

Abstract
In recent years, several landmark studies have provided compelling evidence that cardiomyogenesis occurs in the adult mammalian heart. However, the rate of new cardiomyocyte formation is inadequate for complete restoration of the normal mass of myocardial tissue, should a significant myocardial injury occur, such as myocardial infarction. The cellular origin of postnatal cardiomyogenesis in mammals remains a controversial issue and two mechanisms seem to be participating, proliferation of pre-existing cardiomyocytes and myogenic differentiation of progenitor cells. We will discuss the relative importance of these two processes in different settings, such as normal ageing and post-myocardial injury, as well as the strengths and limitations of the existing experimental methodologies used in the relevant studies. Further clarification of the mechanisms underlying cardiomyogenesis in mammals will open the way for their therapeutic exploitation in the clinical field, with the scope of myocardial regeneration.
Introduction
Cardiomyogenesis (generation of cardiomyocytes) had not been convincingly demonstrated in the adult mammalian heart until very recently; the potential for myocardial regeneration was only recognized in organisms such as fish and amphibians.1,2 However, carefully designed and performed studies have produced compelling evidence for the existence of cardiomyocytes in the adult heart, that were formed well after birth, in rodents and even humans.3 Although the various studies do not agree about the rate of cardiomyocyte renewal in adults, this is clearly low and inadequate to replenish the substantial losses of cells after major injuries such as a myocardial infarction. In addition, controversy surrounds the putative cellular sources of postnatal cardiomyogenesis: do new myocytes arise from proliferation of pre-exisiting ones or from cardiomyogenic differentiation of endogenous progenitors? In this review, we will present the evidence supporting the contribution of these two mechanisms in adult cardiomyogenesis and discuss their relative importance in different settings, such as normal ageing and post-myocardial injury. We will also critically present the existing methodologies that allowed the investigation of these mechanisms, with emphasis on their strengths and limitations.
Postnatal cardiomyogenesis in the mammalian heart
While lower vertebrates (such as newts1 and zebrafish2) and neonatal mice4,5 possess a robust ability for myocardial regeneration, the ability of the mammalian heart to generate myocytes beyond the early neonatal period has been controversial. During the 20th century, the postnatal mammalian heart was viewed as a post-mitotic static organ, in which increases in mass occur exclusively through myocyte hypertrophy (i.e. increase in cell size), rather than hyperplasia (increase in cell number).
The first studies hinting towards postnatal cardiomyogenesis in the human heart can be traced back to the 1970s. By employing histopathological, biochemical and cytophotometric techniques (to measure DNA content and nuclear ploidy), Adler et al. estimated the absolute cardiomyocyte number in explanted human hearts. They found that while normal hearts contained ∼2 billion cardiomyocytes, hearts with pathologic hypertrophy contained up to ∼4.8 billion cardiomyocytes.6–8 These findings suggest that adult human hearts may generate new cardiomyocytes during pathologic hypertrophy. In 1998, the Anversa group examined explanted human hearts obtained from end-stage heart failure patients and from control subjects (who died of non-cardiovascular causes). Fluorescent immunohistochemistry (IHC), for sarcomeric proteins and propidium iodide (PI, a DNA dye which labels the cell nucleus), demonstrated the presence of cardiomyocytes undergoing mitosis (either nuclear division [karyokinesis] or cell division [cytokinesis]) in control hearts (14 mitotic myocytes/million myocytes). In end-stage heart failure hearts, the number of mitotic cardiomyocytes increased 10-fold (∼140 mitotic myocytes/million myocytes).9 These rates of myocyte mitosis (if they translate into genuine proliferation) project to an annual turnover of ∼10% in the healthy adult human heart and ∼107% in the failing human heart. The same group reported even greater rates of cardiomyocyte mitosis early after myocardial infarction. Fluorescent IHC for sarcomeric proteins, PI and Ki67 (a protein expressed in the nucleus of cells in the active phases of the cell cycle [late G1, S, G2, and M phase]) in human hearts obtained from patients who died 4–12 days post myocardial infarction demonstrated Ki67 expression in 4% of myocyte nuclei in the infarct border zone and in 1% of myocytes in the remote myocardium. Mitosis (karyokinesis or cytokinesis) was observed in 0.08% of myocytes (800 mitotic myocytes/million myocytes) in the border zone and 0.03% of myocytes (300 mitotic myocytes/million myocytes) in the remote myocardium.10 If such exceptionally high rates of myocyte cell cycling could be persisted and resulted in genuine proliferation (while no significant myocyte loss occurred post-myocardial infarction beyond the initial ischemic insult), then all myocytes lost after an infarct affecting 30% of the left ventricle could be replaced within as little as 18 days.
More recently, the Anversa group investigated the rates of myocyte turnover in the aging human heart. Fluorescent IHC for sarcomeric proteins, Ki67, phosphorylated histone H3 (H3P, a marker of karyokinesis) and aurora-B-kinase (a marker of cytokinesis) was performed in explanted hearts from human subjects who died of non-cardiovascular causes. The investigators reported that myocyte turnover increases with age, and that female hearts possess a higher regenerative capacity compared to male hearts. In the female heart, myocyte turnover occurs at an annual rate of 10%, 14%, and 40% at 20, 60, and 100 years of age, respectively. The corresponding values in the male heart are 7%, 12%, and 32% per year. The investigators calculated that from 20 to 100 years of age, the myocyte compartment is replaced 15 times in women and 11 times in men.11 Significantly lower myocyte turnover rates were recently reported by the Kuhn lab. Mollova et al. performed fluorescent immunocytochemistry for H3P in dissociated myocytes isolated from explanted hearts from human subjects who died of non-cardiovascular causes. Fluorescent immunohistochemistry (in tissue sections) for mitotic kinesin-like protein was employed for investigation of myocyte cytokinesis. The investigators reported a decrease in myocyte turnover with age: annual myocyte turnover is ∼100% during the first year of life, decreases to ∼1.9% at 20 years of age and drops to 0.04% in subjects older than 40 years. The investigators calculated that during the first two decades of life, the total number of myocytes in the left ventricle increases 3.4 fold. However, it needs to be noted that while cardiomyocyte karyokinesis was detectable throughout life, no instances of myocyte cytokinesis were observed beyond 20 years of age.12
While the aforementioned studies demonstrate that human myocytes can re-enter the cell cycle and may possess some ability to proliferate beyond the early postnatal period, the calculated turnover rates need to be interpreted with caution. Estimations of the total number of myocytes per heart (as performed by Adler et al.6–8) are complicated, require numerous assumptions and can be confounded by problematic discrimination of myocyte versus non myocyte nuclei (which in those studies was performed based on nuclear size and morphology). Quantification of cardiomyocyte cycling with histology (as performed by the Anversa group9–11) is prone to sampling error and can be complicated by the fact that conventional histology has been shown to be problematic for identification of cardiomyocyte nuclei13 (this will be discussed later). Finally, estimation of turnover rates based on exceedingly rare and brief (mitosis in adult rat cardiomyocytes lasts ∼1.8 h in vitro14) events9–12 that may not represent instances of genuine myocyte division (mitotic events involving in karyokinesis and myocyte multinucleation, but not cytokinesis and generation of daughter cells) leaves significant room for error. Ideally, slow processes (like myocyte regeneration) need to be quantified over time, rather than based on a single “snapshot”.
To that end, a more reliable approach to study birth of new myocytes over prolonged periods of time is the “pulse-chase” approach. In “pulse-chase” experiments cells (in our case cardiac myocytes) are exposed to a labeled compound (“pulse”), and then are followed for a period when the labeled compound is no longer administered (“chase”). However, while “pulse-chase” approaches can be readily implemented in experimental animals (where pulsing is typically performed through administration of nucleoside analogues), extraordinary circumstances are required for this approach with humans. Examples of such extraordinary circumstances were the above-ground nuclear testing during the Cold War, which resulted in the temporary release of large quantities of 14C into the atmosphere between 1955 and 1963 (14C “pulse”). After the Limited Nuclear Test Ban Treaty in 1963, atmospheric 14C concentration dropped exponentially (“chase”), resulting in unique “pulse-chase” conditions.15 In a seminal study, Bergmann et al. hypothesized that postnatally generated cardiomyocytes would have incorporated 14C into their DNA after the worldwide 14C pulse.3 The concentration of 14C in DNA of myocytes can be used to retrospectively establish a date mark for when myocytes were born (by identifying the year that atmospheric 14C levels were similar to 14C levels in myocyte DNA). This can be achieved since: a) 14C levels in the human body reflect those of the atmospheric air (14C reacts with oxygen in the atmosphere to form 14CO2, which then enters the biotope through photosynthesis); and b) DNA remains stable following the last cell division. By measuring 14C concentration (with accelerator mass spectrometry) in DNA extracted from fluorescence-activated-sorted myocyte (troponin positive) nuclei isolated from explanted hearts (12 hearts total, 10 from subjects who died of non-cardiovascular causes and 2 from patients who died due to an acute myocardial infarction), Bergmann et al. found compelling evidence for measurable postnatal cardiomyocyte renewal. Mathematical modeling demonstrated that postnatal cardiomyogenesis decreases with age: cardiomyocyte turnover is ∼1%/year at the age of 25 and gradually decreases at 0.45%/year at the age of 75, resulting in an exchange of ∼45% of myocytes during a 50-year span.3 A more recent study by the Anversa group employed similar methods but yielded strikingly different results.16 Kajstura et al. performed carbon dating with accelerator mass spectrometry in DNA extracted from myocytes obtained from 19 healthy hearts and 17 hearts with cardiomyopathy. It should be noted that in this study myocytes were isolated based on density centrifugation, rather than fluorescence-activated cells sorting of troponin positive nuclei (as in the study by Bergmann et al.).3 The investigators found that the healthy adult human heart replaces its entire myocyte compartment ∼8 times between 20 and 78 years of age. Myocyte turnover increases 2-fold in cardiomyopathic heats. Somewhat unexpectedly, the turnover rate of myocytes was found to be similar to that of endothelial cells and cardiac fibroblasts.16 The results of these two studies are difficult to reconcile. It has been argued that staining of myocyte nuclei for troponin (as performed by Bergmann et al.) only identifies nuclei of senescent myocytes17; this could result in significant underestimation of myocyte turnover. However, in a follow-up paper, Bergmann et al. demonstrated that virtually all human cardiomyocytes possess troponin positive nuclei.18
Another unique circumstance allowing for implementation of a “pulse-chase” approach to measure myocyte turnover in the human heart is the use of radiosensitizing nucleoside analogues for therapeutic purposes in cancer patients. Nucleoside analogues are incorporated into newly-synthesized DNA of cycling cells and can therefore serve as markers of DNA synthesis. Kajstura et al. investigated incorportation of the thymidine analogue iododeoxyuridine (IdU) into DNA of myocytes by fluorescent immunohistochemistry in hearts explanted from 8 cancer patients that had previously (8 days – 4 years before death and heart explantation) received infusions of the radiosensitizing agent.17 All 8 hearts had varying degrees of pathology. The investigators found IdU labeling (i.e. DNA synthesis) in 2.5–46% of myocyte nuclei, which projects to an annual myocyte turnover of 22%.17 Concerns have been raised that such high turnover rates are difficult to reconcile with high labeling indices of IdU after long chase periods: rapid cell turnover would presumably translate into death of a significant portion of IdU-labeled myocytes as well as into substantial dilution of IdU (to undetectable levels) due to rapid cell proliferation during the chase period; both processes would result in significantly lower myocyte labeling indices of IdU at the time of death.18 A perhaps more logical explanation for the high rates of IdU myocyte labeling in that study is that a substantial portion of the measured DNA synthesis may represent instances of abortive cell-cyle re-entry, resulting in polypolidization or binucleation, rather than genuine myocyte proliferation (more on that later). While the investigators attempted to rule out polyploidization as a significant confounding factor (they surprisingly reported that >80% of human myocyte nuclei are diploid,17 in contrast to several other studies suggesting that the majority is polyploidy),3,6,12,19,20 no effort was undertaken to quantify to the potential contribution of bi/multinucleation to the measured rates of DNA synthesis.
The estimated rates of myocyte turnover measured in adult mammalian hearts (human and mouse) are depicted schematically in Figure 1. Taken together, even though reported rates of myocyte turnover vary wildly (by more than 1 order of magnitude), the aforementioned studies convincingly establish that the human heart can generate new myocytes beyond the early postnatal period. However, studies in human subjects cannot provide insight into the cellular origins of postnatal mammalian cardiomyogenesis: do newly-generated myocytes arise from division of pre-existing myocytes or from cardiomyogenic differentiation of endogenous progenitors? We will next review studies addressing this challenging question.
Figure 1. .

Annual myocyte turnover in the adult human and mouse heart. The estimated annual rates of turnover and the method of quantification are depicted. Only the turnover rates measured in adult hearts are presented. Note the logarithmic scale of the y axis. The number in parenthesis in the study by Kajstura et al.17 indicates the average rate of turnover measured in that study.
Cellular origins of postnatal cardiomyogenesis in the adult mammalian heart: proliferation of pre-existing myocytes
A landmark study by Soonpaa and Field in 1998 demonstrated convincingly that preformed cardiomyocytes can actively cycle in the adult mouse heart. “Pulse-chase” experiments were performed in transgenic mice, in which a nuclear-localized β-galactosidase reporter gene was expressed in cardiomyocytes (driven by the α-myosin heavy chain [αMHC] promoter). Adult mice received injections of 3H-thymidine and were sacrificed 4 h after the last injection. Heart sections were processed for X-gal reaction (to identify cardiomyocyte nuclei) and autoradiography (to identify incorporation of 3H-thymidine into the DNA, i.e. DNA synthesis). Normal adult hearts had a myocyte labeling index of 0.0006% (1/180000 cardiomyocyte nuclei had incorporated 3H-thymidine). This projects to an annual myocyte turnover of ∼1% (if DNA synthesis is accompanied by genuine cell division). After myocardial injury (focal cauterization), myocyte cycling in the border zone increased ∼14-fold (labeling index of 0.0083% [3/36000 cardiomyocyte nuclei had incorporated 3H-thymidine]).21 With regard to the cellular origins of cycling cardiomyocytes, the short chase period precludes detection of cardiomyogenic contributions by differentiation of endogenous progenitors. Identification of morphologically mature cardiomyocytes that have synthesized their DNA within 4 h after a single injection of a nucleoside analogue22) can only be attributed to DNA synthesis in pre-existing myocytes (as differentiation of progenitors to mature myocytes would take longer).
More recently, an elaborate study by the Lee group investigated the contribution of resident myocyte proliferation to postnatal cardiomyogenesis by a combination of genetic fate-mapping, stable isotope labelling and multi-isotope imaging mass spectrometry.23 The investigators employed an inducible fate mapping approach utilizing bitransgenic αMHC- MerCreMer/ZEG mice.24 In these mice, induction of Cre recombinase activity (driven by αMHC promoter) by 4-OH-tamoxifen results in efficient (∼80%) permanent genetic labeling of myocytes (and their progeny) by green fluorescent protein (GFP). 4-OH-tamoxifen-pulsed adult, healthy bitransgenic mice underwent pulsing with 15N-thymidine over a 10-week period. Multi-isotope imaging mass spectrometry25 (which has a resolution capacity of < 1 μm3 and thus can readily distinguish cardiomyocyte nuclei from adjacent non-cardiomyocyte nuclei) in sections from explanted hearts revealed a 15N myocyte labeling index of ∼0.8% over 10 weeks; this projects to an annual rate of myocyte DNA replication of 4.4%. The investigators undertook extensive efforts to calculate the contribution of abortive cell cycle re-entry (resulting in polyploidization and/or multinucleation, rather than genuine cell division) to the measured DNA synthesis. Perhaps not surprisingly, they found that the majority of DNA synthesis occurred in polyploid and/or multinucleated myocytes. However 17% of the measured DNA synthesis could not be explained away by these confounding factors, leaving generation of new myocytes as the only likely explanation and suggesting an annual myocyte turnover of 0.74% in the healthy adult mouse heart. With regard to the cellular origins of postnatal cardiomyogenesis, 15N+ cardiomyocytes expressed GFP at a similar frequency as 15N- myocytes, suggesting that in the normal heart new myocytes are generated mainly through proliferation of pre-existing myocytes, rather than differentiation of endogenous progenitors. Similar experiments performed in adult mice post-myocardial infarction showed a 20-fold increase in myocyte division in the infarct border zone during the first 8 weeks following injury, which was also attributed to proliferation of pre-existing myocytes rather than contributions of endogenous progenitors to the myocyte pool.23
In a parallel study at the Marbán laboratory, we attempted to quantify postnatal cardiomyogenesis and trace its cellular origins using a combination of genetic fate mapping with long-term pulsing with the nucleoside analogue bromodeoxyuridine (BrdU). Healthy adult bitransgenic αMHC-MerCreMer/ZEG mice were pulsed with 4-OH-Tamoxifen to genetically label pre-existing cardiomyocytes with GFP. Mice subsequently received daily injections of BrdU for up to 5 weeks. Figure 2 depicts two genetically labeled pre-existing cardiomyocytes (pseudocolored in green) that have incorporated BrdU (pseudocolored in white). Measurement of DNA synthesis (BrdU incorporation) in pre-exisiting (GFP+) cardiomyocytes by different methods (flow cytometry of whole cells, flow cytometry of isolated myocyte nuclei, fluorescent immunocytochemistry of dissociated cardiomyocytes and fluorescent immuhistochemistry in cardiac sections) demonstrated that ∼0.4% of pre-existing (GFP+) myocytes synthesized DNA during 5 weeks of BrdU pulsing, suggesting an annual rate of DNA replication in resident myocytes of ∼4%. Actively cycling cardiomyocytes were smaller and more-often mononucleated compared to non-cycling myocytes. Quantification of the contributions of polyploidization and multinucleation to the measured rates of BrdU incorporation demonstrated that abortive cell-cycle re-entry without genuine cell division could explain ∼69% of the observed DNA synthesis; thus the calculated annual rate of myocyte turnover due to cardiomyocyte proliferation in the adult healthy mouse heart was ∼1.3%. Myocardial infarction resulted in a 2-fold increase in the total number of proliferating myocytes in the left ventricle during the first 5 weeks post-injury; this increase was attributed to an upregulation in proliferation of pre-existing myocytes in the border zone (∼10 fold compared to normal heart), but not in the remote myocardium.26
Figure 2. .

Cycling resident cardiomyocytes in the adult mouse heart. Two genetically labeled (with GFP, in green) pre-existing, α-sarcomeric actinin (αSA)-positive cardiomyocytes have incorporated BrdU in their nuclei (arrows). Z-stacks reveal that the BrdU+ nuclei belong to cardiomyocytes (αSA). The image is obtained from Malliaras et al.26
Taken together, the aforementioned studies suggest that mammalian myocytes retain a limited but measurable capacity to proliferate in the healthy adult heart, and that myocyte proliferation increases modestly in the border zone following myocardial infarction.
Cellular origins of postnatal cardiomyogenesis in the adult mammalian heart: myogenic differentiation of progenitor cells
During the past decade several studies supported the notion that the adult mammalian heart contains its own reservoir of stem cells. Numerous populations of putative adult endogenous cardiomyocyte progenitors have been proposed (including c-kit cells,27 Sca-1 cells,28 side population cells,29 cardiosphere-forming cells,30 SSEA-1 cells,31 PDGFRα32 cells and neural-crest derived cells)33 largely based on expression of surface markers or functional properties that have been used to mark progenitors in other organs. The high number of distinct populations of putative endogenous myocyte progenitors is difficult to reconcile with the limited regenerative capacity of the adult mammalian heart. Importantly, while several cell types have been shown to express cardiac proteins in vitro or after delivery into recipient hearts following ex vivo expansion, their physiologic importance and contribution to cardiomyocyte replenishment in the normal or injured adult heart remains controversial.
Genetic fate mapping is a powerful tool that enables study of cardiac regeneration in vivo.34 Perhaps the most compelling evidence indicating postnatal contribution of endogenous progenitors to the adult myocyte pool comes from a landmark study performed at the Lee laboratory.24 Using αMHC-MerCreMEr/ZEG adult bitransgenic mice, Hsieh et al. achieved inducible genetic labeling of ∼80% of preexisting myocytes with GFP, without any detectable labeling of progenitor-like cells (which in their inactive state presumably do not express αMHC). In the normal heart, the percentage of GFP+ myocytes remained unchanged over 1 year of follow-up, indicating that progenitor cells do not contribute significantly to myocyte renewal during normal aging. In contrast, when mice were subjected to myocardial infarction the percentage of GFP+ cardiomyocytes decreased from ∼83% to ∼68% in the border zone and to ∼77% in the remote myocardium over a 3-month follow-up period. Following pressure-overload, the percentage of GFP+ myocytes decreased from ∼83% to 76% over 3 months. These results indicate that post-cardiac injury, unlabeled progenitors undergo cardiomyogenic differentiation, resulting in dilution of GFP+ myocytes by GFP − myocytes (generated from unlabeled precursors) (Figure 3A). Alternatively, the observed dilution of the labeled myocyte pool could be attributed to intrinsic differences between GFP+ and GFP- myocytes, i.e. increased proliferative capacity of GFP- myocytes post-injury or increased susceptibility of GFP+ myocytes to injury (even though the latter hypothesis has not been shown to occur).24,26 BrdU pulsing revealed increased incorporation of BrdU in GFP- myocytes (compared to GFP+ myocytes) post-injury, a finding also compatible with generation of new myocytes from progenitors.24 Based on these findings, the contribution of endogenous precursors to the myocyte pool post-injury appears to be quite substantial: over a 3-month period, ∼15% of myocytes in the border zone and ∼6% of all myocytes in the left ventricle arise from cardiomyogenic differentiation of progenitors. The results of this study (particularly the increased BrdU labeling of GFP- myocytes24,35) do not fully agree with a more recent study from the same group23 (described earlier), in which no increased 15N incorporation into GFP − myocytes could be detected by multi-isotope imaging mass spectrometry, suggesting no significant contribution of endogenous precursors to the myocyte pool post-injury. This discrepancy may be a result of the very small number of mononucleated/diploid 15N+ myocytes (16 15N+ myocytes/ 7063 myocytes analyzed) detected in injured hearts by multi-isotope imaging mass spectrometry (an extremely time-consuming method that precludes analysis of large amounts of tissue); this number may be too small to detect differences in the rate of 15N incorporation in GFP+ and GFP − myocytes.23
Figure 3. .

Fate mapping strategies to study endogenous cardiac regeneration: pros and cons. A: Pre-existing cardiomyocytes are genetically labeled. Any contributions to the myocyte pool arising from cardiomyogenic differentiation of unlabeled progenitors undergo results in dilution of GFP+ myocytes by GFP − myocytes (generated from unlabeled precursors). B: Endogenous progenitors (but not pre-existing myocytes) are genetically labeled in a prospective manner, enabling direct visualization of their future contributions to the myocyte pool.
At the Marbán lab, we attempted to investigate the cellular origins of postnatal cardiomyocytes using similar approaches.26 After inducible genetic labeling of cardiomyocytes by GFP, adult bitrangenic αMHC-MerCreMer/ZEG mice received daily BrdU injections for 5 weeks. Comparison of BrdU incorporation in GFP+ and GFP − cardiomyocytes (by flow cytometry, immunocytochemistry and histology) revealed similar rates of BrdU labeling in both myocyte subsets in healthy mice. These findings indicate that in the normal adult mouse heart, myocyte turnover occurs predominantly through proliferation of resident myocytes, without any measurable progenitor-mediated myocyte formation. However, post-myocardial infarction we could detect a contribution of endogenous precursors to the myocyte pool (∼1% of myocytes in the left ventricle arose from progenitor cell differentiation over a 5-week period post-injury26) (Figure 4).
Figure 4. .
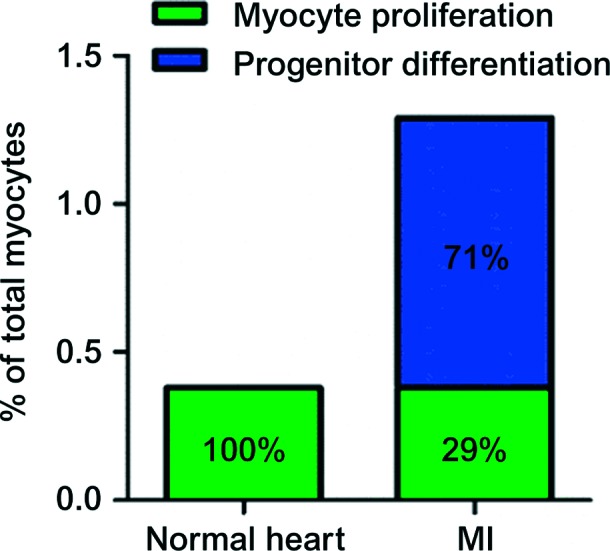
Origins of postnatal cardiomyogenesis in the normal and infarcted heart. Both absolute rates (5 weeks, y axis) and relative magnitudes (percentages inside bars) are presented. In the normal mouse heart, cardiomyocyte turnover occurs through proliferation of resident cardiomyocytes. After MI, cardiomyocyte cycling increases, but the majority of new cardiomyocytes arise from recruited endogenous stem cells. The basal rate of cardiomyocyte proliferation measured in the normal heart is not included in the bar of the infarcted heart; the latter only demonstrates the additional myocytes generated post-myocardial injury. The image is modified from Malliaras et al.26
A different technique (viral gene tagging) to study endogenous cardiac regeneration was employed by the Anversa group. Hosoda et al. injected lentiviruses expressing GFP in the atria and ventricular apex (presumably sites of cardiac stem cell niches) of adult mice. One to 5 months later, nested polymerase chain reaction revealed common viral integration sites in c-kit+ cells, cardiomyocytes, endothelial cells and fibroblasts isolated from the infected hearts. Since lentiviruses are semi-randomly integrated in the host genome of infected cells, these results suggest that postnatally-generated cardiomyocytes, endothelial cells and fibroblasts in the healthy adult mouse heart derive from clonal activation of endogenous stem cells. Six months after viral injection, 25% of myocytes in the mid-portion of the left ventricle (a myocardial region away from the sites of viral injections) were GFP+, presumably arising from migration of GFP+ infected progenitor cells to that region and subsequent differentiation.36 These results indicate a remarkable capacity of the adult mouse heart for stem-cell mediated cardiomyocyte replenishment. However, it needs to be emphasized that viral gene tagging cannot reveal the identity of the parental stem cell. The fact that c-kit cells shared similar viral integration sites as cardiomyocytes, endothelial cells and fibroblasts could be a result of viral infection of a yet unknown progenitor that gives rise to all four celltypes.
Fate mapping of resident myocytes or viral gene tagging cannot reveal the identity of endogenous progenitors, as they indirectly capture the net result of their activation and differentiation. This can only be performed through forward fate mapping experiments in which endogenous progenitors (but not pre-existing myocytes) are genetically labeled in a prospective manner, enabling direct visualization of their future contributions to the myocyte pool (Figure 3b). Such an approach was undertaken at the Fukuda lab in order to investigate the contribution of neural-crest derived cells to postnatal cardiomyogenesis. Tamura et al. used bitransgenic mice in which activation of Cre-recombinase (under the control of protein-0 promoter) induces GFP expression, resulting in genetic labeling of neural-crest derived cells by GFP. In the healthy heart, neural-crest derived (GFP+) cardiomyocytes were undetectable during the first week after birth but appeared at 2 weeks postnatally and increased in number thereafter; however, their absolute contributions to the myocyte pool were minimal (∼0.3% GFP+ cardiomyocytes in the septum, < 0.1% GFP+ cardiomyocytes in the rest of the left ventricle). After myocardial infarction, small GFP+ cardiomyocytes (presumably arising from differentiation of neural-crest-derived cells) first appeared in the border zone 2 weeks post-injury and gradually increased in number thereafter (comprising 3% of total cardiomyocytes in the border zone at 12 weeks post-injury).33 While this study is limited by the fact that the activity of Cre-recombinase was not temporally controlled in an inducible manner (and thus spontaneous activation of the protein-0 promoter in resident cardiomyocytes would result in GFP labeling) it suggests that progenitor cells may contribute to generation of new myocytes (especially post-injury).
Using a similar forward fate mapping approach, Ellison et al recently reported that endogenous c-kit+ cardiac cells are a source of newly formed cardiomyocytes, after myocardial injury, in adult rodents.37 The investigators used a model of a single, high isoproterenol dose that produces severe, albeit spontaneously resolving, myocardial injury. Endogenous c-kit+ cardiac cells were genetically labeled by exogenous administration of a lentivirus expressing Cre-recombinase. under the control of a c-kit promoter, in hearts of RYP reporter mice. In this mouse model, expression of Cre-recombinase in c-kit+ cell results in genetic labeling of infected c-kit+ cells with yellow fluorescent protein (YFP). After isoproterenol injury, a significant fraction of cardiomyocytes were YFP+, indicating that they comprise newly generated cardiomyocytes arising from cardiomyogenic differentiation of infected c-kit+ cells.
While this study suggests an important role of progenitor cell-mediated cardiomyogenesis post-injury, it is in contrast with previous studies, reporting that adult c-kit+ cells have only angiogenic but not cardiomyogenic potential.38,39 In addition, the implemented spontaneously-resolving model of heart failure may not be physiologically relevant. Finally, it needs to be emphasized that c-kit is not a specific marker of stem cells, but it is rather expressed in a variety of cells (e.g. mast cells) and more importantly in myocytes themselves during both proliferation and differentiation.40,41 Thus, the precise expression pattern of c-kit in forward fate mapping models has to be delineated at baseline prior to temporal assessment of possible progeny, before concrete conclusions can be reached. Currently, several studies using forward fate-mapping approaches for putative markers of endogenous progenitors (including c-kit, Sca-1 and Nkx2-5) are ongoing in multiple labs.42
Taken together, the aforementioned studies suggest that endogenous progenitors may generate new myocytes in the adult heart. While their role in postnatal cardiomyogeneis during normal ageing appears to be limited23,24,26,35 (as most studies concur that myocyte turnover in the healthy heart most likely occurs through proliferation of pre-existing myocytes23,26), multiple studies indicate progenitor cell-mediated cardiomyocyte renewal occurs following myocardial injury.24,26,33,35,37 It needs to be emphasized that the two proposed mechanisms of myocyte turnover (cardiomyocyte proliferation and differentiation of endogenous progenitors) are not mutually exclusive. The adult mammalian liver has an impressive capacity to regenerate; following acute 70% partial hepatectomy, the adult liver can fully regenerate within days (in rodents) to weeks (in humans), through division of mature hepatocytes and cholangiocytes, which re-enter the cell cycle and divide.43 However, during chronic liver injuries, hepatic progenitor cells also become activated and contribute to liver regeneration.44
Challenges in studying endogenous cardiac regeneration
Slow regenerative processes are naturally challenging to measure. In addition, inherent characteristics of cardiac tissue architecture and cardiomyocyte biology further complicate quantification of postnatal cardiomyogenesis and tracing of its cellular origins in the mammalian heart. Below, we review the major challenges when studying endogenous postnatal cardiac regeneration.
First, since postnatal cardiomyogenesis in the mammalian heart most likely occurs at very low levels, “pulse-chasing” approaches with long pulsing periods are preferable in experimental animals, as they maximize the chance of capturing rare events of myocyte proliferation. However, long-term administration of radiolabeled thymidine45 or halogenated nucleoside analogues46 (like BrdU) may be toxic and could affect the cycling rates of cardiomyocytes. Nevertheless, we did not observe any differences in the rates of actively-cycling (Ki67+) cardiomyocytes in mice that received BrdU for up to 5 weeks compared to mice that did not receive BrdU.26
Second, the use of conventional histology (which typically employs confocal microscopy for analysis of cardiac sections stained for sarcomeric proteins, DNA [for nuclear identification] and [in some cases] cellular borders) to identify cardiomyocyte nuclei is problematic. Ang et al. performed a careful study to investigate the fidelity of conventional myocyte nuclear identification using confocal microscopy.13 A transgenic mouse in which cardiomyocyte nuclei are genetically labelled (by a nuclear-localized β-galactosidase reporter gene driven by the αMHC promoter) was used as the gold-standard. The investigators demonstrated that conventional histological approaches typically misidentify myocyte nuclei 10% of the time, thus significantly compromising accurate quantification of rare events (like active cycling of myocytes).13 The limitations of conventional microscopy can be overcome by use of transgenic lineage reporters that mark cardiomyocyte nuclei,21,22 implementation of approaches with powerful resolution capacity (e.g. multi-isotope imaging mass spectrometry23), or analysis of enzymatically dissociated cardiomyocytes.26
Third, it needs to be emphasized that DNA incorporation of nucleoside analogues or nuclear expression of cell-cycle proteins, while demonstrating cell-cycle activity, does not necessarily translate into genuine cell division and proliferation; important confounding factors that need to be accounted for include polyploidization (DNA proliferation without karyokinesis and cytokinesis) and bi/multinucleation (DNA proliferation and karyokinesis without cytokinesis), cell fusion and DNA repair (Figure 5). While cell fusion and DNA damage and repair are exceptionally rare events that cannot account for the magnitude of measured turnover rates,3 polypolidization and bi/multinucleation are particularly relevant when studying cardiomyogenesis, as abortive cell-cycle re-entry is quite prevalent in myocytes (most myocytes in the mouse heart are diploid and binucleated, while most myocytes in the human heart are polyploid and mononucleated47). We and others have found that the majority of the measured myocyte cell-cycle activity in the normal or infarcted mouse heart can be attributed to polyploidization and binucleation, rather than true myocyte division.23,26,48 Cardiomyocyte cytokinesis is a brief (the M phase occupies 2% of the cell cycle47) and rare event, and detection of the cleavage furrow between diving myocytes (a hallmark of cytokinesis) is complicated by the compact myocardial architecture as well as by division of adjacent non-myocyte cells (which actively cycle at substantially higher rates compared to myocytes3); thus myocyte division is difficult to visualize convincingly and quantify accurately. A more viable strategy may be to quantify the magnitude of abortive cell-cycle re-entry and subtract it from the total measured DNA synthesis.23,26 To that end, measurements of ploidy levels (by flow cytometry for DNA content or by visualization of the number of sex chromosomes by fluorescence is situ hybridization) and number of nuclei (preferably by analysis of dissociated myocytes) should be performed in cardiomyocytes that have incorporated nucleoside analogues.
Figure 5. .

Confounding factors when studying myocyte turnover. DNA incorporation of nucleoside analogues or nuclear expression of cell-cycle proteins, while demonstrating cell-cycle activity, does not necessarily translate into genuine cell division and proliferation; confounding factors that need to be accounted for include polyploidization (DNA proliferation without karyokinesis and cytokinesis) and bi/multinucleation (DNA proliferation and karyokinesis without cytokinesis) and to a lesser degree cell fusion and DNA repair.
Finally, while genetic fate mapping is the only reliable tool that can be used to trace the cellular origins of organ regeneration in vivo, its implementation in studies of postnatal cardiomyogenesis does not come without problems. While several groups have used the αMHC-MerCreMer/ZEG bitrasngenic mouse for inducible marking of pre-existing myocytes, multiple studies have reported activity of the α-MHC promoter in non-myocyte cells that possess characteristics compatible with endogenous cardiac progenitors.49–51 In addition, development of new lineage-tracing models that could allow for forward fate mapping of cardiac progenitors (in order to prospectively investigate their contributions to the myocyte pool) is complicated by lack of specific cardiac stem cell markers and by potential re-expression of such markers in cardiomyocytes during stress-induced activation of a fetal-gene program52 (Figure 3).
Conclusions
There is now ample and compelling evidence that cardiomyogenesis does occur in the adult mammalian heart, albeit at an insufficient rate to restore cardiac function after substantial cell losses such the ones that occur after a myocardial infarction. Proliferation of pre-existing cardiomyocytes appears as the dominant mechanism of generation of novel cardiomyocytes, at least during normal ageing, although after myocardial injury, differentiation of progenitor cells may also contribute to this phenomenon. The relative importance of these two mechanisms remains controversial. However, both show promising therapeutic potential.
Cell therapies have already reached the stage of clinical trials; most of the cells currently under investigation (with the exception of embryonic stem cells and induced pluripotent stem cells) do not differentiate into functional cardiomyocytes and act through paracrine activation of endogenous reparative53 and regenerative26,35 pathways. Recently, insight has been provided to the mechanisms that control exit and re-entry of cardiomyocytes to the cardiac cycle, together with ways to manipulate the potential of cardiomyocytes for division and proliferation.5,54,55 It is reasonable to expect that therapeutic amplification of endogenous regenerative mechanisms will bolster cardiomyocyte repopulation of injured myocardium and will result in effective therapies for cardiovascular disease and heart failure, in the not so remote future.
Conflict of interest
JT is a consultant for Capricor, Inc, a stem cell company.
References
- 1.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–253. doi: 10.1002/jez.1401870208. [DOI] [PubMed] [Google Scholar]
- 2.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 3.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA. 2013;110:187–192. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler CP. Relationship between deoxyribonucleic acid content and nucleoli in human heart muscle cells and estimation of cell number during cardiac growth and hyperfunction. Recent Adv Stud Cardiac Struct Metab. 1975;8:373–386. [PubMed] [Google Scholar]
- 7.Adler CP, Friedburg H. Myocardial DNA content, ploidy level and cell number in geriatric hearts: post-mortem examinations of human myocardium in old age. J Mol Cell Cardiol. 1986;18:39–53. doi: 10.1016/s0022-2828(86)80981-6. [DOI] [PubMed] [Google Scholar]
- 8.Adler CP, Costabel U. Cell number in human heart in atrophy, hypertrophy, and under the influence of cytostatics. Recent Adv Stud Cardiac Struct Metab. 1975;6:343–355. [PubMed] [Google Scholar]
- 9.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci USA. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001;334:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 11.Kajstura J, Gurusamy N, Ogórek B, Goichberg P, Clavo-Rondon C, Hosoda T, D'Amario D, Bardelli S, Beltrami AP, Cesselli D, Bussani R, del Monte F, Quaini F, Rota M, Beltrami CA, Buchholz BA, Leri A, Anversa P. Myocyte turnover in the aging human heart. Circ Res. 2010;107:1374–1386. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 12.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, Kühn B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA. 2013;110:1446–1451. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ang KL, Shenje LT, Reuter S, Soonpaa MH, Rubart M, Field LJ, Galiñanes M. Limitations of conventional approaches to identify myocyte nuclei in histologic sections of the heart. Am J Physiol Cell Physiol. 2010;298:C1603–C1609. doi: 10.1152/ajpcell.00435.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 15.Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 16.Kajstura J, Rota M, Cappetta D, Ogórek B, Arranto C, Bai Y, Ferreira-Martins J, Signore S, Sanada F, Matsuda A, Kostyla J, Caballero MV, Fiorini C, D'Alessandro DA, Michler RE, del Monte F, Hosoda T, Perrella MA, Leri A, Buchholz BA, Loscalzo J, Anversa P. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–1881. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Kajstura J, Urbanek K, Perl S, Hosoda T, Zheng H, Ogórek B, Ferreira-Martins J, Goichberg P, Rondon-Clavo C, Sanada F, D'Amario D, Rota M, Del Monte F, Orlic D, Tisdale J, Leri A, Anversa P. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Bergmann O, Zdunek S, Alkass K, Druid H, Bernard S, Frisén J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp Cell Res. 2011;317:188–194. doi: 10.1016/j.yexcr.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Meckert PC, Rivello HG, Vigliano C, Gonzalez P, Favaloro R, Laguens R. Endomitosis and polyploidization of myocardial cells in the periphery of human acute myocardial infarction. Cardiovasc Res. 2005;67:116–123. doi: 10.1016/j.cardiores.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 20.Brodsky V, Sarkisov DS, Arefyeva AM, Panova NW, Gvasava IG. Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Arch. 1994;424:429–435. doi: 10.1007/BF00190566. [DOI] [PubMed] [Google Scholar]
- 21.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–H226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 22.Soonpaa MH, Rubart M, Field LJ. Challenges measuring cardiomyocyte renewal. Biochim Biophys Acta. 2013;1833:799–803. doi: 10.1016/j.bbamcr.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinhauser ML, Bailey AP, Senyo SE, Guillermier C, Perlstein TS, Gould AP, Lee RT, Lechene CP. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012;481:516–519. doi: 10.1038/nature10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marbán E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med. 2013;5:191–209. doi: 10.1002/emmm.201201737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 28.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin CM, Meeson AP, Robertson SM, Hawke TJ, Richardson JA, Bates S, Goetsch SC, Gallardo TD, Garry DJ. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev Biol. 2004;265:262–275. doi: 10.1016/j.ydbio.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 30.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004;95:911–921. doi: 10.1161/01.RES.0000147315.71699.51. [DOI] [PubMed] [Google Scholar]
- 31.Ott HC, Matthiesen TS, Brechtken J, Grindle S, Goh SK, Nelson W, Taylor DA. The adult human heart as a source for stem cells: repair strategies with embryonic-like progenitor cells. Nat Clin Pract Cardiovasc Med. 2007;4:S27–S39. doi: 10.1038/ncpcardio0771. [DOI] [PubMed] [Google Scholar]
- 32.Chong JJ, Chandrakanthan V, Xaymardan M, Asli NS, Li J, Ahmed I, Heffernan C, Menon MK, Scarlett CJ, Rashidianfar A, Biben C, Zoellner H, Colvin EK, Pimanda JE, Biankin AV, Zhou B, Pu WT, Prall OW, Harvey RP. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell. 2011;9:527–540. doi: 10.1016/j.stem.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura Y, Matsumura K, Sano M, Tabata H, Kimura K, Ieda M, Arai T, Ohno Y, Kanazawa H, Yuasa S, Kaneda R, Makino S, Nakajima K, Okano H, Fukuda K. Neural crest-derived stem cells migrate and differentiate into cardiomyocytes after myocardial infarction. Arterioscler Thromb Vasc Biol. 2011;31:582–589. doi: 10.1161/ATVBAHA.110.214726. [DOI] [PubMed] [Google Scholar]
- 34.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148:33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Loffredo FS, Steinhauser ML, Gannon J, Lee RT. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell. 2011;8:389–398. doi: 10.1016/j.stem.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosoda T, D'Amario D, Cabral-Da-Silva MC, Zheng H, Padin-Iruegas ME, Ogorek B, Ferreira-Martins J, Yasuzawa-Amano S, Amano K, Ide-Iwata N, Cheng W, Rota M, Urbanek K, Kajstura J, Anversa P, Leri A. Clonality of mouse and human cardiomyogenesis in vivo. Proc Natl Acad Sci USA. 2009;106:17169–17174. doi: 10.1073/pnas.0903089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning B, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B. Adult c-kit pos cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell. 2013;154:827–842. doi: 10.1016/j.cell.2013.07.039. [DOI] [PubMed] [Google Scholar]
- 38.Zaruba MM, Soonpaa M, Reuter S, Field LJ. Cardiomyogenic potential of C-kit(+)-expressing cells derived from neonatal and adult mouse hearts. Circulation. 2010;121:1992–2000. doi: 10.1161/CIRCULATIONAHA.109.909093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, Lee JC, Doran RM, Nikitin AY, Fleischmann BK, Kotlikoff MI. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci USA. 2012;109:13380–13385. doi: 10.1073/pnas.1208114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, Naqvi N, Yahiro E, Liu K, Powell PC, Bradley WE, Martin DI, Graham RM, Dell'Italia LJ, Husain A. c-kit is required for cardiomyocyte terminal differentiation. Circ Res. 2008;102:677–685. doi: 10.1161/CIRCRESAHA.107.161737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tallini YN, Greene KS, Craven M, Spealman A, Breitbach M, Smith J, Fisher PJ, Steffey M, Hesse M, Doran RM, Woods A, Singh B, Yen A, Fleischmann BK, Kotlikoff MI. c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc Natl Acad Sci USA. 2009;106:1808–1813. doi: 10.1073/pnas.0808920106. doi:10.1073/pnas.0808920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garbern JC, Lee RT. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell. 2013;12:689–698. doi: 10.1016/j.stem.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diehl AM, Chute J. Underlying potential: cellular and molecular determinants of adult liver repair. J Clin Invest. 2013;123:1858–1860. doi: 10.1172/JCI69966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulter L, Lu WY, Forbes SJ. Differentiation of progenitors in the liver: a matter of local choice. J Clin Invest. 2013;123:1867–1873. doi: 10.1172/JCI66026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu VW, Black GE, Torres-Duarte A, Abramson FP. 3H-thymidine is a defective tool with which to measure rates of DNA synthesis. FASEB J. 2002;16:1456–1457. doi: 10.1096/fj.02-0142fje. [DOI] [PubMed] [Google Scholar]
- 46.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lió P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 47.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hesse M, Raulf A, Pilz GA, Haberlandt C, Klein AM, Jabs R, Zaehres H, Fügemann CJ, Zimmermann K, Trebicka J, Welz A, Pfeifer A, Röll W, Kotlikoff MI, Steinhäuser C, Götz M, Schöler HR, Fleischmann BK. Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat Commun. 2012;3:1076. doi: 10.1038/ncomms2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zelarayan LC, Noack C, Sekkali B, Kmecova J, Gehrke C, Renger A, Zafiriou MP, van der Nagel R, Dietz R, de Windt LJ, Balligand JL, Bergmann MW. Beta-Catenin downregulation attenuates ischemic cardiac remodeling through enhanced resident precursor cell differentiation. Proc Natl Acad Sci USA. 2008;105:9762–19767. doi: 10.1073/pnas.0808393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey B, Izarra A, Alvarez R, Fischer KM, Cottage CT, Quijada P, Diez-Juan A, Sussman MA. Cardiac stem cell genetic engineering using the alphaMHC promoter. Regen Med. 2009;4:823–833. doi: 10.2217/rme.09.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong F, Harvey J, Finan A, Weber K, Agarwal U, Penn MS. Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation. 2012;126:314–324. doi: 10.1161/CIRCULATIONAHA.111.082453. [DOI] [PubMed] [Google Scholar]
- 52.Steinhauser ML, Lee RT. Regeneration of the heart. EMBO Mol Med. 2011;3:701–712. doi: 10.1002/emmm.201100175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malliaras K, Li TS, Luthringer D, Terrovitis J, Cheng K, Chakravarty T, Galang G, Zhang Y, Schoenhoff F, Van Eyk J, Marbán L, Marbán E. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation. 2012;125:100–112. doi: 10.1161/CIRCULATIONAHA.111.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]