

Abstract
A detailed electronic structure of the Mn4Ca cluster is required before two key questions for understanding the mechanism of photosynthetic water oxidation can be addressed. They are whether all four oxidizing equivalents necessary to oxidize water to O2 accumulate on the four Mn ions of the oxygen-evolving complex, or do some ligand-centered oxidations take place before the formation and release of O2 during the S3 → [S4] → S0 transition, and what are the oxidation state assignments for the Mn during S-state advancement. X-ray absorption and emission spectroscopy of Mn, including the newly introduced resonant inelastic X-ray scattering spectroscopy have been used to address these questions. The present state of understanding of the electronic structure and oxidation state changes of the Mn4Ca cluster in all the S-states, particularly in the S2 to S3 transition, derived from these techniques is described in this review.
Keywords: Photosystem II, Water oxidation, Oxygen evolution, Manganese cluster, X-ray spectroscopy
Introduction
The oxygen-evolving complex (OEC) located in the Photosystem II (PS II) membrane-bound protein complex in plant, algae, and cyanobacteria catalyzes the water-oxidation reaction (Debus 1992; Rutherford et al. 1992; Ort and Yocum 1996; Wydrzynski and Satoh 2005). The OEC couples the 4-electron chemistry of water oxidation with the one-electron photochemistry of the reaction center by sequentially storing oxidizing equivalents through five intermediate S-states (Si, i = 0–4), before one molecule of dioxygen is evolved. The OEC contains four Mn atoms and one Ca (and possibly one Cl); the Mn cluster provides a high degree of redox and chemical flexibility so that several oxidizing equivalents can be stored during the S-state cycle. Thus, nature avoids releasing harmful chemical intermediates such as superoxide or peroxide during the water oxidation.
To understand the mechanism of water oxidation in detail, it is crucial to know whether the extracted electrons are directly derived from bound water, or from the Mn atoms, or from any other parts of the OEC accompanying each S-state transition (Messinger 2004). Several spectroscopic methods, such as electron paramagnetic resonance (EPR) (Styring and Rutherford 1988; Brudvig 1995; Peloquin and Britt 2001; Carrell et al. 2002), Fourier-transformed infra-red (FTIR) (Chu et al. 2001, 2004; Debus et al. 2005; Kimura et al. 2005a, b, c; Strickler et al. 2006), X-ray absorption spectroscopy (XAS) (Yachandra et al. 1996; Iuzzolino et al. 1998; Yachandra 2005), and UV spectroscopy (Dekker 1992), have been used to address the oxidation state of Mn in each S-state (Fig. 1). EPR spectroscopy has shown that the S0 and S2 are characterized by spin S = 1/2 ground states, exhibiting multiline EPR signals (MLS) (Dismukes and Siderer 1980; Hansson and Andrecute;asson 1982; Messinger et al. 1997a, b; Åhrling et al. 1998). The S1 and S3 are characterized by parallel-polarized EPR signals, indicating integral spin ground states (Dexheimer and Klein 1992; Yamauchi et al. 1997; Campbell et al. 1998; Matsukawa et al. 1999). X-ray absorption near-edge structure (XANES) has been used extensively to investigate the oxidation state of Mn during the S-state transition (Roelofs et al. 1996; Messinger et al. 2001; Haumann et al. 2005). There is a consensus that Mn-centered oxidation occurs during the S0 to S1, and S1 to S2 transitions. However, there is still controversy concerning the involvement of Mn oxidation in the S2 to S3 transition. Within the context of localized oxidation, the formal oxidation state of the native S1 state has been assigned to Mn4(III,III,IV,IV) and S2 to Mn4(III,IV,IV,IV). Some groups have proposed Mn4(III,III,III,IV) in the S2 state and therefore Mn4(III,III,III,III) for the S1 state (Kuzek and Pace 2001; Carrell et al. 2002). Uncertainty remains in the case of the S0 and S3 states. In the S0 state, one of the questions is whether Mn(II) is present, in which case the oxidation states are Mn4(II,III,IV,IV) (Messinger et al. 1997b), or whether the oxidation states are Mn4 (III,III,III,IV) (Robblee et al. 2002). In the S3 state, there is conflict about whether a Mn-centered oxidation occurs (Iuzzolino et al. 1998; Haumann et al. 2005), or whether a ligand-centered oxidation takes place before O–O bond formation and release of molecular oxygen (Styring and Rutherford 1988; Roelofs et al. 1996; Liang et al. 2000; Messinger et al. 2001; Siegbahn 2006.
Fig. 1. The S-state cycle and the proposed oxidation states of the Mn cluster in the S-states. The multiline EPR signals for the S0 and S2 states and the spin states identified with S0, S1, S2, and S3 (for spinach) are also shown.
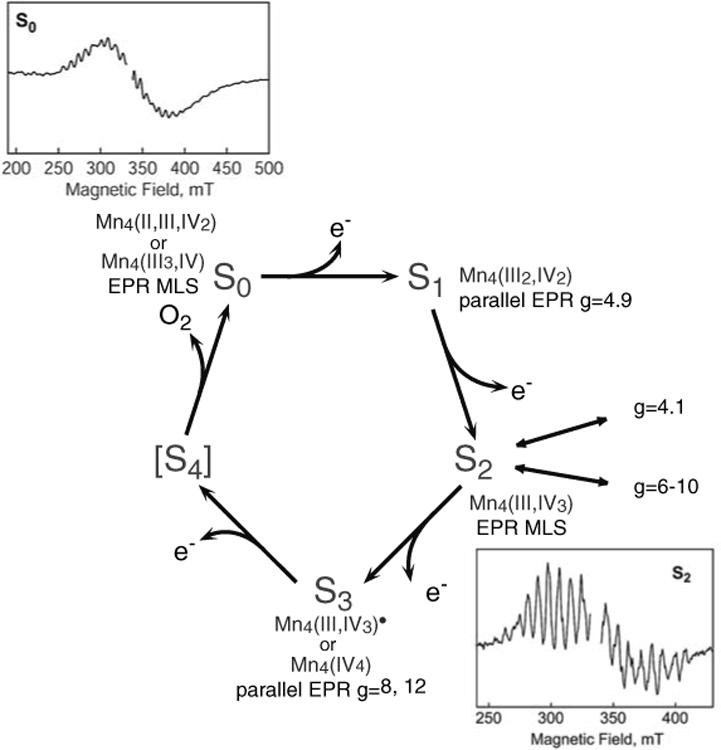
The conflicts concerning the S2 to S3 transition have led to two different types of proposed O2 evolution mechanisms, with one type incorporating the oxidation of ligand or substrate in the S3 state, and the other type invoking Mn oxidation during the S2 to S3 transition (Messinger 2004). Fundamental differences in the chemistry of O–O bond formation and O2 evolution exist between these two types of mechanisms.
As mentioned above, formal oxidation states are commonly used to describe the number of electrons in the metal valence orbitals. Although it is convenient for simplifying the redox states, formal oxidation states do not necessarily coincide with the effective number of electrons in the metal valence shells except for very ionic compounds. In many transition-metal systems the formal oxidation state is an incomplete and in many cases an incorrect description of the electronic structure of the system, because of other important factors, especially metal–ligand covalency (Sarangi et al. 2006). In fact, we have recently shown using resonant inelastic X-ray scattering spectroscopy (RIXS) that the valence electrons are strongly delocalized and, therefore, cannot be assigned to just one metal-center in the OEC (Glatzel et al. 2004). It is very likely that bridging ligands or terminal ligands are actively involved in the catalytic process. In other words, uniqueness of metal catalytic centers in biological system compared to inorganic compounds probably arises from the involvement of these ligands.
In this review, we summarize our current understanding of the oxidation state changes of the OEC, in all S-states and particularly from the S2 to S3 state, studied using various X-ray spectroscopic techniques, mainly; Mn K-edge XANES (1s–4p absorption), Kβ XES (3p–1s emission), and the recently introduced RIXS (1s to 3d/4p absorption followed by 2p to 1s Kα emission). The emphasis is on the results obtained by our group over the years. These results inform us about the more subtle and complicated features involved in the biological system; however, some issues still remain as open questions to be resolved in the future.
Structural or geometric changes in the OEC during S-state transitions
Although the main scope of this review is to focus on the oxidation state changes, geometric and electronic structural changes of the OEC during the S-state transitions are interdependent. Therefore, we briefly summarize the current knowledge of the structural changes of the OEC during the S-state transitions in this section. We have gained much structural information through extensive studies by X-ray crystallography (Zouni et al. 2001; Kamiya and Shen 2003; Ferreira et al. 2004; Loll et al. 2005) and X-ray absorption spectroscopy (Yachandra 2005) over the past several years. Very recently, we have used single-crystal XAS spectroscopy combined with crystallography, to derive a high-resolution structure of the OEC in the dark-stable state (S1) (Yano et al. 2006), under X-ray flux conditions that do not damage the Mn cluster (Yano et al. 2005a). The data suggests that the structure of the Mn4Ca cluster is different from previously proposed models from X-ray crystallography and spectroscopy (Pushkar et al. 2007; Yano et al. 2006).
The structural change as detected by EXAFS from the S1 to S2 state is very subtle (Yachandra 2005; Yano et al. 2005b). In contrast, dramatic structural changes seem to occur during the S2 to S3 transition, as can be observed in the Mn EXAFS spectra of PS II solution samples (Liang et al. 2000). This structural change is also observed from the Ca (or Sr) point of view (unpublished data), suggesting that the S2 to S3 structural change involves the entire cluster. Ligand environment around OEC also undergoes some structural or orientational changes during the S-state cycle as observed in the FTIR studies using site-specific mutants and isotopes (Chu et al. 2004; Debus et al. 2005; Kimura et al. 2005a, b, c; Strickler et al. 2006).
Electronic spin state changes
In terms of the electronic spin, the S0 and S2 states are S = 1/2 ground spin states, each characterized by a EPR multiline spectrum at g = 2 (Fig. 1) (Dismukes and Siderer 1980; Hansson and Andréasson 1982; Messinger et al. 1997a, b; Åhrling et al. 1998). Parallel-polarized EPR signals were observed in the S1 and S3 states instead of the MLS in the perpendicular mode EPR, suggesting integral ground state spin in these two states (Dexheimer and Klein 1992; Yamauchi et al. 1997; Campbell et al. 1998; Matsukawa et al. 1999).
On the basis of the EPR and Electron-Nuclear Double Resonance (ENDOR) studies of the S0 and S2 states and the observation of the Y-shaped OEC electron density from the crystallographic study, spin coupling schemes have been suggested by several groups (Dismukes et al. 1982; Bonvoisin et al. 1992; Kusunoki 1992; Messinger et al. 1997b; Hasegawa et al. 1999; Peloquin et al. 2000; Kulik et al. 2005). Most of these studies support the oxidation state of Mn4(III,IV3) state in the S2 state and Mn4(II,III,IV2) or Mn4(III3,IV) in the S0 state. Summary of the coupling scheme from the S0 and S2 EPR and ENDOR studies is described in detail elsewhere (Peloquin et al. 2000; Kulik et al. 2005).
Another interesting observation is the detection of higher-spin states (g = 4.1 and g = 6–10) in the S2 state in addition to the MLS (S = 1/2) state (Casey and Sauer 1984; Zimmermann and Rutherford 1984; Boussac et al. 1998a, b). The g = 4.1 state, which is best interpreted as S = 5/2, can be created either chemically (Cl– depletion, F– addition, etc.) or by near-infra red (NIR) illumination of native PS II. The g = 6–10 state is also created by NIR illumination and proposed to arise from an S = 5/2 state (Boussac et al. 1998a, b; Horner et al. 1998).
The parallel-polarized EPR signals observed in the S3 state were best explained as an S = 1 spin state (Matsukawa et al. 1999). This could support a Mn-centered oxidation state change upon the S2 to S3 transition (see below). However, if a redox-active organic residue (for example, protein side chain, bound water, or bridging oxygen) is oxidized, the exchange coupling between the Mn cluster with S = 1/2 and the organic residue with S = 1/2 could result in the disappearance of the S2 multiline signal. Additionally, other possible coupling schemes in a multi-spin system could lead to a similar EPR silent state. EPR studies on high spin S2 and S3 states produced by NIR illumination may support the absence of a Mn-centered oxidation during the S2 to S3 transition (Boussac et al. 1996, 2005; Ioannidis et al. 2000, 2002). A similar action spectrum for NIR illumination was observed for the S2 and S3 states. This may indicate similar Mn redox states in the S2 and S3 states, and the possible presence of Mn(III) in the S3 state.
Mn K-edge spectra of PS II
XANES is an element-specific method, and spectra are sensitive to the oxidation state, spin state, and local geometry of the metal site. In general, the rising edge position shifts when the effective number of positive charges (in a simplified way, oxidation state) changes resulting from 1s core hole shielding effects (Yachandra and Klein 1996). In an atom with one electron, for example, the electron experiences the full charge of the positive nucleus. However, in an atom with many electrons, the outer electrons are simultaneously attracted to the positive nucleus and repelled by the negatively charged electrons. The higher the oxidation state of the metal, the more positive the overall charge of the atom, and therefore more energy is required to excite an electron out of an orbital. Conversely, the XANES spectrum shifts to a lower energy when there is more negative charge on the metal.
Figure 2 shows the Mn K-edge spectrum of each S-state of spinach PS II after deconvolution of the spectra obtained from consecutive flash illumination into pure S-state spectra, and their second derivative spectra (Messinger et al. 2001). Traditionally, the inflection point of the rising Mn K main edge (electron 1s to 4p transition) has been used as an indicator of the oxidation states. The edge positions for each of the S-states have been quantitated by measuring the inflection point energy (IPE), given by the zero-crossing of the 2nd derivative. Extensive model compound studies have shown that, when Mn is oxidized by one electron in a set of Mn model compounds with similar ligands, the IPE shifts 1–2 eV to higher energy (Visser et al. 2001).
Fig. 2.

The Mn K-edge spectra of spinach PS II (BBY), from the S0 through S3 states (top left) and their 2nd derivative spectra (bottom left) (Messinger et al. 2001). The magnitude of the inflection point energy shift for the S0 to S1 (2.1 eV) and S1 and S2 (1.1 eV) is much larger than the shift for the S2 to S3 transition (0.3 eV). The pre-edge (1s to 3d transition) from the S-states is enlarged and shown below the Mn K-edge spectra together with the curve fitting result of the S1 pre-edge spectrum (red, experimental; blue, curve fittings; gray, background). The energy level diagram for Mn K-edge XANES and the relevant transitions is shown on the right
In PS II, there is a clear edge shift to higher energy during the S0 to S1 (2.1 ± 0.15 eV shift) and S1 to S2 transitions (1.1 ± 0.05 eV shift), indicating that Mn-centered oxidation occurs during these transitions. On the contrary, the S2 to S3 state transition shows a much smaller shift (0.3 ± 0.05 eV), suggesting that the chemistry of the S2 to S3 transition is different from that of the S0 to S1 or S1 to S2 transition (Roelofs et al. 1996; Messinger et al. 2001).
However, one has to be aware that the edge position cannot be simply an indicator of the oxidation state. Due to the size of the metal 4p orbital, this orbital overlaps with p orbitals of the ligands, either through σ- or π-bonding. Consequently, XANES is sensitive not only to the oxidation state but also to the ligand environment of the metal. Additionally, no definite theory is available for calculating main K-edge spectra for transition-metal complexes, owing to several factors that affect the metal p-density of unoccupied orbitals.
Mn K-edge pre-edge spectra of PS II
In order to obtain a more localized view of the electronic structure at the metal site, it is suitable to probe the lowest unoccupied metal 3d orbitals. The pre-edge spectra arise from excitations of 1s electron into 3d orbitals that are mainly localized around the metal ion. It shows the immediate surrounding of the excited ion through the Coulomb interaction between the core hole and the valence electrons within a short range. This pre-edge feature is a quadrupole-allowed transition; it occurs at a lower energy than the main edge transitions with approximately 1% of the intensity of the dipole-allowed main edge transition. The transition can gain intensity by the metal 4p mixing when the metal–ligand environment is distorted from a centro-symmetric to a non-centro-symmetric coordination. The spectra reflect coordination number, ligand environment, and oxidation state of metals.
As shown in Fig. 2 inset, the pre-edge spectra of PS II noticeably change during the S-state transition (Messinger et al. 2001). There are mainly three peaks (pre-edge fit data shown only for S1), and the low energy component (∼6,540 eV) decreases in intensity during the S0 to S1 and S1 to S2 transitions, and it is not present in the S3 spectrum. In the single crystal XANES of PS II S1 state, these three components show a characteristic dichroism (Yano et al. 2006). In order to understand these pre-edge features and obtain information about the electronic configuration, however, one needs to further investigate the various model compounds and combine experimental data with theoretical calculations based on the ligand field and/or Density-functional theories.
Mn Kβ X-ray emission spectroscopy
X-ray K emission lines are also sensitive to the chemical environment of metals. K (1s core hole) emission spectra arise from the transition of outer shell electrons to a 1s hole, following the formation of the 1s core hole by X-ray absorption (Fig. 3a). Spectral changes in the Kα and Kβ main lines reflect the effective number of unpaired metal 3d electrons through the exchange interaction between the core hole (1s or 2p) and the net electron spin in the metal valence shell. The overall spectral shape of Kβ line is dominated by the (3p, 3d) exchange interaction, while the Kα line is shaped by the 2p spin-orbit splitting. In general, spectral changes for Kβ lines are more pronounced than for Kα, because the 3p and 3d orbitals interact more with each other than the 2p and 3d orbitals.
Fig. 3.
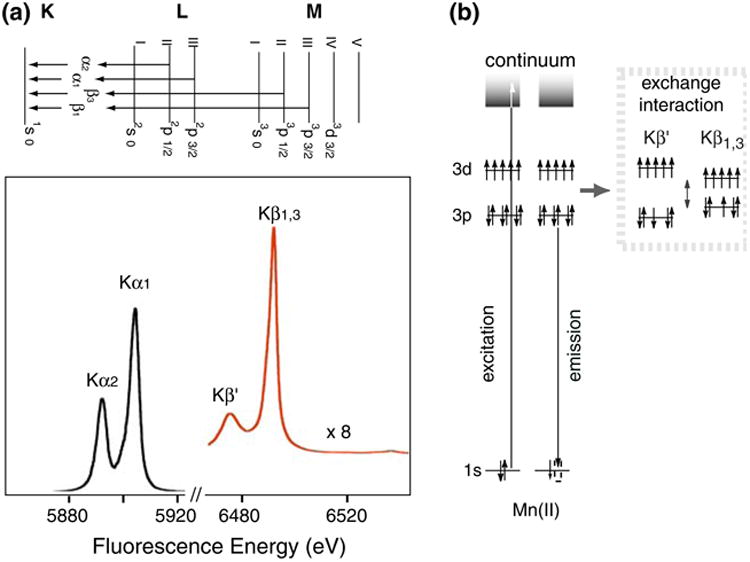
(a) X-ray K-emission from the 2p levels (2p3/2 and 2p1/2), known as Kα1 and Kα2 emission, and from the 3p levels (3p3/2 and 3p1/2) known as Kβ1 and Kβ3 emission. The Kβ lines are approximately 1/8 as intense as the Kα lines. (b) The electronic states and the transitions for Mn Kβ XES. On the right is the exchange interaction between the 3p and 3d levels that gives rise to the Kβ1,3 and Kβ′ states
Kβ XES monitors the X-ray emission from the relaxation of a 3p electron to a 1s hole (Fig. 3b). For the two Kβ main lines, Kβ1 is the emission from 3p3/2 and Kβ3 is from 3p1/2, and these lines cannot be resolved with our spectrometer. Kβ′ feature is due to the (3p, 3d) exchange interaction. In a very simplified model, there are two final spin states; Kβ1,3 is a constructive and Kβ′ is a destructive spin exchange interaction between the unpaired 3p and 3d electrons. The magnitude of the exchange interaction depends on the number of unpaired electrons in the 3p and 3d orbitals. For example, Kβ′ and Kβ1,3 lines move toward each other with decreasing valence spin; i.e., smaller (3p, 3d) interaction. Therefore, the Kβ spectrum is sensitive to the exchange interaction between the core hole (3p) and the net electron spin in the metal 3d valence shell; i.e., it is indirectly sensitive to the effective number of unpaired metal 3d electrons. Thus, the Kβ spectrum serves as an indicator of oxidation state that is different from XANES, which monitors oxidation state through 1s core hole shielding effect.
Figure 4a shows the Kβ emission spectra of a series of Mn oxides, Mn(IV)O2, Mn2(III)O3, and Mn(II)O, which illustrates the sensitivity of Kβ spectra to the oxidation state of Mn. The Kβ′ and Kβ1,3 peaks appear at ∼6,475 eV and ∼6,490 eV, respectively (Bergmann et al. 1998). Separation of these two features is due to the exchange interaction of the unpaired 3d electrons with the 3p hole in the final state of the 3p → 1s fluorescence transition (Fig. 4b). As the oxidation state of Mn increases from Mn(II) to Mn(III) to Mn(IV), fewer unpaired 3d valence electrons are available to interact with the 3p hole; concomitantly, the magnitude of the 3p–3d spin exchange interaction becomes smaller. Accordingly, the Kβ1,3 transition shifts to a lower energy, the Kβ′ transition shifts to a higher energy, and the Kβ′–Kβ1,3 splitting becomes smaller.
Fig. 4.
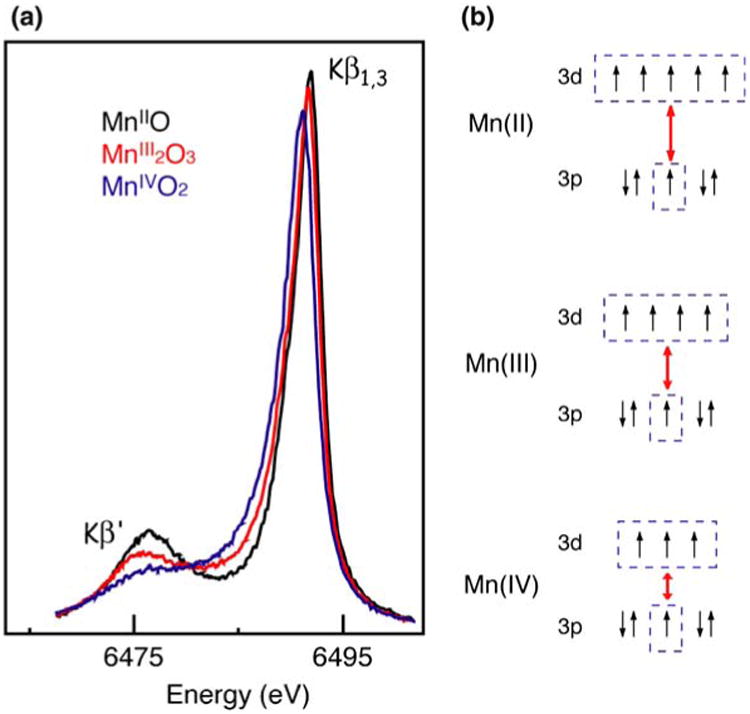
(a) The Mn Kβ emission spectra of Mn oxides in formal oxidation states II, III, and IV (adapted from Messinger et al. 2001). (b) The exchange coupling for each of oxidation states II, III, and IV, showing how the splitting between the Kβ1,3 and K′ depends on the number of unpaired 3d electrons. The splitting increases as the number of unpaired electrons in the 3d states increases
Compared to the 4p orbitals, the 3p orbitals have less overlap with the ligand orbitals, because they are smaller and more buried within the electronic shells. For this reason, Kβ XES is less sensitive to the ligand environment compared to XANES. The Kβ1,3 transition is better resolved than the Kβ′ transition due to a difference in relaxation processes. Hence, we have used Kβ1,3 transition as a indicator of the oxidation state of the metal site for PS II.
The Kβ emission spectra of spinach PS II in S0 through S3 and their difference spectra are shown in Fig. 5 (Messinger et al. 2001). The peak shifts are much smaller than for Mn oxides, because maximally only 1 out of 4 Mn atoms in the OEC is oxidized during each S-state transition. However, the derivative shape of the S0 to S1 and the S1 to S2 difference spectra show that the Kβ1,3 peak shifts to lower energy during these transitions. By contrast, the change is not apparent upon the S2 to S3 transition, suggesting that the change in the metal charge density is much less than for the other transitions. Mn reduction occurs during the S3 to S0 state transition and the Kβ1,3 peak shifts to a higher energy; the difference spectrum reflects a return to the starting oxidation states.
Fig. 5.
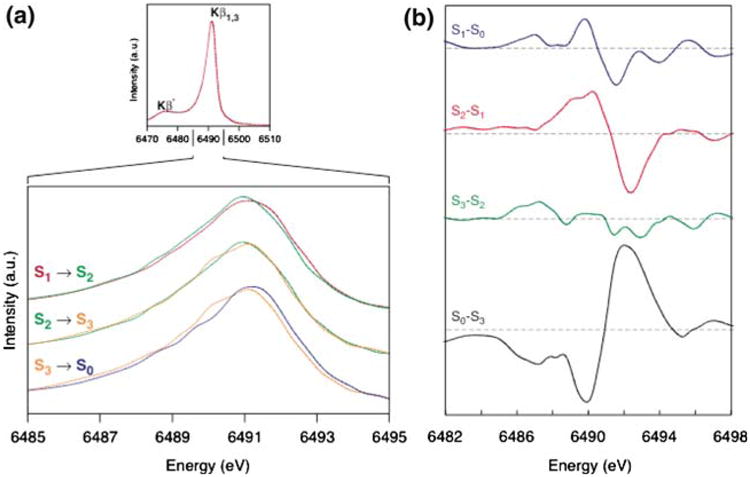
(a) The Mn Kβ emission spectra of the spinach PS II (BBY) S states. (b) The difference spectra between the S1 and S0, S2 and S1 and S0 and S3 are derivative shaped, indicating that the spectral peaks shift in energy. The derivative shape for the S1 and S0, S2 and S1 is the reverse of that for the S0 and S3, indicating oxidation in the first two cases and reduction for the latter case. The difference spectra between the S3 and S2 states show the lack of such a derivative shape, indicating the similarities of the S2 and S3 state spectra and lack of a predominantly metal-centered oxidation. The figure was adapted from Messinger et al. (2001)
The peak shift of the Kβ1,3 emission lines were quantitated using a 1st moment analysis. The 1st moments were calculated for each spectrum using the following equation,
| (1) |
where, Ej and Ij are the energy and fluorescence intensities of the jth data point. The method is suited for very small shifts since the statistics from the entire Kβ1,3 (6,485–6,495 eV) peak is considered rather than just the peak energy.
Figure 6 summarizes the IPE from the XANES and the 1st moments of the Kβ spectra of flash-induced spinach PS II samples. The 1st moment shifts observed in PS II are much smaller than for Mn oxides. As Mn is oxidized from Mn(II), Mn(III) to Mn(IV) in the oxide series, the 1st moments shift to lower energy by ∼0.3 eV step. For the S1 to S2 transitions of PS II, the shift is 0.06 eV, which is approximately three times the value observed for the S2 to S3 transition (0.02 eV). The 1st moment shift of 0.06 eV for the S1 to S2 transition is only one fourth of that seen for the Mn(III) to Mn(IV) oxides.
Fig. 6. Oscillation of XANES inflection point energies (I.P.E.) (B) and that of first moments (E) of the Kβ emission spectra from the 0F to 3F samples of spinach PS II (BBY). The figure was adapted from Messinger et al. (2001).

One point to consider is whether major structural changes can cause the lack of an energy shift in the XES data, even when there is a Mn-centered oxidation state change. A detailed understanding of Kβ XES requires a ligand-field multiplet theory that considers symmetry-dependent perturbations such as (1) spin–orbit coupling, (2) ligand-field splitting, (3) Jahn-Teller distortion, and (4) spin–spin interaction between different metal atoms. Each of these perturbations will split the spin states into a multiplet of states, causing an asymmetric broadening of the observed emission peaks, indicating that there is some dependence of the Kβ XES spectra on the ligand environment. We have compared the XANES and XES spectra of two types of Mn compounds, with very different structures such as the ‘trimers’ (trinuclear complexes) and ‘butterflies’ (tetranuclear), in different oxidation states (Fig. 7) (Pizarro et al. 2004). In the XANES spectra, the Mn oxidation state changes have clear effects on the IPE shifts: the shift is 1.64 eV for Mn3O and 2.20 eV for Mn4O2. However, the magnitude of the shift depends on the type of compounds. Also, the absorption profile of each compound is unique, being affected to different degrees upon changing the Mn oxidation state. On the other hand, the Kβ spectra of each set of compounds have very similar shapes despite differences in core structure and ligand environment. Additionally, the Kβ energy shifts from one oxidation state to another are more or less the same between these two series; the shifts scale with the fractional change in oxidation state: 0.12 eV for Mn3O (1 out of 3) and 0.09 eV for Mn4O2 (1 out of 4). This shows that, compared to XANES, Kβ XES is more susceptible to changes in oxidation state rather than differences in the overall ligand environment.
Fig. 7.
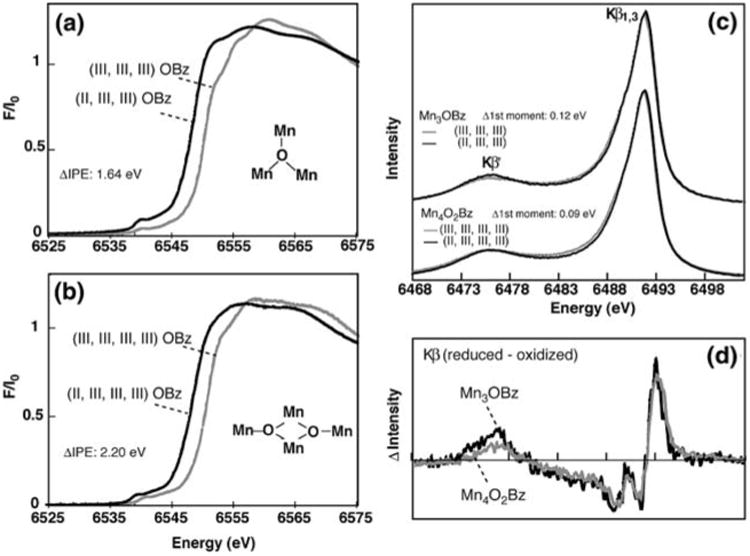
Mn K-edge XANES of the (a) Mn3O (trimers, [Mn3(III,III,III)O(O2Cph)6(ImH)3](ClO4)2(NBu4) and [Mn3(II,III,III)O(O2Cph)6(py)2(H2O)]0.5CH3CN) and (b) Mn4O2 (butterflies, [Mn4(III,III,III,III)O2(O2Cph)7(bipy)2](ClO4) and [Mn4(II,III,III,III)O2(O2Cph)7(bipy)2]) compounds. (c) Kβ X-ray emission spectra (XES) for the Mn3O and Mn4O2 compounds, and (d) Kβ XES difference spectra, the reduced minus oxidized species in each set of compounds. The figure was adapted from Pizarro et al. (2004)
In summary, the X-ray absorption and Kβ emission spectra both indicate Mn-centered oxidation upon S0 to S1 and S1 to S2 transitions. By contrast, both experiments suggest that there are no clear Mn-centered oxidation state changes during the S2 to S3 transition.
Resonant inelastic X-ray scattering (1s2p or Kα RIXS)
The electronic structure of the Mn complex in PS II can be studied in more detail using RIXS (Glatzel et al. 2004; Glatzel and Bergmann 2005). In case of a 3d transition metal ion like Mn, the decay with the highest probability after 1s core hole creation by X-ray absorption is a radiative 2p to 1s transition (therefore, called 1s2p RIXS). This process can be viewed as an inelastic scattering of the incident photon at the Mn atom. In 1s2p RIXS spectroscopy, both the incident X-ray energy (v) and the emission energy (f) are scanned (Fig. 8). The energy difference betweenthe initial state to the intermediate state (v) is equivalent to the K-edge pre-edge transition, and the difference between the initial state and the final state (v–f) is comparable to L-edge spectroscopy. While the K-edge spectrum is a measure of the charge density of the metal, L-edge spectrum has information about the spin state of the metal also, due to the strong (2p, 3d) multiplet interaction. Hence, RIXS is sensitive not only to the metal charge density, but also to the metal spin state. In general, L-edge spectroscopy is difficult for biological samples because of severe radiation damage caused by the higher X-ray absorption at lower energy, and additionally experiments need to be carried out under ultra-high vacuum conditions. In RIXS, L-edge-like spectra are obtained, but the excitation energy is the same as for K-edge spectroscopy. It also allows us measurement of spectra at 10 K in the presence of an exchange gas atmosphere. Hence, RIXS spectroscopy can circumvent some difficulties in the L-edge spectroscopy, and the method is suitable for biological samples.
Fig. 8.

The energy level diagram for the K-pre edge (1s to 3d) and L-edge (2p to 3d) absorption spectra are shown on the right. On the left is the energy level diagram for the RIXS experiment. The excitation is from a 1s to 3d level, and the emission detected is from a 2p to 1s level. The difference in the energy between the ground and final states is equivalent to the Ledge energy levels. Thus by using K-edge X-ray energies one can collect L-edge-like (2p to 3d) spectra
In RIXS spectroscopy, the incident energy is scanned using the beamline double-crystal X-ray monochromator, while the emission energy is scanned using the multi-crystal analyzer mounted on Rowland circles (Fig. 9). By using several single crystal analyzers, we can collect reasonable quality data from a dilute biological sample, such as PS II.
Fig. 9.

The experimental setup used for the RIXS experiment. In RIXS, which is two-dimensional spectroscopy, both the excitation energy is scanned (1s to 3d and 1s to 4p) using the beamline monochromator and the emission energy is scanned across the Kα lines (2p to 1s). Both monochromator and analyzer have ∼1 eV bandwidth
The RIXS spectra are often shown as a surface plot (Fig. 10, top left) or a contour plot (Fig. 10, bottom right). In the surface plot, abscissa is the excitation energy across the 1s–3d energy range of the spectrum and ordinate is the difference between the excitation and emission energy (energy transfer, EB, shown in Fig. 8). RIXS spectra are also presented as line plots by integrating the spectral intensity along the incident energy (constant energy transfer (CET) plot) or the energy transfer axis (constant incident energy (CIE) plot). A CET plot is identical to a normal K pre-edge scan (1s to 3d). A CIE plot is similar to a L-edge spectrum if the intensity is integrated over the entire incident energy range of 2p to 3d transitions.
Fig. 10.
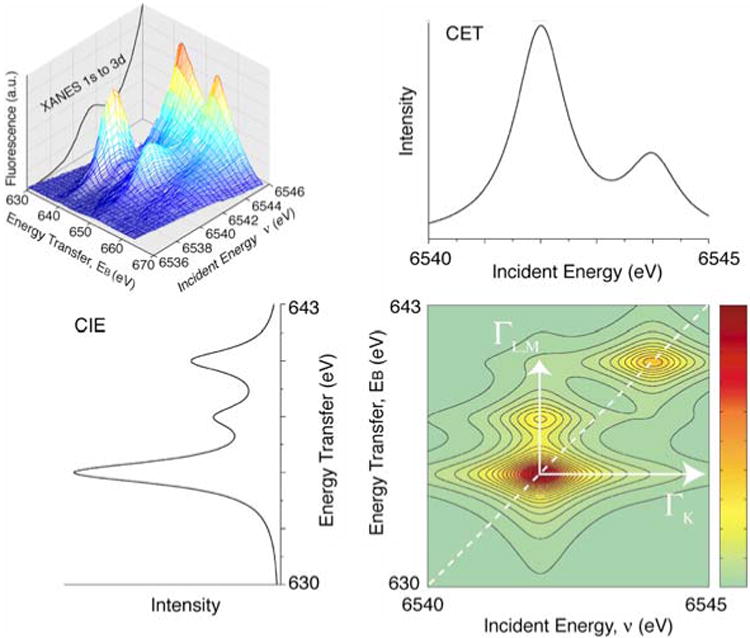
(Top left) A three-dimensional plot showing the theoretical resonant inelastic X-ray spectrum. Abscissa is the excitation energy across the 1s–3d energy range of the spectrum. The 1s–3d K-edge fluorescence excitation spectrum is plotted in the back of the three-dimensional spectrum for reference. Ordinate is the difference between the excitation and emission energy. (Bottom right) The RIXS landscape shown as a contour plot. An integration of the 2D plot parallel to the ordinate yields L-edge like spectra (left bottom), the more intense feature at 640 eV corresponds to transitions to J = 3/2 like states (L3 edges) and transitions to 655 eV correspond to J = 1/2 final states (L2 edges). Integrations parallel to the energy transfer axis sort the spectrum according to the final state (right top). The figure was adapted from Glatzel et al. (2004)
Figure 11 (left) shows RIXS contour plots of two types of Mn compounds: Mn oxides and Mn coordination compounds with formal oxidation states, Mn(II), Mn(III), and Mn(IV) (Glatzel et al. 2004, 2005). The chemical structures of the Mn coordination compounds are shown in Fig. 11 (bottom). The Mn coordination compounds are all six-coordinate, mostly with oxygen ligands. Despite the different ligand environment, similar trends are observed in the Mn oxides and coordination compounds with same oxidation states. The Mn(II)O spectrum shows one broad peak centered at v ∼ 6,540 eV. The spectrum is well explained by the contribution of the crystal field splitting 10Dq between the t2g and eg orbitals (see Glatzel et al. 2004). For Mn in higher oxidation state (Mn(III) and Mn(IV)), the electron (3d–3d) interaction becomes dominant. In Mn(III) compounds, for example, the two strong resonances observed in the contour plots (v = 6,540 and 6,543 eV) are due to the (3d, 3d) multiplet interactions (∼3 eV separation). The separation of two strong features is smaller in Mn coordination compounds (∼2 eV) due to the reduced magnitude of the (3d–3d) interaction; i.e., a more covalent electron configuration decreases the electron– electron interaction.
Fig. 11.
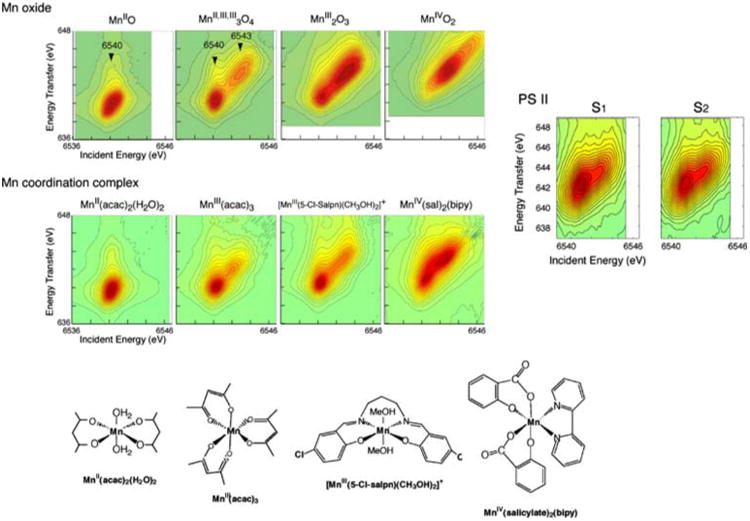
Contour plots of the 1s2p3/2 RIXS planes for four Mn oxides in oxidation states II, III, and IV, the four molecular complexes MnII(acac)2(H2O)2, MnIII(acac)3, [MnIII(5-Cl-Salpn)(CH3OH)2]+, and MnIV(sal)2(bipy), and the S1 and S2 states of PS II. Abscissa is the excitation energy and ordinate is the energy transfer axis. The figure was adapted from Glatzel et al. (2004). Chemical structures of Mn coordination compounds are shown at the bottom
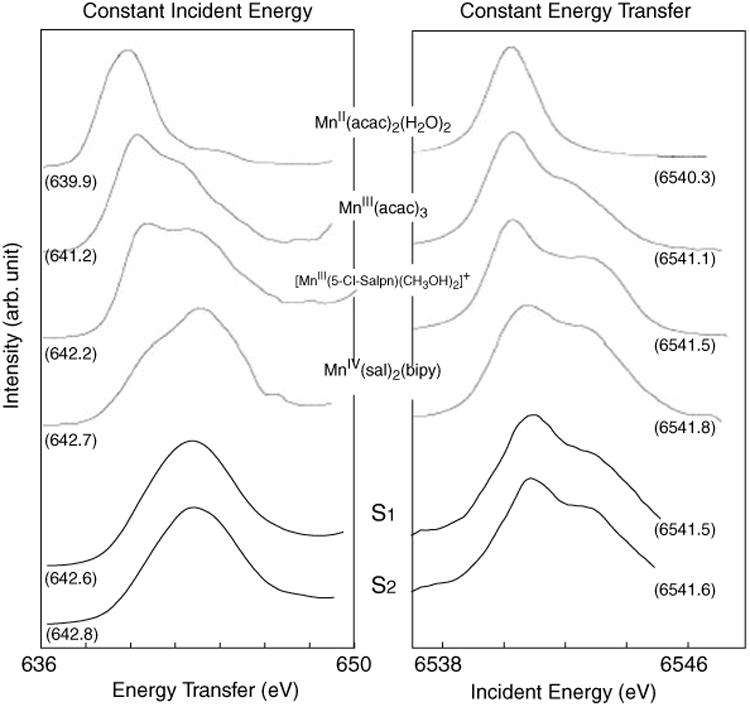
Figure 11 (right) shows the RIXS contours from the S1 and S2 states of PS II (Glatzel et al. 2004). The two types of line plots (CIE and CET) of the S1 and S2 states are also shown in Fig. 12 together with those of Mn coordination compounds. In Fig. 11, the S1 and S2 spectra show mixed features characteristic of Mn (III) and Mn (IV) compounds, having two main peaks. However, the first peak (∼6,541 eV) appears broader than that for the model compounds, and do not have a sharp boundary between the two peaks as in the coordination complexes, owing to the presence of 4 Mn in different oxidation states and different ligand environment.
Fig. 12. Line plots extracted from the RIXS planes for the four coordination complexes and the S1 and S2 states of PS II. First moment value for each compound is also shown.
In RIXS, the 1st moments (Eq. 1) along the incident energy and energy transfer axes define ‘the center of gravity’ energy. As observed in Mn oxides, the 1st moment along the incident energy increases as the Mn oxidation state increases, due to the decrease in charge density on the Mn (Fig. 13a). The interpretation of the 1st moment shift along the energy transfer direction is more complicated, since it is also affected by the final-state interaction. This arises from the (2p, 3d) electron–electron interaction, which is the exchange energy for electrons with parallel spins (see Glatzel et al. 2004). In Fig. 13a, the 1st moment shift for the series of Mn oxides follows the formal oxidation state in a linear manner. This suggests that the effective number of 3d electrons is directly connected to the effective number of unpaired 3d electrons (spins) in these compounds.
Fig. 13.

First moment positions in the 1s2p RIXS plane for (a) the Mn oxides and (b) the coordination complexes and PS II S1 and S2 states (spinach BBY). A linear fit for the Mn oxides is shown as a dotted line in (a). The same Mn oxide line is also indicated in (b) for a comparison to the slope of Mn coordination complexes and PS II data. Along the incident energy axis the plot gives an ordering of the systems in terms of the effective number of 3d electrons. The figure was adapted from Glatzel et al. (2004)
For Mn coordination compounds, the 1st moment position shifts more toward lower energy compared to the Mn oxides (Fig. 13b), which indicates that Mn coordination compounds have much stronger covalency compared to the Mn oxides. The changes per oxidation state in the 1st moment positions are more pronounced between the Mn oxides than those between the Mn coordination compounds. It is worth noting that the two Mn (III) complexes do not have similar 1st moment positions. This can be explained by the presence of one counter ion in Mn(III) (Cl-Salp). The [Mn(III)(Cl-Salp)(CH3OH)2] is positively charged. When the complex contains a counter ion, it behaves like a more ‘oxidized’ form than its formal oxidation state, and therefore, the energy difference between the Mn(III)(Cl-Salp) and Mn(IV)(sal)2(bipy) is much smaller. This in fact confirms that the 1st moment analysis reflects the effective number of electrons on Mn.
The 1st moment of the incident energy for PS II is lower than those for the Mn(IV) coordination complex and larger than or similar to those for the Mn(III) coordination complexes (Fig. 13), which supports the mixed oxidation states of Mn (III) and Mn(IV) in the S1 and S2 states assigned earlier by the EPR and XANES studies. As observed in the PS II S1 and S2 contour plots and line plots (Figs. 11, 12), the spectral changes during the S1 to S2 transition is subtle due to the oxidation state changes of one Mn out of four Mn. The 1st moment shift between S1 and S2 states is a factor of 7–8 smaller than those between Mn(III)(acac)3 and Mn(IV)(Sal)2(bipy), and a factor of 3 smaller than those between Mn(III)(Cl-Salp) and Mn(IV)(Sal)2(bipy). However, the magnitude of the spectral change “per Mn ion” between Mn(III)(Cl-Salp) and Mn(IV)(Sal)2(bipy) is comparable to that between the S1 to S2 states.
We thus find that the electron that is extracted from the OEC in PS II between S1 and S2 is strongly delocalized, consistent with strong covalency for the electronic configuration in the OEC. The data collection of the S0 and S3 RIXS spectra are currently in progress (unpublished data). The preliminary result indicates that the S0 spectrum has a weaker peak compared to the S1 state, having a more pronounced low energy component. In the S2 to S3 transition, peak intensity becomes stronger and the spectral feature is altered. The orbital population change per change in oxidation state between the S2 and S3 states is half as much as that between that between S0 and S1, or S1 and S2 transitions, indicating that the electron is removed from a more covalent form or even more delocalized orbital during this transition.
Summary
The discussion about the oxidation state changes of OEC during the S-state transition has been going on for decades due to its importance in understanding the oxygen-evolving mechanism. The reason for differences in experimental results or in their interpretation, or in the difficulty in the interpretation, particularly in the S2 to S3 oxidation state change, arises very likely from the following points:
A process of deconvolution is involved, in order to obtain and understand the properties of the S3 state using the method of flash illumination. Therefore, one needs careful quantitation of the S-state population of each sample by EPR or other independent methods prior to each X-ray experiment.
-
The effect of radiation damage is also a critical issue to be considered, since reduction of Mn also shifts the XANES spectrum to lower energy. Messinger et al. (2001) have reported the S-state dependence of radiation susceptibility, and have shown that the S3 state is not the most radiation susceptible among the S-states.
Moreover, the following points need to be considered more carefully.
XANES is not an absolute (or unique) way of assigning the oxidation state, particularly when structural changes are involved. In addition, there is no clear theory for simulating the XANES spectra of multinuclear complexes.
As we have emphasized throughout the article, the use of “formal oxidation state” has limited merit, since electron and spin densities on Mn ion do not necessarily correspond to the numbers derived from the oxidation state.
The oxidation state of each S-state has been studied by several experiments including XAS, EPR, and other spectroscopies, and also from the mechanistic point of view. In addition to the points discussed above, one needs to be careful because each experiment or proposal does not necessarily observe or refer to the same phenomenon.
In the current review, we have summarized several X-ray absorption/emission techniques which we applied to tackle the unsettled issue of oxidation state changes in OEC. XANES spectra show that there is a larger shift in the inflection point energy during the S0 to S1 and S1 to S2 transitions compared to that occurring during the S2 to S3 transition. However, the question regarding the Mn-centered oxidation state change on S2 to S3 transition is inconclusive from this method owing to the strong influence of the ligand environment. The Kβ emission spectra, which are less sensitive to the ligand environment, also showed that a Mn-centered oxidation does not occur during the S2 → S3 transition. RIXS results are also consistent with the results obtained from XANES and Kβ XES. On the basis of the overall trend from these three different spectroscopic methods, it is clear that during the S0 to S1, and S1 to S2 transitions there is a change in the oxidation state or charge density population centered on Mn. However, the change is much smaller in the S2 to S3 transition, showing that the nature of this transition is different from that seen for the advancement of the S0 to S1 and S1 to S2 states. At the same time it is worth noting that this is not an all-or-none situation. If Mn is not oxidized, presumably some other species (a protein side-chain ligand or bound water) is oxidized. The delocalization of a small amount of electronic charge from a Mn atom to the ligand could account for the small residual changes seen in the X-ray energies.
We can illustrate the process in an electron density picture by describing an electron cloud that becomes less localized and more distributed between the metal ion and its ligands. As a result, the effective number of 3d electrons and the effective spin state can no longer be derived from the formal oxidation state. These observations do not necessarily conflict with EPR results, in which the data show that the spin state seems to change at each S-state. While EPR probes the spin of the delocalized electron density over the entire OEC cluster, the X-ray spectroscopy sees a superposition of the four local Mn electronic configurations. In other words, Mn X-ray spectroscopy probes a more ‘localized’ aspect of the charge density that is only on Mn.
Acknowledgments
The research from our group presented in this review was supported by the NIH grant (GM 55302), and the DOE, Director, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, under Contract DE-AC02-05CH11231. Synchrotron radiation facilities were provided by SSRL, APS, and ALS, which are supported by DOE, Office of Basic Energy Sciences. The SSRL Biotechnology Program is supported by NIH, National Center of Research Resources, Bio-medical Technology Program, and by DOE, Office of Health and Environmental Research. BioCAT at the APS is a NIH-supported Research Center RR-08630. We thank John Robblee, Johannes Messinger, Uwe Bergmann, Pieter Glatzel, Henk Visser, Carmen Fernandez, Shelly Pizarro, Elodie Anxolabéhère-Mallart, Wen Liang, Theo Roelofs, Yulia Pushkar, and Ken Sauer for contributing to much of the work presented in this review and for many helpful discussions over the years. We dedicate this review to the memory of Ron Guiles (1951–2005), who was a key person in the early development of X-ray spectroscopy techniques as applied to Photosystem II.
Abbreviations
- PS II
Photosystem II
- OEC
oxygen-evolving complex
- EPR
electron paramagnetic resonance
- ENDOR
Electron-Nuclear Double Resonance
- XAS
X-ray absorption spectroscopy
- EXAFS
extended X-ray absorption fine structure
- XANES
X-ray absorption near-edge spectroscopy
- MLS
multiline EPR signal
- NIR
near infra-red
- IPE
Inflection point energy
- XES
X-ray emission spectroscopy
- RIXS
resonant inelastic X-ray scattering
- CIE
constant incident energy
- CET
constant energy transfer
Contributor Information
Junko Yano, Email: JYano@lbl.gov.
Vittal K. Yachandra, Email: VKYachandra@lbl.gov.
References
- Åhrling KA, Peterson S, Styring S. The S0state EPR signal from the Mn cluster arises from an isolated ground state. In: Garab G, editor. Photosynthesis: mechanisms and effects. Kluwer Academic Publishers; Dordrecht: 1998. pp. 1291–1294. [Google Scholar]
- Bergmann U, Grush MM, Horne CR, DeMarois P, Penner-Hahn JE, Yocum CF, Wright DW, Dubé CE, Armstrong WH, Christou G, Eppley HJ, Cramer SP. Characterization of the Mn oxidation states in Photosystem II by Kß X-ray fluorescence spectroscopy. J Phys Chem B. 1998;102:8350–8352. [Google Scholar]
- Bonvoisin J, Blondin G, Girerd JJ, Zimmermann JL. Theoretical study of the multiline EPR signal from the S2 state of the oxygen evolving complex of Photosystem-II – evidence for a magnetic tetramer. Biophys J. 1992;61:1076–1086. doi: 10.1016/s0006-3495(92)81917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussac A, Girerd JJ, Rutherford AW. Conversion of the spin state of the manganese complex in Photosystem II induced by near-infrared light. Biochemistry. 1996;35:6984–6989. doi: 10.1021/bi960636w. [DOI] [PubMed] [Google Scholar]
- Boussac A, Kuhl H, Un S, Rögner M, Rutherford AW. Effect of near-infrared light on the S2-state of the manganese complex of Photosystem II from Synechococcus elongatus. Biochemistry. 1998a;37:8995–9000. doi: 10.1021/bi980195b. [DOI] [PubMed] [Google Scholar]
- Boussac A, Un S, Horner O, Rutherford AW. High-spin states (S ≥ 5/2) of the Photosystem II manganese complex. Biochemistry. 1998b;37:4001–4007. doi: 10.1021/bi9728710. [DOI] [PubMed] [Google Scholar]
- Boussac A, Sugiura M, Kirilovsky D, Rutherford AW. Near-infrared-induced transitions in the manganese cluster of Photo-system II: action spectra for the S2 and S3 redox states. Plant Cell Physiol. 2005;46:837–842. doi: 10.1093/pcp/pci088. [DOI] [PubMed] [Google Scholar]
- Brudvig GW. Structure and function of manganese in Photosystem II. Adv Chem Ser. 1995;246:249–263. [Google Scholar]
- Campbell KA, Peloquin JM, Pham DP, Debus RJ, Britt RD. Parallel polarization EPR detection of an S1-state “multiline” EPR signal in Photosystem II particles from Synechocystis sp. PCC 6803. J Am Chem Soc. 1998;120:447–448. [Google Scholar]
- Carrell TG, Tyryshkin AM, Dismukes GC. An evaluation of structural models for the photosynthetic water-oxidizing complex derived from spectroscopic and X-ray diffraction signatures. J Biol Inorg Chem. 2002;7:2–22. doi: 10.1007/s00775-001-0305-3. [DOI] [PubMed] [Google Scholar]
- Casey JL, Sauer K. EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim Biophys Acta. 1984;767:21–28. [Google Scholar]
- Chu HA, Hillier W, Law NA, Babcock GT. Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim Biophys Acta. 2001;1503:69–82. doi: 10.1016/s0005-2728(00)00216-4. [DOI] [PubMed] [Google Scholar]
- Chu HA, Hillier W, Debus RJ. Evidence that the C-terminus of the D1 polypeptide of Photosystem II is ligated to the manganese ion that undergoes oxidation during the S1 to S2 transition: an isotope-edited FTIR study. Biochemistry. 2004;43:3152–3166. doi: 10.1021/bi035915f. [DOI] [PubMed] [Google Scholar]
- Debus RJ. The manganese and calcium ions of photosynthetic oxygen evolution. Biochim Biophys Acta. 1992;1102:269–352. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- Debus RJ, Strickler MA, Walker LM, Hillier W. No evidence from FTIR difference spectroscopy that aspartate-170 of the D1 polypeptide ligates a manganese ion that undergoes oxidation during the S0 to S1, S1 to S2, or S2 to S3 transitions in photosystem II. Biochemistry. 2005;44:1367–1374. doi: 10.1021/bi047558u. [DOI] [PubMed] [Google Scholar]
- Dekker JP. Optical studies on the oxygen-evolving complex of Photosystem II. In: Pecoraro VL, editor. Manganese redox enzymes. VCH Publishers; New York: 1992. pp. 85–103. [Google Scholar]
- Dexheimer SL, Klein MP. Detection of a paramagnetic intermediate in the S1 state of the photosynthetic oxygen-evolving complex. J Am Chem Soc. 1992;114:2821–2826. [Google Scholar]
- Dismukes GC, Siderer Y. EPR spectroscopic observations of a manganese center associated with water oxidation in spinach chloroplasts. FEBS Lett. 1980;121:78–80. [Google Scholar]
- Dismukes GC, Ferris K, Watnick P. EPR spectroscopic evidence for a tetranuclear manganese cluster as the site for photosynthetic oxygen evolution. Photobiochem Photobiophys. 1982;3:243–256. [Google Scholar]
- Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Glatzel P, Bergmann U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes – electronic and structural information. Coord Chem Rev. 2005;249:65–95. [Google Scholar]
- Glatzel P, Bergmann U, Yano J, Visser H, Robblee JH, Gu WW, de Groot FMF, Christou G, Pecoraro VL, Cramer SP, Yachandra VK. The electronic structure of Mn in oxides, coordination complexes, and the oxygen-evolving complex of Photosys-tem II studied by resonant inelastic X-ray scattering. J Am Chem Soc. 2004;126:9946–9959. doi: 10.1021/ja038579z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzel P, Yano J, Bergmann U, Visser H, Robblee JH, Gu WW, de Groot FMF, Cramer SP, Yachandra VK. Resonant inelastic X-ray scattering (RIXS) spectroscopy at the MnK absorption pre-edge – a direct probe of the 3d orbitals. J Phys Chem Solids. 2005;66:2163–2167. doi: 10.1016/j.jpcs.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson Ö, Andréasson LE. EPR-detectable magnetically interacting manganese ions in the photosynthetic oxygen-evolving system after continuous illumination. Biochim Biophys Acta. 1982;679:261–268. [Google Scholar]
- Hasegawa K, Ono TA, Inoue Y, Kusunoki M. Spin-exchange interactions in the S2-state manganese tetramer in photosynthetic oxygen-evolving complex deduced from g = 2 multiline EPR signal. Chem Phys Lett. 1999;300:9–19. [Google Scholar]
- Haumann M, Muller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H. Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3, S4 → S0) characterized by X-ray absorption spectroscopy at 20 K and room temperature. Biochemistry. 2005;44:1894–1908. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- Horner O, Riviére E, Blondin G, Un S, Rutherford AW, Girerd JJ, Boussac A. SQUID magnetization study of the infrared-induced spin transition in the S2 state of Photosystem II: spin value associated with the g = 4.1 EPR signal. J Am Chem Soc. 1998;120:7924–7928. [Google Scholar]
- Ioannidis N, Schansker G, Barynin VV, Petrouleas V. Interaction of nitric oxide with the oxygen evolving complex of Photosystem II and manganese catalase: a comparative study. JBIC, J Biol Inorg Chem. 2000;5:354–363. doi: 10.1007/pl00010664. [DOI] [PubMed] [Google Scholar]
- Ioannidis N, Nugent JHA, Petrouleas V. Intermediates of the S3 state of the oxygen-evolving complex of Photosystem II. Biochemistry. 2002;41:9589–9600. doi: 10.1021/bi0159940. [DOI] [PubMed] [Google Scholar]
- Iuzzolino L, Dittmer J, Dörner W, Meyer-Klaucke W, Dau H. X-ray absorption spectroscopy on layered Photosystem II membrane particles suggests manganese-centered oxidation of the oxygen-evolving complex for the S0-S1, S1-S2, and S2-S3 transitions of the water oxidation cycle. Biochemistry. 1998;37:17112–17119. doi: 10.1021/bi9817360. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Shen JR. Crystal structure of oxygen-evolving Photosystem II from Thermosynechococcus vulcanus at 3.7-Angstrom resolution. Proc Natl Acad Sci USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Mizusawa N, Ishii A, Nakazawa S, Ono T. Changes in structural and functional properties of oxygen-evolving complex induced by replacement of D1-glutamate 189 with glutamine in Photosystem II – ligation of glutamate 189 carboxylate to the manganese cluster. J Biol Chem. 2005a;280:37895–37900. doi: 10.1074/jbc.M505887200. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Mizusawa N, Ishii A, Ono T. FTIR detection of structural changes in a histidine ligand during S-state cycling of photosynthetic oxygen-evolving complex. Biochemistry. 2005b;44:16072–16078. doi: 10.1021/bi051306r. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Mizusawa N, Yamanari T, Ishii A, Ono T. Structural changes of D1 C-terminal alpha-carboxylate during S-state cycling in photosynthetic oxygen evolution. J Biol Chem. 2005c;280:2078–2083. doi: 10.1074/jbc.M410627200. [DOI] [PubMed] [Google Scholar]
- Kulik LV, Epel B, Lubitz W, Messinger J. Mn-55 pulse ENDOR at 34 GHz of the S0 and S2 states of the oxygen-evolving complex in Photosystem II. J Am Chem Soc. 2005;127:2392–2393. doi: 10.1021/ja043012j. [DOI] [PubMed] [Google Scholar]
- Kusunoki M. A new paramagnetic hyperfine-structure effect in manganese tetramers – the origin of multiline EPR signals from an S2 state of a photosynthetic water-splitting enzyme. Chem Phys Lett. 1992;197:108–116. [Google Scholar]
- Kuzek D, Pace RJ. Probing the Mn oxidation states in the OEC. Insights from spectroscopic, computational and kinetic data. Biochim Biophys Acta. 2001;1503:123–137. doi: 10.1016/s0005-2728(00)00218-8. [DOI] [PubMed] [Google Scholar]
- Liang W, Roelofs TA, Cinco RM, Rompel A, Latimer MJ, Yu WO, Sauer K, Klein MP, Yachandra VK. Structural change of the Mn cluster during the S2 → S3 state transition of the oxygen-evolving complex of Photosystem II. Does it reflect the onset of water/substrate oxidation? Determination by Mn X-ray absorption spectroscopy. J Am Chem Soc. 2000;122:3399–3412. doi: 10.1021/ja992501u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of Photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- Matsukawa T, Mino H, Yoneda D, Kawamori A. Dual-mode EPR study of new signals from the S3-state of oxygen-evolving complex in Photosystem II. Biochemistry. 1999;38:4072–4077. doi: 10.1021/bi9818570. [DOI] [PubMed] [Google Scholar]
- Messinger J. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys Chem Chem Phys. 2004;6:4764–4771. [Google Scholar]
- Messinger J, Nugent JHA, Evans MCW. Detection of an EPR multiline signal for the S0 state in Photosystem II. Biochemistry. 1997a;36:11055–11060. doi: 10.1021/bi9711285. [DOI] [PubMed] [Google Scholar]
- Messinger J, Robblee J, Yu WO, Sauer K, Yachandra VK, Klein MP. The S0 state of the oxygen-evolving complex in photosystem II is paramagnetic: detection of an EPR multiline signal. J Am Chem Soc. 1997b;119:11349–11350. doi: 10.1021/ja972696a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger J, Robblee JH, Bergmann U, Fernandez C, Glatzel P, Visser H, Cinco RM, McFarlane KL, Bellacchio E, Pizarro SA, Cramer SP, Sauer K, Klein MP, Yachandra VK. Absence of Mn-centered oxidation in the S2 to S3 transition: implications for the mechanism of photosynthetic water oxidation. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ort DR, Yocum CF, editors. Oxygenic photosynthesis: the light reactions. Kluwer Academic Publishers; Dordrecht: 1996. [Google Scholar]
- Peloquin JM, Britt RD. EPR/ENDOR characterization of the physical and electronic structure of the OEC Mn cluster. Biochim Biophys Acta. 2001;1503:96–111. doi: 10.1016/s0005-2728(00)00219-x. [DOI] [PubMed] [Google Scholar]
- Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD. 55Mn ENDOR of the S2-state multiline EPR signal of Photosystem II: implications on the structure of the tetranuclear cluster. J Am Chem Soc. 2000;122:10926–10942. [Google Scholar]
- Pizarro SA, Glatzel P, Visser H, Robblee JH, Christou G, Bergmann U, Yachandra VK. Mn oxidation states in tri- and tetra-nuclear Mn compounds structurally relevant to Photosystem II: Mn K-edge X-ray absorption and Kβ X-ray emission spectros-copy studies. Phys Chem Chem Phys. 2004;6:4864–4870. doi: 10.1039/B407513G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkar Y, Yano J, Glatzel P, Messinger J, Lewis A, Sauer K, Bergmann U, Yachandra VK. Structure and orientation of the Mn4Ca cluster in plant Photosystem II membranes studied by polarized range-extended X-ray absorption spectroscopy. J Biol Chem. 2007;282:7198–7208. doi: 10.1074/jbc.M610505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robblee JH, Messinger J, Cinco RM, McFarlane KL, Fernandez C, Pizarro SA, Sauer K, Yachandra VK. The Mn cluster in the S0 state of the oxygen-evolving complex of Photosystem II studied by EXAFS spectroscopy: are there three di-μ-oxo-bridged Mn2 moieties in the tetranuclear Mn complex? J Am Chem Soc. 2002;124:7459–7471. doi: 10.1021/ja011621a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs TA, Liang MC, Latimer MJ, Cinco RM, Rompel A, Andrews JC, Sauer K, Yachandra VK, Klein MP. Oxidation states of the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex. Proc Natl Acad Sci USA. 1996;93:3335–3340. doi: 10.1073/pnas.93.8.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford AW, Zimmermann JL, Boussac A. Oxygen evolution. In: Barber J, editor. The photosystems: structure, function, molecular biology. Elsevier; Amsterdam: 1992. pp. 179–229. [Google Scholar]
- Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. X-ray absorption edge spectroscopy and computational studies on LCuO2 species: superoxide-Cu-II versus peroxide-Cu-III bonding. J Am Chem Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn PEM. O-O bond formation in the S4 state of the oxygen-evolving complex in Photosystem II. Chem Eur J. 2006;12:9217–9227. doi: 10.1002/chem.200600774. [DOI] [PubMed] [Google Scholar]
- Strickler MA, Hillier W, Debus RJ. No evidence from FTIR difference spectroscopy that glutamate-189 of the D1 polypep-tide ligates a Mn ion that undergoes oxidation during the S0 to S1, S1 to S2, or S2 to S3 transitions in Photosystem II. Biochemistry. 2006;45:8801–8811. doi: 10.1021/bi060583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styring SA, Rutherford AW. The microwave power saturation of SIIslow varies with the redox state of the oxygen-evolving complex in Photosystem II. Biochemistry. 1988;27:4915–4923. [Google Scholar]
- Visser H, Anxolabéhère-Mallart E, Bergman U, Glatzel P, Robblee JH, Cramer SP, Girerd JJ, Sauer K, Klein MP, Yachandra VK. Mn K-edge XANES and Kβ XES studies of two Mn-oxo binuclear complexes. Investigation of three different oxidation states relevant to the oxygen-evolving complex of Photosystem-II. J Am Chem Soc. 2001;123:7031–7039. doi: 10.1021/ja004306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wydrzynski T, Satoh S. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Springer; Dordrecht: 2005. [Google Scholar]
- Yachandra VK. The catalytic manganese-cluster: organization of the metal ions. In: Wydrzynski T, et al., editors. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Springer; Dordrecht: 2005. pp. 235–260. [Google Scholar]
- Yachandra VK, Klein MP. X-Ray absorption spectroscopy: determination of transition metal site structures in photosynthesis. In: Amesz J, et al., editors. Biophysical techniques in photosynthesis. Kluwer Academic Publishers; Dordrecht, Netherlands: 1996. pp. 337–354. [Google Scholar]
- Yachandra VK, Sauer K, Klein MP. Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Mino H, Matsukawa T, Kawamori A, Ono T. Parallel polarization electron paramagnetic resonance studies of the S1-state manganese cluster in the photosynthetic oxygen-evolving system. Biochemistry. 1997;36:7520–7526. doi: 10.1021/bi962791g. [DOI] [PubMed] [Google Scholar]
- Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. X-ray damage to the Mn4Ca complex in Photosystem II crystals: a case study for metallo-protein X-ray crystallography. Proc Natl Acad Sci USA. 2005a;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Pushkar Y, Glatzel P, Lewis A, Sauer K, Messinger J, Bergmann U, Yachandra VK. High-resolution Mn EXAFS of the oxygen-evolving complex in Photosystem II: structural implications for the Mn4Ca Cluster. J Am Chem Soc. 2005b;127:14974–14975. doi: 10.1021/ja054873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Kern J, Sauer K, Latimer M, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK. Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann JL, Rutherford AW. EPR studies of the oxygen-evolving enzyme of photosystem II. Biochim Biophys Acta. 1984;767:160–167. [Google Scholar]
- Zouni A, Witt HT, Kern J, Fromme P, Krauß N, Saenger W, Orth P. Crystal structure of Photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]