) therapy in France [24]. From Germany, Wichter et al. published the largest series on pharmacological therapy in ARVC, first published in 1992 and updated in 2005, comparing multiple antiarrhythmic drugs and concluding in their 2005 update that sotalol at a dosage of 320–480 mg/d was the most effective drug resulting in a 68% overall efficacy [25,26]. The same group in 2004, concluded from their single-center experience that there is a marked improvement in long-term prognosis with ICD therapy in high-risk ARVC patients [27]. The Greek association between the cutaneous manifestations and the cardiac manifestations in Naxos disease had lead to the association of ARVC with desmosomal protein abnormalities (Figure 2) [13]. In Italy, an ECG-based pre-participation screening program for athletes has been going for more than 30 years. This screening program has shown marked efficacy in detecting cardiac pathologies leading to SCD in the young, with a drop of mortality rates among athletes in the Veneto region from 3.5/100,000 in 1979 to 0.5/100,000 in 2003 (Figure 3) [28]. This impressive drop was largely attributable to the successful detection of ARVC and hypertrophic cardiomyopathy among the screened individuals, as early reports accounted ARVC as the second most common cause of death in the young in the Veneto region [11]. Moreover, in 2003, Corrado et al. in the DARVIN I study recruited 132 ARVC patients from 22 institutions in North Italy and one in the United States, of whom 80% received an ICD implant for sudden cardiac arrest or sustained ventricular tachycardia. The group demonstrated that 48% of the patients were saved by at least one appropriate ICD intervention during the study period of 39 ± 25 months (Figure 4) [29]. Spanish football fans were overwhelmed with the tragic death of FC Sevilla midfielder Antonio Peurta in 2007 due to ARVC (Figure 5). This sad incident reflected the study results on the causes of deaths among athletes in Spain from the period 1995 to 2001, with arrhythmic cardiomyopathies accounting for 16% of all deaths [30]. A notable paper was published in 2009 on the etiology of sudden cardiac death in athletes in the UK from 1996 to 2008. It was found that cardiomyopathies are the most common cause of SCD in young British athletes accounting for 62% with ARVC attributed to 14% of deaths [31].
Figure 2. .

Clockwise from left: The Greek island of Naxos. A nuclear Naxos disease family. Analysis reveals the presence of a mutant allele (one bold band) in the homozygous boy and the wild-type (two light bands) coexisting with the mutant allele in the heterozygous carrier parents. Classic symptoms of Naxos disease, keratoderma striate in palmar and diffuse in plantar areas in the homozygous boy and wooly hair [41].
Figure 3. .

Annual incidence rates of SCD per 100,000 person-years among screened competitive athletes and unscreened non-athletes aged 12 to 35 in the Veneto region, Italy. Note the sharp decline of the screened athlete line in red [28].
Figure 4. .

Kaplan-Meier analysis from DARVIN I study of actual patient survival (upper line) compared with survival free of ventricular fibrillation/flutter (lower line) that in all likelihood would have been fatal in the absence of the ICD. The divergence between the lines reflects the estimated mortality reduction by ICD therapy of 24% at 3 years of follow up [29].
Figure 5. .

Antonio Jose Puerta Perez (1984–2007), a Spanish midfielder who used to play for FC Sevilla was affected with ARVC, 3 days after suffering a series of cardiac arrests during a Spanish league game against Getafe.
Along the Mediterranean basin, Naxos disease was described in one family in Turkey [32] and three Arab Israeli families [33]. With the increased vigilance and physician awareness, many new ARVC cases have been diagnosed yearly in Egypt (Figure 6). The South African ARVC registry started in 2004, and published its first results in 2009 on the clinical course of patients with ARVC and PKP 2 mutations.
Figure 6. .

Above: Surface ECG precordial lead recordings of a 50 year old Egyptian/Sudanese patient showing Epsilon wave in V1, T wave inversion in V1-3, and relative QRS prolongation in right precordial leads. Below: Cardiac magnetic resonance imaging demonstrating delayed gadolinium enhancement in the right ventricular free wall denoting extensive fibrosis. The patient suffered two episodes of sustained ventricular tachycardias with left bundle branch block pattern with altered level of consciousness. An ICD was implanted for secondary prevention. (Images courtesy of Mohamed Donya, MD, FRCR, Aswan Heart Centre, Aswan, Egypt).
The characteristics of ARVC in South Africa are similar to those from the French cohort showing a mean age of presentation of around 40, with male predominance of about 70%, and palpitations and syncope being the main presenting symptoms. South African researchers discovered five new PKP2 gene mutations causing ARVC (Figure 7). One of these gene mutations was found in four different unrelated Caucasian families. Dr. Mayosi from the South African group explains this finding as a founder gene effect, attributed most likely to one of the early Dutch settlers arriving to The Cape of Good Will [34]. The South African results were in concordance with the results of the Dutch paper on the prevalence of PKP 2 mutations in patients with ARVC in the Netherlands published in 2006. PKP2 gene mutations were found in nearly half of all Dutch patients with established ARVC diagnosis. It also accounted for more than 70% of familial ARVC in the Netherlands [35]. The matching findings of the two studies miles apart highlight the importance of taking into consideration all historical, ethnic and anthropological factors when dealing with a genetically determined disease.
Figure 7. .

Results from the South African registry; Schematic representation of the PKP2 mutations found in the study. Black boxes indicate the 14 exons of the PKP2 gene. Disease causing mutations are shown above the exons and polymorphisms are shown below [34].
Moving to the Far East, Komura and colleagues studied the clinical course of ARVC in a Japanese cohort of 35 patients. The study subdivided the patients to a group presenting with VT and another group presenting with congestive HF. The group with congestive HF presented at a later age of 66 and carried a worse prognosis with all three patients dying after only 20 months of follow up. The VT group presented at a younger age of 44 years and carried a better prognosis with only two patients dying after more than 60 months of follow up. Also, concerning the management of ARVC, patients who were inserted ICD's and were controlled on drug therapy did much better than patients sent for RF ablation, with a higher rate of recurrence of VT within the RF ablation group (Figure 8) [36].
Figure 8. .
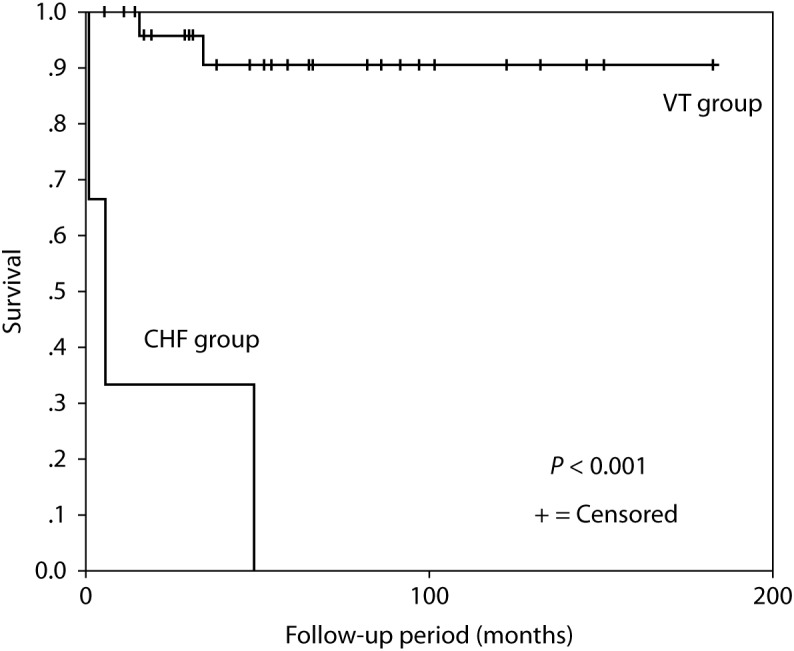
The Kaplan-Meier survival curve from the Japanese study. The CHF group showed a significantly higher mortality rate in comparison to the VT group [36].
A Chinese group published a cohort of 39 patients with ARVC, sharing almost the same clinical picture as the South African, French, and Japanese cohorts also including the same outcome of therapy. Of these patients, 33 were males and 6 were females and the age at first presentation was 34.9 ± 9.8 years. The most common symptoms were palpitation (62%) and syncope (44%). An interesting observation of the Chinese cohort was that all seven patients who were inserted an ICD had at least one appropriate shock [37].
On the other side of the ocean, a large United States registry on ARVC was published in 2005. With a cohort of 100 patients from the Johns Hopkins registry, the American Working Group concluded that ARVC patients present between the second and fifth decades of life either with symptoms of palpitations and syncope associated with ventricular tachycardia or with SCD. Due to genetic heterogeneity, signs and symptoms can widely vary and diagnosis is often delayed. However, once diagnosed and treated with an ICD, mortality is low (Figure 9) [38].
Figure 9. .

Schematic outline and quantification of the presentation, treatment, arrhythmia, and outcome in the entire patient population in the John Hopkins Registry. Sx indicates symptoms; Asx, asymptomatic; VF, ventricular fibrillation; Abl, catheter ablation of VT; Rec VT, recurrent VT; HF, heart failure; and w/o, without. *These patients died of noncardiac causes of death; autopsy revealed findings consistent with ARVD. †These patients experienced fatal VF leading to SCD; they could not be resuscitated. ‡This patient died because of biventricular heart failure at the age of 27 years; an ICD was in place at the time of death and the patient was awaiting a heart transplant [38].
ARVC has also been observed in South America. Carvajal et al. from Ecuador published in 1998 a series of 12 patients presenting with cardiocutaneous syndrome of palmoplanter keratosis and DCM. This is known as Carvajal syndrome and it has been associated with desmoplakin mutations and severe left ventricular involvement [14,39]. The magnetic resonance image (MRI) picture of Carvajal syndrome often mimics that of LV non- compaction. Noteworthy, there have been case reports of patients with Carvajal syndrome from all over the world which shows that the condition is not exclusive to the South American population (Figure 10) [40].
Figure 10. .

A: Cutaneous features of Carvajal syndrome, including wooly hair, palmoplantar keratosis [39]. B: Cine true fast imaging with steady-state precession image in the short-axis orientation shows dilatation and a trabecular disarray of both ventricles with the appearance of a noncompaction syndrome. This patient was found to have Carvajal syndrome postmortem. He was living in Germany at the time of study however, his family origins were from the city Giresun of Tirebolu near Azerbaijan [40].
In Australia, there is an ongoing major national registry on cardiomyopathies that was initiated in 2009. Another exciting research focus in Australia is the Poll Hereford cattle. It was recently found that this particular Australian cattle have the same phenotype of Naxos/Carvajal disease with woolly hair and biventricular fibrosis and sudden death, representing a natural animal model for further studies on ARVC (Figure 11) [41,42].
Figure 11. .

Cardiomyopathy/wooly haircoat syndrome (CWH) in Poll Hereford calves. (A) Distinctive wooly haircoat. (B, upper) Cross section of the heart reveals subepicardial myocardial loss with fibrous replacement of the right ventricular free wall. (B, lower) Cross section of the heart reveals the same pathologic process in both ventricles associated with calcification. (C) Right ventricular free wall with extensive subepicardial myocyte loss and replacement fibrosis embedding surviving myocytes (Masson's trichrome stain) [41].
The need to set up international registries for ARVC has been recognized in recent years and many attempts have been accomplished after the publication of the diagnostic Task Force criteria in 1994 [43,44]. A multidisciplinary collaborative European study has been designed with the aim to investigate the clinical, pathological and genetic features of ARVC. The completed European registry had been coordinated in Padua and benefited from the contributions of researchers from France, Germany, Greece, the Netherlands, Poland and the United Kingdom [45]. Simultaneously, another major registry in North America was coordinated by Dr. Frank Marcus in collaboration with European expertise coordinated by Dr. Gaetano Thiene [46].
The labor of these large registries has bared fruit in the form of the updated modified Task Force criteria for the diagnosis of ARVC, which was published in 2010. With the international community sharing two decades of knowledge and expertise, the modified Task Force maintained the approach of classifying the structural, histological, electrocardiographic, arrhythmic, and genetic features of ARVC into major and minor criteria [43,47]. The new modifications emphasized on quantifying the extent of RV dysfunction by echocardiography or MRI. Moreover, it was also proposed to characterize the tissue of the RV wall by evaluating the residual myocytes by morphometric analysis with less than 60% of residual myocytes as a major criterion. In addition, definite genetic linkage to ARVC–by identification of a pathogenic mutation categorized as associated or most likely associated with ARVC in the patient under evaluation–is now considered a major criterion [47].
Summary
In summary, the scientific research on ARVC clinical course and management has much progressed over the last 30 years. International collaboration has led to the establishment of diagnostic criteria and a management plan for patients with ARVC. However, more national and multinational registries and effective screening programs are still needed to overcome the risk of sudden cardiac death in this globally recognized cardiomyopathy (Figure 12).
Figure 12. .

The global perspective of ARVC: ARVC has been reported and studied in Europe, Middle East, South Africa, India, China, Japan, Australia, North America and South America.
References
- [1].Basso C, Corrado D, Marcus F, Nava A, Thiene C. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- [2].Basso C, Pilichou K, Carturan E, Rizzo S, Bauce B, Thiene G. Pathobiology of Arrhythmogenic Cardiomyopathy. Cardiac Electrophysiology Clinics. 2011;3:193–204. [Google Scholar]
- [3].Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97:530–539. doi: 10.1136/hrt.2010.193276. [DOI] [PubMed] [Google Scholar]
- [4].Thiene G, Corrado D, Basso C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet journal of rare diseases. 2007;2:45. doi: 10.1186/1750-1172-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiology in review. 1999;7:127–135. doi: 10.1097/00045415-199905000-00009. [DOI] [PubMed] [Google Scholar]
- [6].Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? Journal of the American College of Cardiology. 2003;42:1959–63. doi: 10.1016/j.jacc.2003.03.002. [DOI] [PubMed] [Google Scholar]
- [7]. Lancisi, G. De motu cordis et anuerysmatibus. 1738.
- [8].Marcus F, Fontaine G, Guiraudon G, Frank R, Laurenceau J, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–98. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- [9].Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, Scampardonis G. Cardiac abnormalities in familial palmoplantar keratosis. British heart journal. 1986;56:321–326. doi: 10.1136/hrt.56.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Martini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia: a study involving nine families. Journal of the American College of Cardiology. 1988;12:1222–1228. doi: 10.1016/0735-1097(88)92603-4. [DOI] [PubMed] [Google Scholar]
- [11].Thiene G, Nava A, Corrado D, Rossi L. Cardiomyopathy and sudden death in young people. New England Journal of Medicine. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- [12].Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Human molecular genetics. 1994;3:959–962. doi: 10.1093/hmg/3.6.959. [DOI] [PubMed] [Google Scholar]
- [13].McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- [14].Norgett E, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh M, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Human molecular genetics. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- [15].Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. American journal of human genetics. 2002;71:1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. European heart journal. 2005;26:1666–1675. doi: 10.1093/eurheartj/ehi341. [DOI] [PubMed] [Google Scholar]
- [17].Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz J, McKenna W. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. American journal of human genetics. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nature genetics. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- [19].Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- [20].Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. American journal of human genetics. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, Bohms B, Schulz U, Zu Knyphausen E, Vorgerd M, Gummert J, Milting H. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Human molecular genetics. 2010;19:4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- [22].Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, Danieli GA, Lorenzon A, Nava A, Bauce B, Thiene G, Basso C, Calkins H, Gear K, Marcus F, Towbin JA. Multidisciplinary Study of Right Ventricular Dysplasia Investigators. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, De Bortoli M, Rigato I, Mazzotti E, Steriotis A, Marra MP, Towbin JA, Thiene G, Danieli GA, Rampazzo A. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart rhythm. 2010;7:22–29. doi: 10.1016/j.hrthm.2009.09.070. [DOI] [PubMed] [Google Scholar]
- [24].Hulot J, Jouven X, Empana J, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879–1884. doi: 10.1161/01.CIR.0000143375.93288.82. [DOI] [PubMed] [Google Scholar]
- [25].Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29–37. doi: 10.1161/01.cir.86.1.29. [DOI] [PubMed] [Google Scholar]
- [26].Wichter T, Paul TM, Eckardt L, Gerdes P, Kirchhof P, Böcker D, Breithardt G. Arrhythmogenic right ventricular cardiomyopathy. Antiarrhythmic drugs, catheter ablation, or ICD? Herz. 2005;30:91–101. doi: 10.1007/s00059-005-2677-6. [DOI] [PubMed] [Google Scholar]
- [27].Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, Tjan TD, Soeparwata R, Block M, Borggrefe M, Scheld HH, Breithardt G, Böcker D. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004;109:1503–1508. doi: 10.1161/01.CIR.0000121738.88273.43. [DOI] [PubMed] [Google Scholar]
- [28].Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. Journal of the American Medical Association. 2006;296:1593–1601. doi: 10.1001/jama.296.13.1593. [DOI] [PubMed] [Google Scholar]
- [29].Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, Zanotto G, Verlato R, Vergara G, Delise P, Turrini P, Basso C, Naccarella F, Maddalena F, Estes NA, 3rd, Buja G, Thiene G. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. doi: 10.1161/01.CIR.0000103130.33451.D2. [DOI] [PubMed] [Google Scholar]
- [30].Suárez-Mier P, Aguilera B. Causes of sudden death during sports activities in Spain. Revista española de cardiología. 2002;55:347–358. [PubMed] [Google Scholar]
- [31].de Noronha S, Sharma S, Papadakis M, Desai S, Whyte G, Sheppard M. Aetiology of sudden cardiac death in athletes in the United Kingdom: a pathological study. Heart. 2009;95:1409–1414. doi: 10.1136/hrt.2009.168369. [DOI] [PubMed] [Google Scholar]
- [32]. Narin N, et al. ,. Arrhythmogenic right ventricular cardiomyopathy (Naxos disease): report of a Turkish boy Pacing and clinical electrophysiology 2003. 26 2326 2329 [DOI] [PubMed] [Google Scholar]
- [33].Djabali K, Martinez-Mir A, Horev L, Klapholz L, Glaser B, Christiano A. Evidence for extensive locus heterozeneity in Naxos disease. J Invest Dermatol. 2002;118:557–560. doi: 10.1046/j.0022-202x.2001.01627.x. [DOI] [PubMed] [Google Scholar]
- [34].Watkins DA, Hendricks N, Shaboodien G, Mbele M, Parker M, Vezi BZ, Latib A, Chin A, Little F, Badri M, Moolman-Smook JC, Okreglicki A, Mayosi BM. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the arrhythmogenic right ventricular cardiomyopathy registry of South Africa. Heart Rhythm. 2009;6:S10–S17. doi: 10.1016/j.hrthm.2009.08.018. [DOI] [PubMed] [Google Scholar]
- [35].van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- [36].Komura M, Suzuki J, Adachi S, Takahashi A, Otomo K, Nitta J, Nishizaki M, Obayashi T, Nogami A, Satoh Y, Okishige K, Hachiya H, Hirao K, Isobe M. Clinical course of arrhythmogenic right ventricular cardiomyopathy in the era of implantable cardioverter-defibrillators and radiofrequency catheter ablation. International heart journal. 2010;51:34–40. doi: 10.1536/ihj.51.34. [DOI] [PubMed] [Google Scholar]
- [37].Ma KJ, Li N, Wang HT, Chu JM, Fang PH, Yao Y, Ma J, Hua W, Zhang S, Wang FZ, Li Z, Pu JL. Clinical study of 39 Chinese patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Chinese Medical Journal. 2009;22:1133–1138. [PubMed] [Google Scholar]
- [38].Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- [39].Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. Journal of the American Academy of Dermatology. 1998;39:418–420. doi: 10.1016/s0190-9622(98)70317-2. [DOI] [PubMed] [Google Scholar]
- [40].Prompona M, Kozlik-Feldmann R, Mueller-Hoecker J, Reiser M, Huber A. Images in cardiovascular medicine. Magnetic resonance imaging characteristics in Carvajal syndrome (variant of Naxos disease) Circulation. 2007;116:e524–530. doi: 10.1161/CIRCULATIONAHA.107.704742. [DOI] [PubMed] [Google Scholar]
- [41].Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovascular pathology. 2004;13:185–194. doi: 10.1016/j.carpath.2004.03.609. [DOI] [PubMed] [Google Scholar]
- [42].Simpson M, Cook R, Solanki P, Patton M, Dennis J, Crosby A. A mutation in NFkappaB interacting protein 1 causes cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle. Animal genetics. 2009;40:42–46. doi: 10.1111/j.1365-2052.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- [43].McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society. Heart. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G, Wichter T. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Need for an International Registry. Circulation. 2000;101:e101–e106. doi: 10.1161/01.cir.101.11.e101. [DOI] [PubMed] [Google Scholar]
- [45].Basso C, Wichter T, Danieli GA, Corrado D, Czarnowska E, Fontaine G, McKenna WJ, Nava A, Protonotarios N, Antoniades L, Wlodarska K, D'Alessi F, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: clinical registry and database, evaluation of therapies, pathology registry, DNA banking. European heart journal. 2004;25:531–534. doi: 10.1016/j.ehj.2003.12.025. [DOI] [PubMed] [Google Scholar]
- [46].Marcus F, Towbin JA, Zareba W, Moss A, Calkins H, Brown M, Gear K, ARVD/C Investigators Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C): a multidisciplinary study: design and protocol. Circulation. 2003;107:2975–2978. doi: 10.1161/01.CIR.0000071380.43086.29. [DOI] [PubMed] [Google Scholar]
- [47].Marcus F, McKenna W, Sherrill D, Basso C, Bauce B, Bluemke D, Calkins H, Corrado D, Cox M, Daubert J, Fontaine G, Gear K, Hauer R, Nava A, Picard M, Protonotarios N, Saffitz J, Sanborn D, Steinberg J, Tandri H, Thiene G, Towbin J, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. European heart journal. 2010;31:806–814. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]