

Abstract
Lower vertebrates, such as newt and zebrafish, retain a robust cardiac regenerative capacity following injury. Recently, our group demonstrated that neonatal mammalian hearts have a remarkable regenerative potential in the first few days after birth. Although adult mammals lack this regenerative potential, it is now clear that there is measurable cardiomyocyte turnover that occurs in the adult mammalian heart. In both neonatal and adult mammals, proliferation of pre-existing cardiomyocytes appears to be the underlying mechanism of myocyte turnover. This review will highlight the advances and landmark studies that opened new frontiers in cardiac regeneration.
Introduction
Cardiovascular disease is the leading cause of morbidity and mortality in the developed world.1 Thus, numerous attempts at unraveling the regenerative potential of the mammalian heart have been pursued.2 Earlier advancements on the regenerative capabilities of lower organisms have paved the road for a new understanding of the ability of the mammalian heart to regenerate following injury. Recent evidence identified an inherent regenerative capacity of the mammalian heart during the neonatal period, resembling their evolutionary ancestors.3–5 Here, we will discuss the recent rapid advancements in the heart regeneration field, with focus on cardiomyocyte proliferation.
Regeneration in lower vertebrates
Tissue regenerative capacity varies widely between species, with some lower organisms displaying a remarkable ability to regenerate various organs and extremities. For example, adult zebrafish effectively regenerate multiple organs and structures, including amputated fins, injured retinae, transected optic nerves and spinal cord.6–8 Zebrafish also displays a robust natural capacity for heart regeneration, making it a very useful model system.9,10 Zebrafish heart regeneration was studied by examining the effects of removing 20% of the ventricle by surgical resection. This resulted in an initial fibrotic response, and cardiomyocyte proliferation within first two week following amputation. This was followed by complete regeneration within 60 days post-injury.10 To address the source of the newly formed cardiomyocytes, genetic fate-mapping studies were performed to trace the lineage of cardiomyocytes using the Cre/lox system. These studies demonstrated that the vast majority of heart muscle cells formed during the process of zebrafish heart regeneration arise from proliferation of the pre-existing cardiomyocytes, rather than progenitor cells.11,12
Since then several zebrafish cardiac injury models, that yield distinct outcomes of myocardial regeneration, have been described. For example, the infarct models of zebrafish after cryoinjury to the ventricular apex, resulted in necrosis of approximately 25% of ventricular tissue, followed by myocyte proliferation and regeneration of heart within 130 days post injury.13,14 However, perfect ventricular shape was not restored, which highlights the heterogeneity of the regenerative response after different types of injury. In addition, Wang and colleagues generated an elegant Z-CAT (zebrafish cardiomyocyte ablation transgene) model using a 4-hydroxytamoxifen (4-HT)–inducible Cre recombinase (CreER) system to facilitate cell type–specific ablation. Intriguingly, the mechanism of regeneration in all of these models was myocyte proliferation despite the varying degrees of cell death and regenerative response (Table 1). Moreover, recent hypoxic cardiac injury models in zebrafish also demonstrated evidence of cardiac oxidative stress, inflammation, and proliferation; which mimics ischemic injury in mammalian heart.15,16 Ultimately, in these models of heart regeneration, cardiomyocyte proliferation was the key mediator of the regenerative process.
Table 1 .
Cardiac Regenerative Capacity in Different Models.
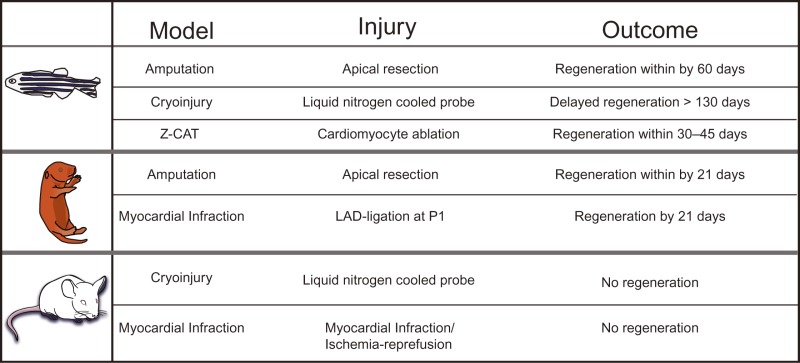 |
Outline of cardiac regenerative potential of zebrafish, neonatal and adult mouse hearts following various types of injury.
Similarly, newts are able to renew lost or injured body parts including jaw, spinal cord, gut, limbs, brain, and heart.17–22 The newt heart was one of the first models of heart regeneration described.23 As in zebrafish, myocyte proliferation appears to be the underlying mechanism of newt heart regeneration.24 More recently, resection injuries at the base of the newt heart has been reported to completely regenerate,25 providing a good model of complete myocardial regeneration. These pioneering studies in lower vertebrates indicate that activating cardiomyocyte proliferation is the key mediator of heart regeneration.
Neonatal heart regeneration
In contrast to lower vertebrates, adult mammalian heart cardiomyogenesis is very limited and is insufficient to restore normal cardiac function following injury. Studies in the late 1990s elegantly mapped the DNA synthesis and cell cycle dynamics of the mammalian heart during development and after birth,26 where they showed that DNA synthesis drops significantly around birth with low-level DNA synthesis few days after birth. Around P5 to P7, cardiomyocytes undergo a final round of DNA synthesis that correspond to binucleation, and the majority of cardiomyocytes then permanently exit the cell cycle (Figure 1A). Therefore, due to the similarities between the immature mammalian heart and lower vertebrates (Table 1),9,27 it became important to determine whether higher mammals have similar regenerative abilities.
Figure 1. .

Proliferative Window of Postnatal Cardiomyocytes A) Cardiomyocyte DNA synthesis (left index) significantly drops around birth with low-level synthesis few days after birth with corresponding increase in the binucleation (right index) (Adapted from Soonpaa et al.26). This correlates with the cardiac growth being hyperplastic and hypertrophic before and after birth respectively (lower panel). E, embryonic; P, postnatal days. B) Schematic of neonatal heart 21 days post myocardial regeneration (MI) at P1 showing complete full thickness regeneration with minimal fibrosis (blue), while decreased regenerative capacity following MIs induced at P7, and no detectable regeneration with more fibrosis following MIs induced at P14 was seen (lower panel).
Recently, we demonstrated that removal of up to 15% of the apex of the left ventricle of postnatal day 1 (P1) mice results in complete regeneration within three weeks without any measurable fibrosis and cardiac dysfunction.3 This response was well characterized by an early transient extracellular matrix deposition, and a robust cardiomyocyte proliferation with gradual restoration of normal cardiac morphology. Fibrotic scarring and hypertrophy, both typical hallmarks of the injured adult mammalian heart, were absent in the neonatal heart following regeneration. In addition to the histological evidence of proliferating myocytes, genetic fate-mapping studies confirmed that the majority of newly formed cardiomyocytes are derived from the proliferation of preexisting cardiomyocytes.3 This robust regenerative response occurred in the brief window of time after birth during which cardiomyocytes are mononucleated and are able to proliferate. However, this regenerative capacity is lost by P7, after which injury results in the typical cardiomyocyte hypertrophy scar-formation characteristic of the adult mammalian heart. Not surprisingly, loss of this regenerative potential coincides with the binucleation and cell cycle exit of cardiomyocytes.26,27
More recently, we established an ischemic injury model where the left anterior descending coronary artery was ligated in P1 neonates.5 The injury response was similar to the resection model, with robust cardiomyocyte proliferation throughout the myocardium, as well as restoration of normal morphology and systolic function (Figure 1B). This regenerative phenomenon provides an invaluable tool for unraveling the molecular networks that regulate mammalian heart regeneration, and the mechanism of post-natal cardiomyocyte cell cycle arrest. Insights from this transition could provide important clues on how to reactivate myocyte cell cycle in the adult heart.
This close temporal relationship between cardiomyocyte proliferation and heart regeneration amongst animals raises the question about the extent of cardiomyocyte proliferation in humans. During postnatal development, the human heart was believed to grow in size through cardiomyocyte hypertrophy rather than cardiomyocyte hyperplasia. However, recent studies showed that cardiomyocyte proliferation contributes to developmental heart growth in young humans.28 This report showed that the percentages of cardiomyocytes in mitosis were highest at 0.04% in infants and decreased to 0.009% by 20 years. Similarly, cytokinesis of cardiomyocytes decreases from 0.016% at birth to 0.005% by adolescent humans and completely absent in adults. Interestingly, the number of cardiomyocytes in the left ventricle increased by 3.4 fold from birth to over a period of 20 years of life, which intriguingly was consistent with measured cardiomyocyte cell cycle activity.28 Clinical studies have also shown that in newborns who underwent surgical repair of the anomalous left coronary artery (LCAPA) showed that the majority of improvement in their ejection fraction occurred when the defect was corrected in the first 3 months following birth, but not after.29 This suggests that myocyte hyperplasia may play a role in the ability of the infant heart to recover. Together, these studies imply that infants may in fact be capable of regenerating myocardial tissue following injury, and that stimulating cardiomyocyte proliferation would be a promising approach for older patients.
Myocyte turnover in adult mammalian heart
In sharp contrast to lower vertebrates and neonatal mammals, the adult mammalian heart has a limited regenerative capacity following injury and responds to cardiac tissue damage by scarring. However, over the past decade, it has become clear that the adult mammalian heart is not a terminally differentiated organ, and that there is constant cardiomyocyte turnover occurring within the adult mammalian, murine and human hearts throughout life.30–34
Cardiomyocyte turnover in the murine heart was established through quantitative studies of DNA synthesis using autoradiographic measurement, which indicated that turnover rates of cardiomyocytes are around 0.0006% in uninjured hearts, while following injury the rate was 0.0083%.35,36 This mapping of the limited, but measurable ability of adult murine cardiomyocytes to enter the cell cycle generated a promising potential for therapeutic manipulation of these proliferative cardiomyocytes.
Similarly, several other reports have documented the formation of new cardiomyocytes in the human heart. Data from the nuclear fallout during the cold war by using 14C dating to measure cardiomyocyte age indicated that turnover rates of cardiomyocytes are about 1% per year at the age of 20 years.30 These rate further decreases to 0.4% around 75 years of age, and approximately 45% of cardiomyocytes are replaced over the normal human lifespan.30
These findings suggested that the adult mammalian heart maintains a measurable capacity for cardiomyocyte turnover throughout life, which provides a rationale for manipulating the cell cycle activity of cardiomyocytes as a strategy for myocardial regeneration. However, several questions needed to be addressed; in particular, identifying the source of newly formed cardiomyocytes, and whether this inherent capacity can be augmented to replenish lost cardiomyocytes.
Recently, a landmark study described the dynamics of cardiomyocyte turnover in the adult mammalian heart.37 Senyo and colleagues adapted a novel multi isotope mass spectrometry (MIMS) analysis to have better understanding of the origin and regeneration rate of cardiomyocytes in the adult murine heart. By using nonradioactive stable nitrogen isotope 15N label that marks DNA of cells undergoing mitosis, they showed that in young adult mice, the cardiomyocyte turnover rate per year was 0.76%, and this turnover rate decreases with aging mice. Intriguingly, this is similar to the cardiomyocyte turnover dynamics described previously in humans30. More importantly, since cardiomyocytes can replicate their DNA without completing the cell cycle and resulting in polyploidy and multinucleation as has been previously shown38; the authors show that a higher number of the 15N+ cardiomyocytes are diploid, indicative of cell division in the uninjured adult heart. In addition, analysis of GFP labeled cardiomyocytes and genetic fate mapping studies in double transgenic MerCreMer/ZEG mice on induction with 4-OH-tamoxifen showed that the newly formed cardiomyocytes are derived from preexisting cardiomyocytes, in both during aging and following myocardial infarction. Interestingly, the authors showed an increase in cardiomyocyte proliferation at the border zone following myocardial infarction, in contrast to a previous study that showed increased ploidy rather than cell division39. The authors do not exclude the possibility that a progenitor population contributed to the newly formed cardiomyocytes following injury through differentiation without proliferation, which might explain earlier confounding results40. Therefore, conclusive genetic fate mapping studies suggest that pre-existing cardiomyocytes are indeed the source of cardiomyocyte turnover in both the neonatal and adult hearts, which sets the stage for therapeutic strategies aimed at manipulating cardiomyocyte cell cycle as a strategy for heart regeneration.
Forced Cardiomyocyte Proliferation in Mouse
Lower vertebrate cardiomyocytes are smaller in size and mononucleated with less myofibrils that ease cell cycle reentry following injury.41 In contrast, mammalian cardiomyocytes mainly proliferate during embryonic development and a short period after birth. Following postnatal switch, cardiomyocytes turnover at an extremely low rate as described above and cardiac hypertrophy becomes the major growth mechanism of cardiomyocytes.
The mammalian cell cycle is tightly regulated by a combination of positive and negative regulators such as cyclins, cyclin-dependent kinases (CDKs), CDK inhibitors (CDKIs), CDK activating kinase (CAK) and the E2F family of transcription factors. Cyclin/CDK complexes show a periodic accumulation and degradation pattern, and require activation by CAKs through phosphorylation.42–45 Induction of cell cycle progression by Cyclin-D/CDK4-6 and Cyclin-E/CDK2 complexes allow progression through G1/S transition phase. Cyclin-E/CDK2 complex play a significant role in the phosphorylation of Rb and activation of E2F transcription factors for cell cycle progression. However, these two complexes are under regulation of CDKIs such as Cip/Kip family (p21Cip1, p27Kip1, p57Kip2) and Ink4 family (p15Ink4b, p16Ink4a, p18Ink4c, p19Ink4d) which directly bind and inhibit cyclin-D/E/A dependent kinases and CyclinD-CDK4/6 complex respectively.30,46 One other important role of the Cyclin-D/CDK4-6 complex in addition to activation of E2F is sequestering p21, thereby preventing its interaction with Cyclin-E/CDK2 complex and which inhibits cell cycle progression.
In adult mammalian hearts, cardiomyocytes show increased levels of CDKIs and decreased activity of positive regulators of cell cycle.47–52 Henceforth, reactivation of the cardiomyocyte cell cycle is possible by suppression of cell cycle inhibitors or by over-expressing cell cycle activators. Over-expression studies of viral oncoprotein SV40 T-Antigen, known to stimulate entry of quiescent or differentiated cells into S phase, induced continuous cycling of cardiomyocytes in both embryonic and adult hearts.53,54 Similarly, transgenic mice over-expressing cell cycle regulators such as Cyclin D2, Cyclin A4 and Cyclin D1 in postnatal heart also demonstrate evidence of mitosis and induction of CDK2/4 levels associated with DNA synthesis.26,55–59 In addition, improved cardiac function after MI was detected following overexpression of the cell cycle activator Cyclin D2 in cardiomyocytes.56 Conversely, deletion of CDKI p27Kip1 or immunodepletion of p21Cip1 in cardiomyocytes is associated with a trend towards progression to S-phase.47,48 Furthermore, E2F-1, which plays important role in cell cycle control, has been shown to induce cardiomyocyte DNA synthesis in vitro and in vivo.60
Studies have also demonstrated induction of cardiomyocyte cell cycle in vivo in response to mitogens such as periostin, neuregulin-1, and FGF1, a response that seems to be limited to a subpopulation of mononucleated cardiomyocytes.61–64 Although, some of the effects of periostin on cell cycle could not be reproduced.65 More recently, activation of the evolutionary conserved Hippo signaling pathway has a remarkable effect on increasing cardiomyocyte proliferation beyond the postnatal proliferative window in mice.66–68 Thus, the approach to coax adult cardiomyocytes to reenter the cell cycle holds a lot of potential for cardiac regeneration.
While these important studies demonstrated proof of principal that induction of myocyte cell cycle is possible, the molecular mechanisms responsible for postnatal cardiomyocyte cell cycle arrest remained unclear. Recently we identified the bHLH transcription factor Meis1 as a key transcriptional regulator of the cardiomyocyte cell cycle upstream of two synergistic CDKI inhibitors (p21, p16). Meis1 mRNA levels, and nuclear localization significantly increase in cardiomyocytes at the time of cell cycle arrest (Figure 2). Moreover, Meis1 deletion in mouse cardiomyocytes is sufficient for extension of the postnatal proliferative window of cardiomyocytes, and for re-activation of cardiomyocyte mitosis in the adult heart with no deleterious effect on cardiac function.69 In addition, Meis1 KO results in upregulation of Ccnd2, MCM3, Chek1 and downregulation of APbb1, TP53, Gpr132, all of which have a critical role in cell cycle regulation. Although the mechanism of activation of Meis1 in the postnatal heart is not fully understood, these findings provide evidence for the possibility of reactivating dormant regenerative programs in adult mammals at the molecular level that can be potential therapeutic targets. Most importantly, rigorous analysis is required to identify the evolutionary advantages for cardiomyocyte cell cycle exit, and to what extent can we harness the benefits of cardiomyocyte proliferation.
Figure 2. .

Meis1 Regulates Cardiomyocyte Cell Cycle Arrest Expression profile of Meis1 demonstrates perinuclear in P1, nuclear localization at P7 and P14 cardiomyocytes that corresponds to active cell cycle in P1 and arrest in P7 and P14. Conversely, this is switched in P14 Meis1 knock out models with reactivation of cell cycle and myocyte proliferation.
MicroRNAs and Cardiomyocyte Proliferation
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression by repression of their target gene transcripts.70 Although miRNAs are fine tuners of gene expression, they can target multiple members of a common pathway and thus regulate multiple biological processes. Thus, searching for miRNAs that regulate cardiomyocyte cell cycle has been pursued.71 Members of the miR-15 family were identified as being highly upregulated during the postnatal period when mammalian cardiomyocytes binucleate and become terminally differentiated.72 Myocardial-specific overexpression of miR-195, a member of the miR-15 family, decreased cardiomyocyte proliferation,72 and inhibits neonatal heart regeneration.5 In addition, pharmacological inhibition of miR-15 family members using LNA-modified antimiRs at birth until adulthood increases cardiomyocyte proliferation and improved left ventricular systolic function following ischemia-reperfusion (I/R) injury.5 Protection from I/R injury and reduced apoptosis was also observed following antimiR administration in adult mice.73 Similarly, miRNAs regulate zebrafish heart regeneration, where miR-133 was shown to be downregulated during regeneration,74 while overexpression of miR-133 reduced cardiomyocyte proliferation and inhibited zebrafish cardiac regenerative. These data combined indicate that miRNAs may be negative regulators of cardiomyocyte proliferation and heart regeneration.
Conversely, some miRNAs were found to be positive regulators of heart regeneration. To identify miRNAs that induce cardiomyocyte proliferation, a large-scale screen representing 988 mature miRNAs was performed that could specifically enhance the proliferation of neonatal rodent cardiomyocytes in vitro.75 Surprisingly, more than 40 miRNAs that could independently trigger cardiomyocyte mitosis were identified.75 Of these miRNAs, miR-199a and miR-590 were shown to induce cell-cycle re-entry and cytokinesis of adult cardiomyocytes in vitro. In addition, in vivo induction of miR-199a as well as miR-590 in the heart by adeno-associated viruses led to increased cardiomyocyte proliferation, and improvement of function following myocardial infarction. A more recent study identified the miR-17-92 cluster as another positive regulator of cardiomyocyte proliferation in postnatal and adult hearts.76 Deletion of miR-17-92 reduces cardiomyocyte proliferation in embryonic, postnatal and adult stages. In addition, overexpression of miR-17-92 increases adult cardiomyocyte proliferation and improved heart function following injury. These studies suggest that stimulating cardiomyocyte proliferation is a powerful tool for replacement of lost myocardium following injury.
Concluding Remarks
The cardiac regeneration field has evolved rapidly over the past two decades. Although it was previously thought that the heart was a post mitotic organ, it is now clear that there is measurable myocyte turnover that occurs in the adult mammalian and human hearts. The excitement created by the realization that the heart is not a terminally differentiated organ has undoubtedly created a gold rush approach to cardiac regeneration therapy. While the mechanism of myocyte turnover in the adult mammalian heart is not well understood, a plausible strategy to achieve the elusive goal of cardiac regeneration may lie within cardiomyocytes themselves. It is important here to draw a distinction between the endogenous regenerative capacity of the myocardium, and the prospect of cell therapy as a treatment for systolic dysfunction, which may activate alternative repair mechanisms. The studies discussed in this review leave little doubt that pre-existing cardiomyocytes are indeed the source of cardiomyocyte turnover in lower vertebrates as well as neonatal and adult hearts, and have become a major focus of cardiac regenerative medicine (Figure 3). These studies have opened the door for exploration of future therapeutic strategies focused on harnessing the power of myocyte proliferation for heart regeneration. One critical question, which likely holds the key to heart regeneration, remains unanswered; Why, and not how, does the mammalian heart permanently turn off this remarkable regenerative capacity shortly after birth?
Figure 3. .
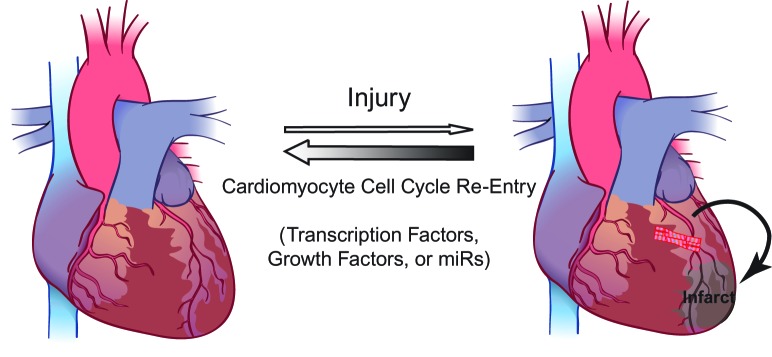
Endogenous Heart Regeneration Schematic of endogenous cardiac regenerative mechanism, which occurs through proliferation of preexisting cardiomyocytes. Future therapeutic strategies could focus on a number of pathways to enhance this phenomenon through maniplulation of direct cell cycle regulators, transcription factors, growth factors or microRNAs.
Acknowledgements
We apologize to whose work was not cited in this review and for the omission of some discussion points related to myocyte regeneration owing to space constraints. This work was supported by grants from the American Heart Association (Grant in Aid) (Sadek), the Gilead Research Scholars Program in Cardiovascular Disease (Sadek), Foundation for Heart Failure Research, NY and the NIH (1R01HL115275-01) (Sadek).
Footnotes
# These authors contributed equally to this work.
References
- 1.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. doi:10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 2.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. doi:10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. doi:10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahmoud AI, Porrello ER. Turning back the cardiac regenerative clock: lessons from the neonate. Trends Cardiovasc Med. 2012;22:128–133. doi: 10.1016/j.tcm.2012.07.008. doi:10.1016/j.tcm.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187–192. doi: 10.1073/pnas.1208863110. doi:10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroehne V, Freudenreich D, Hans S, Kaslin J, Brand M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development. 2011;138:4831–4841. doi: 10.1242/dev.072587. doi:dev.072587 [pii] 10.1242/dev.072587. [DOI] [PubMed] [Google Scholar]
- 7.Poss KD, Keating MT, Nechiporuk A. Tales of regeneration in zebrafish. Dev Dyn. 2003;226:202–210. doi: 10.1002/dvdy.10220. doi:10.1002/dvdy.10220. [DOI] [PubMed] [Google Scholar]
- 8.Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000;44:289–307. doi: 10.1002/1097-4695(20000905)44:3<289::aid-neu1>3.0.co;2-h. doi:10.1002/1097-4695(20000905)44:3 < 289:AID-NEU1>3.0.CO;2-H [pii] [DOI] [PubMed] [Google Scholar]
- 9.Poss KD. Getting to the heart of regeneration in zebrafish. Semin Cell Dev Biol. 2007;18(1):36–45. doi: 10.1016/j.semcdb.2006.11.009. doi:10.1016/j.semcdb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. doi:10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 11.Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–609. doi: 10.1038/nature08899. doi:10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–605. doi: 10.1038/nature08804. doi:10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chablais F, Veit J, Rainer G, Jazwinska A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev Biol. 2011;11:21. doi: 10.1186/1471-213X-11-21. doi:1471-213X-11-21 [pii] 10.1186/1471-213X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Rosa JM, Martin V, Peralta M, Torres M, Mercader N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development. 2011;138:1663–1674. doi: 10.1242/dev.060897. doi:10.1242/dev.060897. [DOI] [PubMed] [Google Scholar]
- 15.Jopling C, Sune G, Faucherre A, Fabregat C, Izpisua Belmonte JC. Hypoxia induces myocardial regeneration in zebrafish. Circulation. 2012;126:3017–3027. doi: 10.1161/CIRCULATIONAHA.112.107888. doi:CIRCULATIONAHA.112.107888 [pii] 10.1161/CIRCULATIONAHA.112.107888. [DOI] [PubMed] [Google Scholar]
- 16.Parente V, Balasso S, Pompilio G, Verduci L, Colombo GI, Milano G, Guerrini U, Squadroni L, Cotelli F, Pozzoli O, Capogrossi MC. Hypoxia/reoxygenation cardiac injury and regeneration in zebrafish adult heart. PLoS One. 2013;8:e53748. doi: 10.1371/journal.pone.0053748. doi:10.1371/journal.pone.0053748 PONE-D-12-29126 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brockes JP, Kumar A. Appendage regeneration in adult vertebrates and implications for regenerative medicine. Science. 2005;310:1919–1923. doi: 10.1126/science.1115200. doi:10.1126/science.1115200. [DOI] [PubMed] [Google Scholar]
- 18.Eguchi G. Cellular and molecular background of wolffian lens regeneration. Cell Differ Dev. 1988;25:147–158. doi: 10.1016/0922-3371(88)90111-6. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, Thorogood P, Ferretti P. Regenerative capability of upper and lower jaws in the newt. Int J Dev Biol. 1994;38:479–490. [PubMed] [Google Scholar]
- 20.Keefe JR. An analysis of urodelian retinal regeneration. IV. Studies of the cellular source of retinal regeneration in Triturus cristatus carnifex using 3 H-thymidine. J Exp Zool. 1973;184:239–258. doi: 10.1002/jez.1401840209. doi:10.1002/jez.1401840209. [DOI] [PubMed] [Google Scholar]
- 21.Minelli G, Franceschini V, Del Grande P, Ciani F. Newly-formed neurons in the regenerating optic tectum of Triturus cristatus carnifex. Basic Appl Histochem. 1987;31:43–52. [PubMed] [Google Scholar]
- 22.O'Steen WK, Walker BE. Radioautographic studies or regeneration in the common newt. III. Regeneration and repair of the intestine. Anat Rec. 1962;142:179–187. doi: 10.1002/ar.1091420210. [DOI] [PubMed] [Google Scholar]
- 23.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187(2):249–253. doi: 10.1002/jez.1401870208. doi:10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- 24.Flink IL. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anat Embryol (Berl) 2002;205(3):235–244. doi: 10.1007/s00429-002-0249-6. doi:10.1007/s00429-002-0249-6. [DOI] [PubMed] [Google Scholar]
- 25.Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol. 2011;354:67–76. doi: 10.1016/j.ydbio.2011.03.021. doi:S0012-1606(11)00185-0 [pii] 10.1016/j.ydbio.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 26.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–H2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 27.Walsh S, Ponten A, Fleischmann BK, Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo–an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365–373. doi: 10.1093/cvr/cvq005. doi:10.1093/cvr/cvq005. [DOI] [PubMed] [Google Scholar]
- 28.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kühn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110. doi:10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michielon G, Di Carlo D, Brancaccio G, Guccione P, Mazzera E, Toscano A, Di Donato RM. Anomalous coronary artery origin from the pulmonary artery: correlation between surgical timing and left ventricular function recovery. Ann Thorac Surg. 2003;76:581–588. doi: 10.1016/s0003-4975(03)00344-8. discussion 588, doi:S0003497503003448 [pii] [DOI] [PubMed] [Google Scholar]
- 30.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. doi:10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergmann O, Zdunek S, Frisén J, Bernard S, Druid H, Jovinge S. Cardiomyocyte renewal in humans. Circ Res. 2012;110:e17–e18. doi: 10.1161/CIRCRESAHA.111.259598. author reply e19–21, doi:110/1/e17 [pii] 10.1161/CIRCRESAHA.111.259598. [DOI] [PubMed] [Google Scholar]
- 32.Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002;90:634–640. doi: 10.1161/01.res.0000014822.62629.eb. [DOI] [PubMed] [Google Scholar]
- 33.Nadal-Ginard B. Generation of new cardiomyocytes in the adult heart: prospects of myocardial regeneration as an alternative to cardiac transplantation. Rev Esp Cardiol. 2001;54:543–550. doi: 10.1016/s0300-8932(01)76354-3. [DOI] [PubMed] [Google Scholar]
- 34.Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. doi:10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 35.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–H226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 36.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 37.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. doi:nature11682 [pii] 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marbán E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med. 2013;5:191–209. doi: 10.1002/emmm.201201737. doi:10.1002/emmm.201201737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hesse M, Raulf A, Pilz GA, Haberlandt C, Klein AM, Jabs R, Zaehres H, Fügemann CJ, Zimmermann K, Trebicka J, Welz A, Pfeifer A, Röll W, Kotlikoff MI, Steinhäuser C, Götz M, Schöler HR, Fleischmann BK. Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat Commun. 2012;3:1076. doi: 10.1038/ncomms2089. doi:10.1038/ncomms2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. doi:10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rumyantsev PP. Interrelations of the proliferation and differentiation processes during cardiact myogenesis and regeneration. Int Rev Cytol. 1977;51:186–273. [PubMed] [Google Scholar]
- 42.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–1054. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 43.Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci. 2002;59:126–142. doi: 10.1007/s00018-002-8410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowell JD, Field LJ, Pasumarthi KB. Cell cycle regulation to repair the infarcted myocardium. Heart Fail Rev. 2003;8:293–303. doi: 10.1023/a:1024738104722. [DOI] [PubMed] [Google Scholar]
- 45.Lafontant PJ, Field LJ. The cardiomyocyte cell cycle. Novartis Found Symp. 2006;274:196–207. doi: 10.1002/0470029331.ch12. discussion 208–113, 272–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. doi:1208619 [pii] 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 47.Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85:117–127. doi: 10.1161/01.res.85.2.117. [DOI] [PubMed] [Google Scholar]
- 48.Poolman RA, Gilchrist R, Brooks G. Cell cycle profiles and expressions of p21CIP1 AND P27KIP1 during myocyte development. Int J Cardiol. 1998;67:133–142. doi: 10.1016/s0167-5273(98)00320-9. [DOI] [PubMed] [Google Scholar]
- 49.Poolman RA, Brooks G. Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J Mol Cell Cardiol. 1998;30:2121–2135. doi: 10.1006/jmcc.1998.0808. doi:S0022-2828(98)90808-2 [pii] 10.1006/jmcc.1998.0808. [DOI] [PubMed] [Google Scholar]
- 50.Li JM, Poolman RA, Brooks G. Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol. 1998;275:H814–H822. doi: 10.1152/ajpheart.1998.275.3.H814. [DOI] [PubMed] [Google Scholar]
- 51.Brooks G, Poolman RA, McGill CJ, Li JM. Expression and activities of cyclins and cyclin-dependent kinases in developing rat ventricular myocytes. J Mol Cell Cardiol. 29:2261–2271. doi: 10.1006/jmcc.1997.0471. doi:S0022-2828(97)90471-5 [pii] 10.1006/jmcc.1997.0471. [DOI] [PubMed] [Google Scholar]
- 52.Brooks G, Poolman RA, Li JM. Arresting developments in the cardiac myocyte cell cycle: role of cyclin-dependent kinase inhibitors. Cardiovasc Res. 1998;39:301–311. doi: 10.1016/s0008-6363(98)00125-4. doi:S0008636398001254 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Field LJ. Atrial natriuretic factor-SV40 T antigen transgenes produce tumors and cardiac arrhythmias in mice. Science. 1988;239:1029–1033. doi: 10.1126/science.2964082. [DOI] [PubMed] [Google Scholar]
- 54.Sen A, Dunnmon P, Henderson SA, Gerard RD, Chien KR. Terminally differentiated neonatal rat myocardial cells proliferate and maintain specific differentiated functions following expression of SV40 large T antigen. J Biol Chem. 1988;263:19132–19136. [PubMed] [Google Scholar]
- 55.Jackson T, Allard MF, Sreenan CM, Doss LK, Bishop SP, Swain JL. The c-myc proto-oncogene regulates cardiac development in transgenic mice. Mol Cell Biol. 1990;10:3709–3716. doi: 10.1128/mcb.10.7.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–118. doi: 10.1161/01.RES.0000152326.91223.4F. doi:10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 57.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1) EMBO J. 1999;18:5310–5320. doi: 10.1093/emboj/18.19.5310. doi:10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao HS, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, Lorell BH, Izumo S. Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res. 2001;88:443–450. doi: 10.1161/01.res.88.4.443. [DOI] [PubMed] [Google Scholar]
- 59.Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99:2644–2654. doi: 10.1172/JCI119453. doi:10.1172/JCI119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, Schneider MD. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997;100:2722–2728. doi: 10.1172/JCI119817. doi:10.1172/JCI119817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. doi:10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kühn B, del Monte F, Hajjar RJ, Chang Y-S, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. doi:10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 63.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. doi:10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 64.Braun T, Dimmeler S. Breaking the silence: stimulating proliferation of adult cardiomyocytes. Dev Cell. 17:151–153. doi: 10.1016/j.devcel.2009.07.022. doi:S1534-5807(09)00306-2 [pii] 10.1016/j.devcel.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 65.Lorts A, Schwanekamp JA, Elrod JW, Sargent MA, Molkentin JD. Genetic manipulation of periostin expression in the heart does not affect myocyte content, cell cycle activity, or cardiac repair. Circ Res. 2009;104:e1–e7. doi: 10.1161/CIRCRESAHA.108.188649. doi:10.1161/CIRCRESAHA.108.188649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. doi: 10.1126/science.1199010. doi:10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.von Gisea A, Lina Z, Schlegelmilchc K, Honora LB, Pana GM, Bucka JN, Maa Q, Ishiwatag T, Zhoua B, Camargoc FD, Pua WT. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–2399. doi: 10.1073/pnas.1116136109. doi:10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. doi: 10.1126/scisignal.2002278. doi:10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253. doi: 10.1038/nature12054. doi:nature12054 [pii] 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. doi:10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Porrello ER. microRNAs in cardiac development and regeneration. Clin Sci (Lond) 2013;125:151–166. doi: 10.1042/CS20130011. doi:10.1042/CS20130011. [DOI] [PubMed] [Google Scholar]
- 72.Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW, 2nd, van Rooij E, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–679. doi: 10.1161/CIRCRESAHA.111.248880. doi:10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110:71–81. doi: 10.1161/CIRCRESAHA.111.244442. doi:10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin VP, Lepilina A, Smith A, Poss KD. Regulation of zebrafish heart regeneration by miR-133. Dev Biol. 2012;365:319–327. doi: 10.1016/j.ydbio.2012.02.018. doi:10.1016/j.ydbio.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. doi:10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 76.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang DZ. mir-17-92 Cluster is Required for and Sufficient to Induce Cardiomyocyte Proliferation in Postnatal and Adult Hearts. Circ Res. 2013;112(12):1557–1566. doi: 10.1161/CIRCRESAHA.112.300658. doi:10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]