

Abstract
Objectives
The aim of this study was to evaluate the role of c-Src inhibition on connexin43 (Cx43) regulation in a mouse model of myocardial infarction (MI).
Background
MI is associated with decreased expression of Cx43, the principal gap junction protein responsible for propagating current in ventricles. Activated c-Src has been linked to Cx43 dysregulation.
Methods
MI was induced in 12-week-old mice by coronary artery occlusion. MI mice were treated with c-Src inhibitors (PP1 or AZD0530), PP3 (an inactive analogue of PP1), or saline. Treated hearts were compared to sham mice by echocardiography, optical mapping, telemetry ECG monitoring, and inducibility studies. Tissues were collected for immunoblotting, quantitative PCR, and immunohistochemistry.
Results
Active c-Src was elevated in PP3-treated MI mice compared to sham at the scar border (280%, p=0.003) and distal ventricle (346%, p=0.013). PP1 treatment restored active c-Src to sham levels at the scar border (86%, p=0.95) and distal ventricle (94%, p=1.0). PP1 raised Cx43 expression by 69% in the scar border (p=0.048) and by 73% in distal ventricle (p=0.043) compared to PP3 mice. PP1-treated mice had restored conduction velocity at the scar border (PP3: 32 cm/s, PP1: 41 cm/s, p < 0.05) and lower arrhythmic inducibility (PP3: 71%, PP1: 35%, p < 0.05) than PP3 mice. PP1 did not change infarct size, ECG pattern, or cardiac function. AZD0530 treatment demonstrated restoration of Cx43 comparable to PP1.
Conclusions
c-Src inhibition improved Cx43 levels and conduction velocity and lowered arrhythmia inducibility after MI, suggesting a new approach for arrhythmia reduction following MI.
Keywords: Src, sudden death, connexin43, gap junctions, myocardial infarction
Introduction
The estimated incidence of myocardial infarction (MI) is 525,000 new and 190,000 recurrent events every year in the United States (1). Following MI, patients are at increased risk for ventricular tachy-arrhythmia and sudden cardiac death (2). This risk continues after resolution of the MI. In chronic ischemic cardiomyopathy, ventricular tachycardia (VT) is most often attributable to reentrant circuits formed near the scar border (3, 4). These reentrant circuits have lead to ablation strategies to cure recurrent monomorphic VT with limited success (5, 6).
Reentrant arrhythmias are favored by slow conduction in the circuit. Gap junctions are the low resistance channels that facilitate cell-to-cell current propagation. Connexin43 (Cx43) is the primary gap junction protein responsible for conduction in the ventricles. Slow conduction and consequent increased arrhythmic risk after MI are, in part, the result of Cx43 downregulation, resulting in decreased conduction velocity (CV) and creating the substrate for arrhythmia (7, 8).
Recently, activation of the proto-oncogene tyrosine-protein kinase cellular-Src (c-Src) has been linked to the dysregulation of Cx43 in the heart (9–11). Cx43 is known to interact with the scaffolding protein zonula-occludens-1 (ZO-1), which is a crucial regulator of gap junction size, stabilization, and function (12–17). ZO-1 has complicated effects on gap junctions. Overexpression can inhibit Cx43 incorporation into gap junctions (14), but displacement of ZO-1 from gap junctions leads to their internalization (18). Phosphorylation of c-Src on Tyr416 (p-Src) creates an active form of the kinase that can displace the ZO-1/Cx43 interaction (12, 13, 16, 18, 19). p-Src membrane localization results in internalization and degradation of cardiac Cx43 (9–11).
c-Src is a non-receptor tyrosine kinase of the Src family of kinases that has been implicated in cell growth, differentiation, cell adhesion, and tumorigenesis (20). Inhibitors of c-Src activation, such as PP1, have been developed that have proven beneficial in slowing tumorigenesis (21–25). Newer c-Src inhibitors, including AZD0530, are in clinical development and have proven tolerable in human cancer studies (26, 27). Recently, we studied mice with a cardiac-specific activated renin-angiotensin system and showed that PP1 inhibition of c-Src activation restores Cx43 expression and conduction velocity and decreases arrhythmias and sudden cardiac death (11), suggesting usefulness of c-Src inhibition in preventing arrhythmias associated with heart failure. Since c-Src has been shown to be activated in animal models of MI (10), we tested the hypothesis that c-Src inhibition could ameliorate Cx43 degradation, increase conduction velocity, and decrease arrhythmic risk after MI.
Methods
Detailed methods are available in the Online Supplement. Briefly, 12-wk old male C57BL/6 mice underwent either sham surgery or coronary artery ligation to induce MI. Two weeks after surgery, heart function was evaluated using echocardiography as previously described (28). MI animals meeting inclusion criteria (ejection fraction < 45%) were randomized into treatment groups including the c-Src inhibitor PP1 (n=49), the inactive analogue PP3 (n=42), saline (n=12), or the c-Src inhibitor AZD0530 (Saracatinib, AstraZeneca) (n=12). Animals were treated for two weeks and compared to sham mice (n=24). After two weeks of treatment, cardiac function was evaluated again using echocardiography. Then, animals underwent optical mapping or inducibility studies and were euthanized for tissue collection. Optical mapping was performed on 17 mice (7 PP1, 6 PP3, and 4 sham) and conduction velocity calculated as previously described (29). Arrhythmia inducibility was evaluated using epicardial burst pacing protocols on sham (n=6), PP1 (n=17), and PP3 (n=17) mice. Harvested tissue was evaluated by immunoblotting, immunohistochemistry, and quantitative PCR as previously described (11). Telemetry ECG analysis was performed for four weeks following surgery on sham (n=4), PP1 (n=4), and PP3 (n=4) mice.
Data are expressed as mean ± standard error of the mean. Statistical tests were considered significant at p <0.05. The animal experiments were handled according to the National Institutes of Health Guide for Care and Use of Experimental Animals, and use was approved by the University of Illinois Institutional Animal Care and Use Committee.
Results
Left ventricular function after MI was unaffected by c-Src inhibition
Following coronary artery ligation, mice in both the PP1 and PP3 treatment groups developed large anterior infarcts, increased reactive oxygen species (ROS) production, and decreased left ventricular (LV) function. There were no differences in MI size by area (PP3: 35 ± 3 %, PP1: 34 ± 4%, p=0.82) between treatment groups. ROS production, as measured by nitrotyrosine staining, was elevated comparably in both the PP3 and PP1-treated MI groups as compared to sham (PP3 scar border: 172 ± 7 % [p<0.0001], PP3 distal ventricle: 164 ± 10 % [p<0.0001], PP1 scar border: 163 ± 8 % [p<0.0001], PP1 distal ventricle: 153 ± 7 % [p=0.001]). Echocardiography data, taken before and after treatment, demonstrated decreased ejection fraction in both the PP3- and PP1-treated groups compared to shams. PP3- and PP1-treated mice had increased end systolic and diastolic volumes as well as decreased fractional shortening compared to shams (Figure 1, Table 1). Treatment with PP1 did not affect ejection fraction, fractional shortening or volume measurements compared to the inactive PP3. The lack of difference between treatment groups in infarct size and LV function implied that any arrhythmic changes were not likely related to an influence on cell necrosis or LV function.
Figure 1. Wall motion defects and evidence of ROS production after MI.
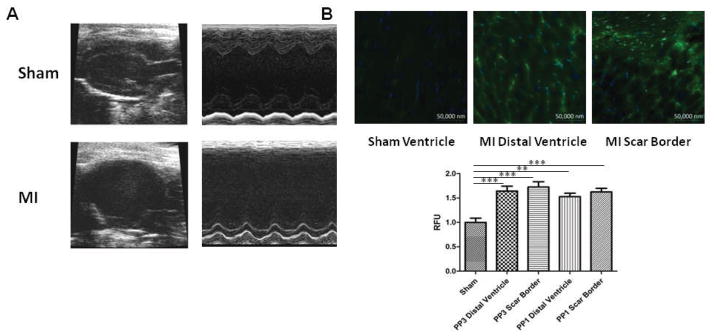
(A) Representative trichrome staining and echocardiography images of sham and MI mice, demonstrating scar formation and decreased anterior wall movement in the MI model. (B). Representative images of fluorescent nitrotyrosine stains indicative of increased ROS in the distal ventricle and scar border of PP3 and PP1-treated post-MI mice. Quantification of relative fluorescent units (RFUs) demonstrates significantly elevated ROS in the distal ventricle and scar border of both PP3 and PP1 groups compared to sham. There were no differences between treatment groups.
Table 1.
Echocardiography, electrophysiology, and scar size.
| Two weeks following MI | |||
|---|---|---|---|
| Sham | PP3 | PP1 | |
| Ejection Fraction (%) | 59 ± 1 | 38 ± 1* | 38 ± 1* |
| End Systolic Volume (μL) | 34 ± 2 | 84 ± 5* | 78 ± 4* |
| End Diastolic Volume (μL) | 79 ± 4 | 133 ± 6* | 125 ± 5* |
| Stroke Volume (μL) | 45 ± 3 | 49 ± 2 | 48 ± 2 |
| Fractional Shortening (%) | 29 ± 1 | 18 ± 1* | 19 ± 1* |
| Heart Rate (bpm) | 561 ± 24 | 579 ± 32 | 609 ± 40 |
| RR-Interval (ms) | 109 ± 2 | 106 ± 4 | 100 ± 2 |
| QRS duration (ms) | 8 ± 1 | 10 ± 1* | 10 ± 1 |
| QT interval (ms) | 20 ± 2 | 27 ± 2 | 25 ± 1 |
|
Four weeks following MI
| |||
| Ejection Fraction (%) | 60 ± 2 | 38 ± 2* | 38 ± 1* |
| End Systolic Volume (μL) | 37 ± 4 | 90 ± 7* | 87 ± 7* |
| End Diastolic Volume (μL) | 91 ± 6 | 141 ± 8* | 137 ± 9* |
| Stroke Volume (μL) | 53 ± 2 | 52 ± 3 | 50 ± 3 |
| Fractional Shortening (%) | 32 ± 2 | 19 ± 1* | 18 ± 1* |
| Heart Rate (bpm) | 569 ± 30 | 587 ± 20 | 556 ± 26 |
| RR-Interval (ms) | 100 ± 2 | 105 ± 4 | 100 ± 2 |
| QRS duration (ms) | 9 ± 1 | 11 ± 1* | 11 ± 1* |
| QT interval (ms) | 26 ± 2 | 29 ± 2 | 25 ± 2 |
| Infarct Size (%) | 35 ± 3 | 34 ± 4 | |
Values given as mean ± standard error. Infarct size calculated was calculated as percentage of total circumferential length occupied by scar by averaging three cross-sections through the LV. There were no significant changes between PP1 and PP3 groups.
p < 0.05 vs. Sham.
c-Src was activated following MI and inhibited by PP1 treatment
PP1 treatment prevented c-Src activation in both regions of the heart following MI. At the scar border of PP3-treated MI mice, p-Src expression was elevated to 280 ± 42% of sham levels (p=0.003). The scar border of PP1-treated mice showed no significant change in p-Src compared to sham (86 ± 16%, p=0.95). In the distal ventricle of PP3-treated mice, p-Src expression was increased by 346 ± 75% of that of sham mice (p=0.013). The distal ventricle of PP1-treated mice had no significant change in p-Src compared to sham (94 ± 57%, p=1.0). PP1 treatment maintained p-Src at basal expression levels in both the scar border and distal ventricle (Figure 2).
Figure 2. c-Src inhibition with PP1 improves Cx43 levels after MI.
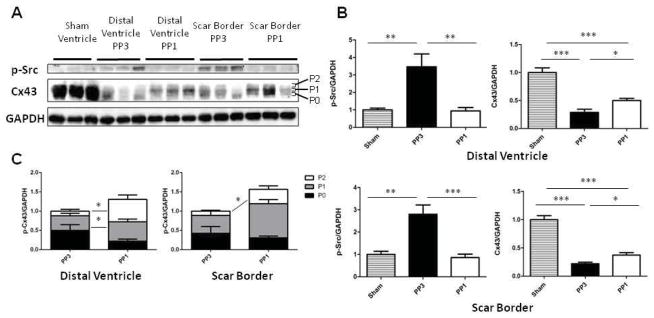
(A) Representative images of p-Src, Cx43, and GAPDH Western blots. Cx43 blots are subcategorized into P0, P1, or P2 bands reflecting increasing levels of phsophorylation. (B) Quantification of band intensity of p-Src and Cx43 normalized to GAPDH expression and relative to sham expression levels. (C) Distribution of Cx43 phosphorylation states according to treatment group.
c-Src inhibition lessened Cx43 downregulation following MI
Since gap junction proteins are reduced after MI and are major determinants of CV, we measured Cx43 changes after MI with and without c-Src inhibition (Figure 2). In the scar border of PP3-treated mice, Cx43 expression was reduced to 22 ± 3% of sham levels (p<0.0001). PP1 improved scar border Cx43 levels to 37 ± 4% of sham expression (p=0.0001). This change represents a 69% increase in Cx43 expression attributable to c-Src inhibition (p=0.048). In the distal ventricle, PP3 mice expressed 29 ± 6% of the Cx43 of sham mice (p<0.0001), while PP1 mice expressed 50 ± 4% of sham Cx43 (p<0.0001). PP1 mice had a 73% increase in distal ventricle Cx43 compared to PP3 (p=0.043). Cx43 mRNA levels were not statistically significantly changed between treatment groups in the distal ventricle, but there was a trend towards increased Cx43 mRNA in the PP1-treated tissue (p=0.08, Supplemental Figure 2). Immunohistochemical staining showed no evidence of improvement in Cx43 lateralization with c-Src inhibition (data not shown), suggesting that redistribution of Cx43 was not a major cause of the Cx43 changes observed.
c-Src inhibition appeared to enhanced Cx43 phosphorylation following MI
Ischemia is associated with dephosphorylation of Cx43 (30). Generally, higher degrees of phosphorylation are associated with increased Cx43 function and with reduced electrophoretic mobility (31). Cx43 mobility was evaluated in MI groups treated with PP3 and PP1 by categorizing bands as P0, P1, or P2 based on band migration as has been done before (Figure 2) (32). At the scar border, PP1 treated mice had significantly higher P2 (p=0.03) and P1 (p=0.01) band concentrations than PP3 treated mice, with no change in P0 expression (p=0.54). In the distal ventricle, the P2 band was elevated (p=0.008) compared to PP3, with no significant changes in P1 (p=0.22) or P0 (p=0.12). This suggests that PP1 treatment enhanced Cx43 phosphorylation in both regions of the MI heart.
c-Src inhibition enhanced conduction velocity and reduced arrhythmic inducibility following MI
Regional CV was measured in sham, PP1, and PP3 groups via optical mapping. At a 160 ms pacing cycle, CV was 47 ± 2 cm/s in sham ventricle. CV was reduced to 32 ± 2 cm/s in the scar border of PP3-treated mice (p<0.01 compared to sham) and reduced only to 41 ± 2 cm/s in PP1-treated mice (p<0.05 compared to sham). This represents a significant restoration of CV by PP1 treatment compared to PP3 (p<0.05). Comparable restoration of CV by PP1 was seen in the distal ventricle at various cycle lengths (Figure 3). The CV changes are consistent with the changes predicted by cable theory for the increase in Cx43 observed (33). Additionally, there were no significant changes in action potential duration between groups (data not shown), and the changes in CV could not be explained by changes in sodium channels, the other major determinant of CV. Sodium channel expression was evaluated in sham and distal ventricle of PP3 and PP1-treated mice, and there were no significant changes between any groups (Supplemental Figure 2).
Figure 3. c-Src inhibition improves conduction velocity after MI.
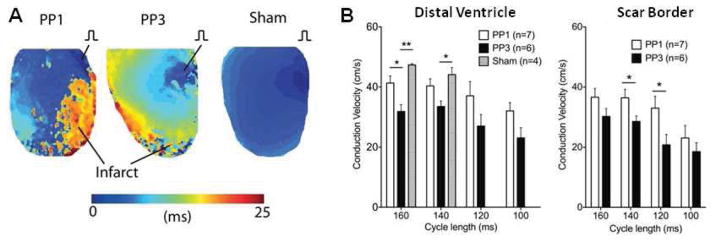
(A) Representative images of activation maps of sham, PP1 and PP3-treated mice. (B) Conduction velocities of PP1, PP3, and sham mice at the distal ventricle and scar border. Sham mice could not be consistently captured at cycle lengths faster than 140 ms.
c-Src inhibition reduced ventricular arrhythmic inducibility (Figure 4). Ventricular epicardial burst pacing protocols demonstrated no inducibility in the sham group (0 of 6 mice) and 71% inducibility in placebo-treated mice (12 of 17 mice). c-Src inhibition with PP1 reduced inducibility of MI mice to 35% (6 of 17 mice; p = 0.04 compared to PP3).
Figure 4. c-Src inhibition reduces arrhythmia inducibility after MI.

(A) Representative ECG at the initiation of stimulus protocol. Stimulus artifacts are labeled as “Stim.” (B) Representative ECG patterns with stimulus pacing. (C) The number of mice in each treatment arm that were inducible or not. NSVT, PMVT, and SMVT stand for nonsustained VT, polymorphic VT, and sustained, monomorphic VT, respectively.
AZD0530 prevents p-Src activation and Cx43 degradation following MI
While PP1 is not used in humans, AZD0530, a clinically relevant p-Src inhibitor, has proven safe in patients (27). Daily gavage administration of AZD0530 showed prevention of p-Src activation and protection of Cx43 expression compared to a saline-treated control group (Supplemental Figure 1). p-Src expression was elevated in saline-treated mice compared to sham levels at the scar border (335 ± 77 %, p=0.024) and distal ventricle (422 ± 92 %, p=0.01) but was normalized in AZD0530-treated mice (scar border: 135 ± 73 %, p=0.88, distal ventricle: 167 ± 91 %, p=0.74).
In the scar border of saline-treated mice, Cx43 expression was reduced to 14 ± 1% of sham levels (p<0.0001). AZD0530-treated mice had Cx43 levels at 59 ± 11% of sham expression (p=0.021). This change represents a 321% increase in Cx43 expression attributable to c-Src inhibition (p=0.012). In the distal ventricle, saline-treated mice expressed 31 ± 5% of the Cx43 of sham mice (p=0.0002), while AZD0530-treated mice expressed 51 ± 4% of sham Cx43 (p=0.004). We did not find a significant change between saline and AZD0530-treated mice in Cx43 expression (p=0.25).
Discussion
MI and ischemic cardiomyopathy are associated with increased risk of sudden death (2). This increase in malignant arrhythmias is thought to be secondary to reduced conduction velocity with its attendant increase in risk of reentry (4, 8). Conduction slowing has been shown to be worse at the scar border and at least in part a result of Cx43 downregulation (8). Restoration of myocardial Cx43 by gene-therapy and cell engraftment have both been shown to lower VT inducibility following MI (34, 35). In this work, we show that c-Src inhibition lessened the reduction in Cx43, improved conduction velocity, and reduced arrhythmia inducibility following MI. The effect of c-Src inhibition was independent of changes in ROS production, fibrosis, ejection fraction, ECG parameters, action potential duration, or sodium channel expression.
The effects of c-Src inhibition in MI were consistent with those seen with RAS activation, suggesting that inhibition of a signaling pathway activated by angiotensin II may explain the effect of c-Src inhibitors (11). RAS activation has been associated with increased ROS, and increased ROS production has been linked to c-Src phosphorylation in other models (36, 37). Since c-Src inhibition affected Cx43 without affecting ROS, our data are consistent with a model where increased ROS after MI activates c-Src resulting in Cx43 degradation and decreased CV.
p-Src levels were significantly elevated, and Cx43 levels were reduced in both the scar border and distal ventricle of the post-MI heart. Inhibition of p-Src using the c-Src inhibitors PP1 and AZD0530 resulted in a return to near basal p-Src levels and increased Cx43 expression (Figure 2, Supplemental Figure 1). Previous work in Cx43 knockout and heterozygous mice has shown that half the normal Cx43 expression is sufficient to support normal current propagation (38). In our model, c-Src inhibition restored the gap junction protein to 50% in the distal ventricle of the post-MI heart and significantly raised expression at the scar border. Therefore, the improvement in CV and the decrease in arrhythmic inducibility with c-Src inhibition were consistent with the changes in Cx43 levels observed. Though Cx43 restoration was sufficient to explain the beneficial effects after c-Src inhibition, the data suggest that there is an undiscovered, second process preventing full restoration of Cx43 levels. Moreover, the data suggest that the processes causing Cx43 degradation are evenly activated in the LV myocardium, rather than being localized to the peri-infarct region.
We observed an increased in slowly migrating forms of Cx43 with c-Src inhibition. These forms are commonly considered to represent increasingly phosphorylated Cx43 that have been associated with increased gap junction function (39, 40). Specific phosphorylation sites on the channel have various activities (40), and in this work, we did not evaluate changes in specific Cx43 phosphorylations sites. Nevertheless, our data are consistent with previous findings that increased Cx43 forms with decreased electrophorectic mobility are associated with enhance gap junctional activity. The mechanism linking p-Src inhibition to these Cx43forms remains unclear.
While PP1 is not a clinically relevant drug, AZD0530 has already been proven safe in phase 1 and 2 clinical trials (24–27). The efficacy of AZD0530 was similar to that of PP1 and suggests that translation of these findings to trials in man may be achievable.
Limitations
This study focused on Cx43 regulation two to four weeks following MI. While we found salutary effects of c-Src inhibition when starting the drug two weeks after MI, it remains to be seen if starting c-Src inhibition immediately after MI would be advantageous. In a myocardial cryoinjury model, treatment with a peptide mimetic of the Cx43 carboxy terminus immediately following injury resulted in increased Cx43 phosphorylation and a lower incidence of inducible arrhythmia (41). Conversely, limiting gap junction communication by using carbenoxolone or Cx43 heterozygous mice has shown a beneficial effect by limiting scar size following ischemia (42, 43), suggesting an initial benefit of Cx43 downregulation. A mouse with modified Cx43 preventing inactivation has shown increased infarct size (44). Long-term effects of c-Src inhibition have not been evaluated fully in humans. Finally, recent work has raised concerns about QT prolongation in animals treated with tyrosine kinase inhibitors (45). We did not observe QT prolongation via telemetry analysis or significant increases in action potential duration but further study of repolarization may be warranted.
Conclusions
Inhibition of c-Src following MI increases Cx43, improves CV, reduces arrhythmia inducibility, and may represent a new approach to arrhythmia reduction in ischemic cardiomyopathy.
Supplementary Material
Acknowledgments
Grants and support: National Institutes of Health grants P01 HL058000, R01 HL1024025, R01 HL106592, Veterans Administration Merit Award, and R41 HL112355 to SCD. National Center for Advancing Translational Sciences of the National Institute of Health TL1 TR000049 to CAR. National Institutes of Health R01 HL114395 and RO1 HL085369 to IRE. British Heart Foundation Travel Fellowship FS/11/69/29017 to FSN.
Abbreviations List
- MI
Myocardial infarction
- VT
Ventricular tachycardia
- Cx43
Connexin43
- CV
Conduction velocity
- c-Src
Tyrosine kinase cellular-Src
- p-Src
phosphorylated, active c-Src
- PP1
4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]-pyrimidine
- PP3
4-amino-7-phenylpyrazol[3,4-d]-pyrimidine
- AZD0530
Saracatinib=N-(5-chlorobenzo[d][1,3]dioxol-4-yl)-7-(2-(4-methylpiperazin-1-yl)ethoxy)-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine
- ZO-1
zonula-occludens-1
Footnotes
Disclosure: SCD is the inventor on patent applications: 1) Method for Modulating or Controlling Connexin43 (Cx43) Level of a Cell and Reducing Arrhythmic Risk 13/507,319 and Activation of the Renin-Angiotensin System (RAS) and Sudden Cardiac Death 13/032,629.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Executive summary: heart disease and stroke statistics--2013 update: a report from the american heart association. Circulation. 2013;127:143–52. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 2.Wellens HJJ, Bär FWHM, Vanagt EJDM, Brugada P. Medical treatment of ventricular tachycardia: Considerations in the selection of patients for surgical treatment. Am J Cardiol. 1982;49:186–93. doi: 10.1016/0002-9149(82)90293-4. [DOI] [PubMed] [Google Scholar]
- 3.Wellens HJ, Lie KI, Durrer D. Further observations on ventricular tachycardia as studied by electrical stimulation of the heart. Chronic recurrent ventricular tachycardia and ventricular tachycardia during acute myocardial infarction. Circulation. 1974;49:647–53. doi: 10.1161/01.cir.49.4.647. [DOI] [PubMed] [Google Scholar]
- 4.de Bakker JM, van Capelle FJ, Janse MJ, et al. Reentry as a cause of ventricular tachycardia in patients with chronic ischemic heart disease: electrophysiologic and anatomic correlation. Circulation. 1988;77:589–606. doi: 10.1161/01.cir.77.3.589. [DOI] [PubMed] [Google Scholar]
- 5.Soejima K, Suzuki M, Maisel WH, et al. Catheter ablation in patients with multiple and unstable ventricular tachycardias after myocardial infarction: short ablation lines guided by reentry circuit isthmuses and sinus rhythm mapping. Circulation. 2001;104:664–9. doi: 10.1161/hc3101.093764. [DOI] [PubMed] [Google Scholar]
- 6.Marchlinski FE, Callans DJ, Gottlieb CD, Zado E. Linear ablation lesions for control of unmappable ventricular tachycardia in patients with ischemic and nonischemic cardiomyopathy. Circulation. 2000;101:1288–96. doi: 10.1161/01.cir.101.11.1288. [DOI] [PubMed] [Google Scholar]
- 7.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–75. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 8.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–96. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 9.Sorgen PL, Duffy HS, Sahoo P, et al. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 10.Kieken F, Mutsaers N, Dolmatova E, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–12. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sovari AA, Iravanian S, Dolmatova E, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–9. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–4. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 13.Toyofuku T, Yabuki M, Otsu K, et al. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–31. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- 14.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–28. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–98. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giepmans BN, Hengeveld T, Postma FR, Moolenaar WH. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J Biol Chem. 2001;276:8544–9. doi: 10.1074/jbc.M005847200. [DOI] [PubMed] [Google Scholar]
- 17.Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008;582:4039–46. doi: 10.1016/j.febslet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilleron J, Fiorini C, Carette D, et al. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008;121:4069–78. doi: 10.1242/jcs.033373. [DOI] [PubMed] [Google Scholar]
- 19.Toyofuku T, Akamatsu Y, Zhang H, et al. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–8. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 20.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 21.Hanke JH, Gardner JP, Dow RL, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 22.Das J, Chen P, Norris D, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49:6819–32. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- 23.Pham NA, Magalhaes JM, Do T, et al. Activation of Src and Src-associated signaling pathways in relation to hypoxia in human cancer xenograft models. Int J Cancer. 2009;124:280–6. doi: 10.1002/ijc.23912. [DOI] [PubMed] [Google Scholar]
- 24.Trevino JG, Summy JM, Lesslie DP, et al. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am J Pathol. 2006;168:962–72. doi: 10.2353/ajpath.2006.050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trevino JG, Summy JM, Gallick GE. SRC inhibitors as potential therapeutic agents for human cancers. Mini Rev Med Chem. 2006;6:681–7. doi: 10.2174/138955706777435724. [DOI] [PubMed] [Google Scholar]
- 26.Hennequin LF, Allen J, Breed J, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 27.Baselga J, Cervantes A, Martinelli E, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clin Cancer Res. 2010;16:4876–83. doi: 10.1158/1078-0432.CCR-10-0748. [DOI] [PubMed] [Google Scholar]
- 28.Jeong EM, Monasky MM, Gu L, et al. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2013;56:44–54. doi: 10.1016/j.yjmcc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laughner JI, Ng FS, Sulkin MS, Arthur RM, Efimov IR. Processing and analysis of cardiac optical mapping data obtained with potentiometric dyes. Am J Physiol Heart Circ Physiol. 2012;303:H753–H765. doi: 10.1152/ajpheart.00404.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beardslee MA, Lerner DL, Tadros PN, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–62. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 31.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–74. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remo BF, Qu J, Volpicelli FM, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011;108:1459–66. doi: 10.1161/CIRCRESAHA.111.244046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–41. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 34.Greener ID, Sasano T, Wan X, et al. Connexin43 gene transfer reduces ventricular tachycardia susceptibility after myocardial infarction. J Am Coll Cardiol. 2012;60:1103–10. doi: 10.1016/j.jacc.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roell W, Lewalter T, Sasse P, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–24. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 36.Aikawa R, Komuro I, Yamazaki T, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23:4598–610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–4. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 39.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–42. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011;108:704–15. doi: 10.1161/CIRCRESAHA.110.235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shintani-Ishida K, Unuma K, Yoshida K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circ J. 2009;73:1661–8. doi: 10.1253/circj.cj-09-0079. [DOI] [PubMed] [Google Scholar]
- 43.Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol. 2003;41:681–6. doi: 10.1016/s0735-1097(02)02893-0. [DOI] [PubMed] [Google Scholar]
- 44.Maass K, Chase SE, Lin X, Delmar M. Cx43 CT domain influences infarct size and susceptibility to ventricular tachyarrhythmias in acute myocardial infarction. Cardiovasc Res. 2009;84:361–7. doi: 10.1093/cvr/cvp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Z, Wu CY, Jiang YP, et al. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 2012;4:131ra50. doi: 10.1126/scitranslmed.3003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.