

Abstract
Texaphyrins are pentaaza expanded porphyrins with the ability to form stable complexes with a variety of metal cations, particularly those of the lanthanide series. In biological milieus, texaphyrins act as redox mediators and mediate the production of reactive oxygen species (ROS). In this review, newer studies involving texaphyrin complexes targeting several different applications in anticancer therapy are described. In particular, the preparation of bismuth- and lead texaphyrin complexes as potential alpha core emitters for radiotherapy is detailed, as are gadolinium texaphyrin functionalized magnetic nanoparticles with features that make them of interest as dual-mode MRI contrast agents and as constructs with anticancer activity mediated through ROS-induced sensitization and concurrent hyperthermia. Also discussed are gadolinium texaphyrin complexes as possible carrier systems for the targeted delivery of platinum payloads.
Keywords: Porphyrins, Expanded Porphyrins, MRI Contrast Agents, Radiosensitizers, Radiation Sensitizers, Nanoparticles, Metal-Based Anticancer Drugs, Cisplatin Analogs
The combined use of chemotherapy and radiation therapy has led to clinical breakthroughs in the controlled treatment and cure of several cancerous diseases. Today, the three main types of radiation therapy are classified as external beam radiation therapy (EBRT or more commonly X-ray therapy, XRT), brachytherapy (sealed source radiation therapy), and systematic radioisotope therapy (unsealed source radiotherapy). However, the search for efficient radiation sensitizers, i.e., compounds that actively support radiation therapy through different mechanisms, remains a critical, albeit elusive goal in anticancer therapy. Active, or so-called sensitized, radiation therapy could prove particularly beneficial when treating solid tumors. Solid tumors usually outgrow their blood supply, causing a low-oxygen state known as hypoxia. As revealed by modern detection techniques, these hypoxic regions are often characterized by reduced XRT efficiencies. In the absence of oxygen, DNA is repaired more efficiently. In contrast, oxygenated tissues are generally two to three times more sensitive towards radiation. From an operational perspective, hypoxic cells are difficult to destroy completely using XRT alone.1,2 Applying radiation sensitizers could allow modulation of the radiation response and lead to an improvement in local tumor control. Here, the idea is to administer radiosensitizers that would enhance or support the effects of radiation at cancerous sites, reduce cytotoxic side effects for normal tissues, or both.
Oxygen derived species, such as superoxide, singlet oxygen, hydroxyl radicals and hydrogen peroxide, are prominent cytotoxic substances and have been implicated in the etiology of a wide array of human diseases, including cancer. When administered in a cancer-selective manner, drugs that are able to produce reactive oxygen species (ROS) can give rise to manifest benefits. Several classes of anticancer drugs, such as quinonebased agents, have been studied as a means to promote the generation of ROS at tumor sites.3 The mechanism is believed to involve a redox cycling process that relies in part on chemical reduction in vivo by biological reductants, such as NADPH; reoxidation with oxygen produces ROS that can inter alia damage DNA.
Many strategies to enhance the efficacy of radiation therapy involve diminishing the activity of natural ROS defense mechanisms. Often enzymes, such as superoxide dismutase (SOD), glutathione peroxidase, and catalase, are involved. Many other endogeneous species, including glutathione (GSH), thioredoxin (TRX)/thioredoxin reductase (TRXR), ascorbate (vitamin C), and α-tocopherol (vitamin E), are also able to serve as ROS scavengers. Agents that either compromise these defense mechanisms or which are able to produce actively enhanced levels of ROS are thus attractive since they could lead to more efficient anticancer treatments.
Texaphyrin, a Redox-Active Expanded Porphyrin
Several classes of FDA-approved anticancer drugs, including quinone-based agents, are believed to exhibit radiation sensitizing effects as the result of producing reactive oxygen species, such as superoxide and hydrogen peroxide. These latter entities are able to damage DNA and promote cell death. Texaphyrins are experimental drugs that are known to localize to cancerous lesions and to produce reactive oxygen species. This is discussed further below.
Texaphyrins are pentaaza Schiff-base macrocycles with a strong, but “expanded” similarity to traditional porphyrins.4-6 They also bear resemblance to the five-pointed star in the state flag of Texas, a feature that accounts for their name. From a chemical perspective, texaphyrins are characterized by the presence of an inner coordination core that is roughly 20% larger than that present in porphyrins. The formal charge on the deprotonated texaphyrin ligand is −1, as compared to −2 for a porphyrin. To date, the texaphyrins have been demonstrated to form stable 1:1 complexes with a wide variety of metal cations, particularly with those of the trivalent lanthanide series (cf. Figure 2).4,7,8
Figure 2.

Stable texaphyrin complexes with all metals shown in green are known.
One particular functionalized gadolinium(III) texaphyrin, motexfin gadolinium 1 has been studied in detail by the Sessler group and was developed for clinical study under the aegis of Pharmacyclics, Inc.3,9 In a series of physical chemical and mechanistic studies, it was shown that the gadolinium species 1 is easy to reduce in comparison to, e.g., typical porphyrins and can act as a redox mediator producing ROS in the presence of suitable reductants and molecular oxygen (Scheme 1). In the intracellular environment, it has been proposed that complex 1 accepts an electron from, and catalyzes the oxidation of, various reducing metabolites, such as ascorbate, reduced nicotinamide-adenine dinucleotide phosphate (NADPH), thioredoxin reductase, glutathione, and dihydrolipoate. This electron transfer event leads to the formation of a reduced texaphyrin radical that then reacts with oxygen to produce superoxide in a rapid equilibrium process, which in turn regenerates compound 1. In vitro, and presumably in vivo, this superoxide is converted quickly into hydrogen peroxide,10 a species that is known to be a potent apoptosis trigger.
Scheme 1.

Mechanistic representation of how motexafin gadolinium is thought to act as a redox mediator.
In an effort to determine whether the centrally coordinated metal cation plays a role in regulating the ability of texaphyrins to function as oxidation catalysts for ascorbate, several transition metal complexes were prepared and characterized. A summary of representative stable texaphyrin species, including various lanthanide complexes, is given in Figure 3.4,11-17
Figure 3.

Summary of representative stable texaphyrin complexes.4,11-17
The role of the chelated metal center was found to be substantial. While the Mn(II) complex of texaphyrin ligand 6 displayed an initial rate that was approximately three times slower than 1 under identical experimental conditions (Vo = 3.0 μM/min vs. 8.7 μM/min, respectively), the Co(II) and Fe(III) (as the μ-oxo dimer) complexes of texaphyrin ligand 6 gave initial rate values (Vo = 23.8 and 30.6 μM, respectively) that were substantially larger.18 This proved true in spite of the fact that these species are harder to reduce than 1 (E1/2 = −571 for 6 as Co(II)-complex vs. −294 for 1; vs. Ag/AgCl in DMSO).19 In this instance, it is thought that the redox active metal centers participate in ascorbate decomposition. Unfortunately, the Co(II) complex and the Fe(III) complex of 6 were considered too lipophilic to be attractive in terms of further drug development, at least for the XRT sensitization indications for which motexafin gadolinium was being tested.
Synthesis of Texaphyrins, Physical Properties and MRI Activity
The synthesis of the first texaphyrins benefited from an efficient synthesis of a symmetric tripyrrane dialdehyde key precursor. This intermediate, shown as compounds 15 and 16 in Scheme 2, was obtained via the condensation of two pyrrole subunits, 7 or 8 (obtained via Paal-Knorr reactions) and 9 (prepared using the Barton-Zard procedure), respectively, followed by further functional group elaboration. These latter reactions included ester deprotection, decarboxylation and formylation. Reduction of the side chain terminal ester to the corresponding alcohol was also carried out during the sequence of steps leading to 16.
Scheme 2.

Synthesis of the texaphyrin key precursors 15 and 16.
The nonaromatic form of the texaphyrin ligand, is synthesized by a hydrogen chloride catalyzed 1:1 Schiff base condensation between a tripyrrane dialdehyde, such as 15 or 16, with an appropriately derivatized o-phenylenediamine under conditions of high dilution. This procedure is similar to the one employed by Mertes et al. for the formation of the so-called “accordion” macrocycle.20,21
Oxidation of the nonaromatic texaphyrin ligand in the presence of an appropriate metal salt, molecular oxygen (air) and an organic base (e.g., triethylamine) generally affords the aromatic texaphyrin macrocycle as its metal complex in good yield (Scheme 3). The metal cation is thought to stabilize the macrocycle as a result of a presumed thermodynamic template effect.22
Scheme 3.
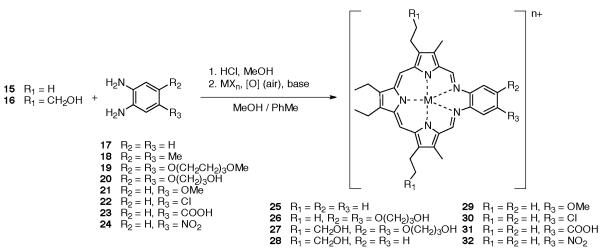
General synthesis of texaphyrin.
Thus, once formed these metal complexes are extremely stable, except under acidic conditions, which readily lead to hydrolysis of the macrocycle.23
The UV-visible spectrum of compound 1 is dominated by two absorption bands. The higher energy Soret-like band at 474 nm is analogous to the ~400 nm band of porphyrins and is characteristic of the absorption bands seen for other vividly pigmented porphyrin-moieties. The Soret-like band is flanked by N- and Q-like bands at higher and lower energies, respectively, with the lowest energy Q-band for motexafin gadolinium being seen at 740 nm (cf. Figure 4).
Figure 4.

UV-visible spectrum of motexafin gadolinium 1, 25 μM in methanol.
Interestingly, there is a steady shift in the Q-like band from red to blue (Δ = 15 nm) as the Ln(III) cation under study progresses from lanthanum to lutetium.24 This shift in the Q-like bands appears to follow the contraction of the metal cations in the lanthanide series. A plot of the wavelength (in nm) of the Q-like band versus the ionic radius of the Ln(III) ion gives a linear relationship.24
Another spectral feature of certain metallated texaphyrins, especially those containing diamagnetic cations, is their ability to fluoresce. The resulting Q-type emission bands, like the Q-type absorption bands, are substantially red-shifted (by >100 nm) compared to typical porphyrins.25, 26 This combination of spectral and redox features made texaphyrins attractive for study in the context of certain biomedical applications.
Some of the first biological tests with compound 1 involved magnetic resonance imaging (MRI) studies. It was found to be easily visualized by this modality and to enhance the contrast of MR images substantially. These attractive findings were ascribed to the centrally coordinated paramagnetic metal cation gadolinium(III)27, which serves to enhance the effective spin lattice (T1) relaxation. On the basis of initial MRI analyses, motexafin gadolinium was found to localize well in tumors. No appreciable localization in adjacent normal tissue was observed.28 Additional MRI studies conducted by Viala et al. provided further evidence for the proposed tumor selectivity of 1.29 The ratio of motexafin gadolinium in tumor cells to that in surrounding normal cells was reported to be up to 9:1.30 As inferred from MR images, this ratio increases to 50:1 in the case of metastatic brain tumors.31 The uptake in target lesions was higher after ten daily injections than after the first dose. This finding was interpreted in terms of an ability to accumulate and persist in brain metastases. In clinical tests, the response to treatment at successive MRI examinations could be evaluated as well, since either the gadolinium texaphyrin or the gadolinium(III) cation, originally contained in its core, was found to remain in tumorous lesions for several months. This could be of practical benefit in the context of a treatment regimen.29
Initially compound 1 was developed by Pharmacyclics, Inc. as an experimental drug that was considered attractive for use in treating patients suffering from non-small-cell lung cancer (NSCLC) with brain metastases. However, after a Phase III study revealed tantalizing signs of efficacy, but without meeting the pre-negotiated statistical endpoints, motexafin gadolinium failed to obtain FDA approval in December 2007.32 Although limited clinical studies of motexafin gadolinium are ongoing, this failure has served as an incentive to define new research goals for texaphyrins and to explore other cancer-related opportunities for this class of compounds. The following summaries are designed to provide synopses of three projects developed as the result of these refocusing efforts.
Bismuth and Lead Coordinated Texaphyrins
One area wherein texaphyrins could see further biomedical application involves their use in supporting complexes of main group elements. In porphyrin chemistry, complexes with posttransition elements, such as Ga, In, Tl, Pb, and Bi, are rare as compared to those of the transition elements.33 Yet the chemistry of bismuth has become of increasing interest since its 212Bi and 213Bi isotopes show promise for use as α-emitters in radiotherapy.34,35 Due to the high linear energy transfer radiation produced (100 keV/μm), these isotopes demonstrate a strong anticancer cell effect under hypoxic conditions.36 This ultimately leads to double-strand DNA breaks at levels that preclude efficient cell repair and survival. However, the short halflife of these two isotopes (60.55 min and 45.65 min for 212Bi and 213Bi, respectively) and the difficulties of administrating salts in a biocompatible, disease-specific manner provides an incentive to develop complexing agents that can coordinate the Bi(III) cation quickly and which would then impart a degree of tumor-specific targeting.
Also attractive is the concept of an in situ generator for either 212Bi or 213Bi. One approach would involve the initial complexation of lead.37 One particular lead isotope, 212Pb, has a half-life of 10.64 hours and produces 212Bi as its primary decay product along with a β-particle. Thus, if this precursor isotope (212Pb) could be complexed readily, it would allow for the effective production of the corresponding 212Bi complex.
Finding suitable ligands for bismuth or lead has proved challenging. An ideal ligand would be one that is able to form stable complexes with both bismuth and lead rapidly and to do so under mild conditions. Complexes of bismuth and lead that possess inherent tumor selectivity would be further advantageous since they would allow the radioactive species in question, namely 212Bi, 213Bi, or 212Pb, to be delivered selectively to cancerous tissues. This led us to suggest that texaphyrin would be an ideal ligand for these metals. As noted above, texaphyrins have been shown to localize to, or be retained selectively in, rapidly growing tissues, including cancerous lesions; they are thus attractive as carriers for these radioisotopes.38
As demonstrated recently, texaphyrin is indeed able to complex the Bi(III) and Pb(II) cations rapidly (reaction in methanol at 75 °C completed after 34 minutes in the case of Bi(III) and 98 minutes in the case of Pb(II)).39 Specifically, spectroscopic and mass spectrometric evidence was put forward to support the formation of the first lead(II) texaphyrin complexes 33 and 35 (cf. Figure 5). Similar methods were used to confirm the formation of the first discrete binuclear μ-oxo bismuth(III) macrocyclic complex 34, a system that was further characterized via a single crystal X-ray diffraction analysis.39
Figure 5.

Lead and bismuth texaphyrins 33–36, and views of the single crystal X-ray structure of complex 34.
These newly prepared Pb(II) and Bi(III) texaphyrin complexes proved chemically stable despite the μ-oxo bond present in the latter complex. This allowed the water soluble derivatives to be studied in vitro using the A2780 ovarian cancer cell line. On this basis, it was concluded that the Pb(II) texaphyrin 35 and the Bi(III) texaphyrin 36 gave IC50 values of 2.9 and 2.2 μM, respectively. This represents a two to three fold increase in cytotoxicity relative to motexafin gadolinium (6.3 μM).40 Based on these findings and considering the tumor selectivity properties of texaphyrins, we suggest that the texaphyrins could emerge as useful complexants for 212Bi, 213Bi, or 212Pb and, as such, warrant further study as candidates for radiotherapy.
Texaphyrin Functionalized Magnetic Nanoparticles
Achieving high accuracy and precision are the main challenges in a variety of imaging techniques, including MRI. Typical MRI contrast agents are comprised of either paramagnetic materials for T1 weighted scans (i.e., to depict differences in the spin-lattice relaxation time of various tissues) or superparamagnetic nanoparticles for T2 weighted scans (i.e., to depict differences in the spin-spin relaxation time).41-44 However, such single mode contrast agents are far from ideal, particularly when accurate imaging of small biological targets is required.45,46 One of us (J.C.) put forward a potential solution to this problem via the development of magnetic nanoparticles that can act as dualmode MRI contrast agents (DMCA).47 The so-called “magnetically decoupled” core-shell design of these nanoparticles consists of a T2 active core (e.g., MnFe2O4) and a T1 active material (Gd2O(CO3)2) located on the shell.
The initial goal of this project was thus to use gadolinium(III) texaphyrins as the T1 contrast material in a DMCA system. With this consideration in mind, gadolinium(III) texaphyrin 37-conjugated magnetic nanoparticle constructs (GdTx-MNP) consisting of a zinc doped iron oxide T2 core coated with a layer of silicon dioxide functioning as a separating layer, were prepared. In this case, the final conjugation step results in the formation of constructs where the texaphyrin macrocycles are covalently linked to the surface of the nanoparticles.48
The elaborated nanoparticle systems were then tested as dualmode MRI contrast agents. While contrast agents used clinically, such as Magnevist® and Feridex®, display either only bright T1 or dark T2 contrast, in an MRI phantom study, GdTx-MNP was found to give rise to intense MRI signals in both modes (cf. Figure 6). Simultaneous T1 bright and T2 dark contrast effects are ascribable to the gadolinium texaphyrin (T1 active material) and magnetic nanoparticle (T2 active material) portions of the constructs, respectively. In contrast, MR images associated with the control groups and the commercially available contrast agents Magnevist® and Feridex® display either only bright T1 contrast or dark T2 contrast, but not both.
Figure 6.

Dual-Mode MRI contrast enhancements (T1 and T2 modes shown; note that a bright contrast in the T1 mode and a dark contrast in the T2 mode are desired in MR images of tumorous tissues) and anticancer activity that is ascribed to a combination of sensitization (ROS production) and hyperthermia.48
Additionally, we demonstrated that the GdTx-MNP construct can effectively sensitize cancer cells (here: MDA-MB-231, a breast cancer cell line) in vitro and in vivo, making them highly vulnerable to apoptotic magnetic hyperthermia at low temperatures (Figure 6).48 This enhancement was ascribed to the ability of the texaphyrins to produce ROS under the conditions of the experiment.
The in vivo studies involved xenograft mouse models. These models were produced by injecting MDA-MB-231 cells into the right hind leg of nude mice in a series of experimental groups (n = 3). A dispersion of GdTx-MNPs (75 μg, dispersed in 50 μL normal saline) was directly injected into the tumor tissue (100 mm3). The mouse was then placed in a water-cooled magnetic induction coil (Figure 7 (a)) and an AC magnetic field (500 kHz at 30 kA m−1) was applied for 30 minutes to maintain a constant temperature at the tumor (43 ± 1 °C). This hyperthermia treatment was applied once and the tumor size was monitored for 14 consecutive days. In the mice making up the untreated control group, the average tumor size increased approximately sevenfold by day 14 (Figure 7 (b) and (c)). However, for the group receiving hyperthermia treatment with GdTx-MNPs, the tumors were absent after eight days (Figure 7 (b) and (c)). For comparison, another group of mice was subjected to hyperthermia treatment after administration of unfunctionalized MNPs at an identical dosage. Although the size of the tumors regressed initially, a significant amount of tumor mass remained at day eight (V/Vinitial = 0.6) and the tumors started to regrow at day 12.48
Figure 7.

In vivo magnetic hyperthermia: (a) Injection of GdTx-MNPs into right hind leg of nude mice and application of an AC magnetic field for 30 min. (b) Plot of tumor volume (V/Vinitial) versus the number of days after treatment. Three different groups were either untreated, treated with unfunctionalized magnetic nanoparticles (MNPs) or treated with GdTx-MNPs hyperthermia. (c) Images of xenografted tumors (MDA-MB-231) on nude mice before treatment (left column) and 14 days after treatment (right column). Note the different outcomes for untreated control and the mice subjected to hyperthermia with MNPs and GdTx-MNPs. Each scale bar indicates 5 mm.48
Until now, attempts to use low temperature magnetic hyperthermia for cancer therapy have proved challenging due to the development of thermal tolerance. The dramatic reduction in tumor burden seen in vivo and the high degree of efficacy seen in vitro using the texaphyrin-functionalized nanoparticles are ascribed to the sensitization effect arising from ROS production as noted above. The efficient heat generation produced by GdTx-MNPs is also advantageous because lower concentrations of nanoparticles are required to achieve the same biological effect at low temperatures (43 °C). The pathway of cell death involves predominantly apoptosis, a mode of action that is considered beneficial for ultimate clinical use. Given these features, we propose that double effector nanoparticles, such as the texaphyrin-bearing systems produced to date, could emerge as a new approach to achieving apoptotic magnetic hyperthermia.
Texaphyrin-Platinum Conjugates
Building on appreciation that texaphyrins display tumor selective localization features, our group became intrigued by the possibility that texaphyrins could act as active delivery vehicles for other known cancer therapeutics. We considered this approach for drug delivery to be attractive relative to other potentially cometing strategies (i.e., pegylation, liposomal formulation, etc.) in that the carrier (i.e., texaphyrin) itself is welltolerated and effective at cancer targeting; it also shows some promise as an anticancer agent (vide supra). To test this potential, an effort was made to create conjugates containing platinum(II) centers. The hope was that this would allow certain mechanisms of platinum resistance to be overcome.
While active in several cancer types and included in front line therapy by oncologists, platinum anticancer agents display acquired resistance in many cancers, which limits their clinical utility. The cause of this resistance is multifactorial and includes both pharmacologic mechanisms (e.g., decreased drug uptake, increased glutathione, and increased DNA adduct repair) and molecular mechanisms of resistance (e.g., loss of p53 function, increase in survivin, and an increase in Bcl2).49-51
A major incentive for using texaphyrin as a “carrier” involved the challenge of overcoming platinum-drug resistance, particularly as applied to ovarian cancer. The FDA-approved platinum drugs, cisplatin 39, carboplatin 40, and oxaliplatin 41 (cf. Figure 8) are widely used cancer therapeutic agents.52-55 Cisplatin and carboplatin, however, are the main agents used in ovarian cancer.56 The mode of action of platinum based agents is the formation of platinum-DNA adducts, which in turn activate several signal transduction pathways eventually leading to apoptosis. In several cell lines, platinum resistance has become a major factor, recapitulating a key limitation in terms of the clinical use of platinum-based drugs. In the clinic, resistance serves to compound the inherent limitations of the platinum drugs, including systemic (and often dose limiting) toxicity that reflects, at least in part, a lack of tumor-specific tissue distribution.
Figure 8.

FDA-approved platinum drugs and texaphyrin-Pt(II) conjugates 42 and 43.
We began exploring the hypothesis that conjugation of platinum to a tumor localizing texaphyrin would serve to overcome some platinum resistance pathways, such as reduced accumulation and fewer platinum-DNA lesions, and thus ultimately reactivate p53 mediated apoptosis via increased accumulation of intracellular platinum. Towards this end, we designed and synthesized a novel texaphyrin platinum conjugate (cisTEX 42, Figure 8). A pair of ovarian cancer models, consisting of a platinum sensitive A2780 cell line and its isogenic platinum resistant 2780CP cell line, were chosen to determine whether this conjugate was effective in overcoming resistance.40
Cell proliferation assays were used initially to assess cytotoxicity and probe anti-resistance benefits (Table 1). Conjugate 42 provided cytotoxicity profiles similar to that of carboplatin and other controls in the ovarian A2780 model. In addition, complex 42 provided higher cytotoxicity than compound 1. However, conjugate 42 provided greater cytotoxicity (i.e., lower IC50) than carboplatin against platinum resistant 2780CP cells. In terms of the associated resistance factor (reflecting the difference between resistant and sensitive cell lines), conjugate 42 provided the lowest value in its class and proved to be about 32-55% lower relative to cisplatin 39 and carboplatin 40. This finding was considered indicative of partial circumvention of cisplatin resistance. It was later determined that the decrease in resistance factor of conjugate 42 is due to increased intracellular platinum provided by conjugation to texaphyrin (cf. Figure 9).56
Table 1.
IC50 values of platinum complexes with cisplatin sensitive A2780 ovarian and its isogenic cisplatin resistant cell line (2780CP). Data are shown as mean ± SD.
| Complex | IC50 (μM) A2780 |
IC50 (μM) 2780CP |
Resistance Factor |
|---|---|---|---|
| cisTEX 42 | 1.4 ± 0.3 | 14.4 ± 1.7 | 10.3 ± 1.3 |
| carboplatin 40 | 1.6 ± 0.3 | 26.3 ± 4.1 | 16.4 ± 5.2a |
| cisplatin 39 | 0.31 ± 0.06 | 7.1 ± 0.9 | 22.9 ± 5.3a |
| oxaliTEX 43 | 0.55 ± 0.06 | 0.65 ± 0.09 | 1.2 ± 0.18 |
| oxaliplatin 41 | 0.15 ± 0.05 | 0.30 ± 0.05a | 2.0 ± 0.29 |
| complex 1 | 6.3 ± 0.6 | 13.7 ± 0.8 | 2.2 ± 0.38 |
p<0.05 by Student’s t-test vs. resistance factor for conjugate 42.
Figure 9.

Cellular uptake of platinum drugs. Levels of intracellular platinum in A2780 and 2780CP were determined by FAAS after a four-hour incubation with 200 μM of the respective complex (concentrations confirmed by FAAS). p<0.05 by Student’s t-test for platinum uptake of cisplatin and oxaliplatin in 2780CP vs. A2780.
In fact, a 12-fold increase in intracellular platinum from conjugate 42 was detected relative to carboplatin. Additionally, no reduction was seen in the uptake of platinum between the A2780 and 2780CP cell lines with conjugate 42 whereas a >50% reduction was observed in platinum based controls car-boplatin and cisplatin. This significant increase in intracellular platinum with conjugate 42 resulted in increased formation of platinum-DNA adducts in both the A2780 and 2780CP cell lines, presumably accounting for the reduced resistance as compared to control complexes. However, it was found that while intracellular platinum accumulation was increased and a relatively increased number of platinum-DNA lesions was seen, the type of platinum delivered and the resultant adduct was not capable of reactivating p53 activity in resistance cells. This was evidenced by DNA damage tolerance with levels of cisTEX being similar to that of cisplatin in both A2780 and 2780CP.56
To address this, we then focused on two major cisplatin resistance mechanisms, reduced drug uptake and attenuated wildtype p53 function. Specifically, we sought to target these mechanisms via a novel platinum drug design. With this goal in mind, we designed the second generation conjugate 46 (oxaliTEX).57 The focus on this design reflected a desire to target the tumor suppressor p53 and derived from an appreciation that cisplatin has a greater curative rate in ovarian cancer when p53 is present in its wild-type state than in the mutant form.50,51
Paradoxically, about a half of advanced ovarian cancers that harbor wild-type p53 are resistant, primarily as a result of failure of upstream DNA damage signaling to stabilize and activate p53. Furthermore, in these resistant cancers, the presence of wild-type p53 can lead to a “gain-of-resistance” phenotype, where the resistance is greater than those with mutant p53.50,51 Thus, loss of function of wild-type p53 is one of the most formidable molecular mechanism of resistance. However, we have reported that a panel of resistant ovarian tumor models respond to diaminocyclohexyl (DACH)-based platinum drugs through distinctly different DNA damage signaling processes that serve to restore p53 function and cellular apoptotic activity.58-60 Such a restoration of activity was considered likely to hold in the case of (DACH)-based oxaliplatin, and was specifically confirmed using the resistant 2780CP cell line as detailed below.
To test our hypothesis we synthesized and studied conjugate oxaliTEX 43 by cell proliferation assays with our ovarian cancer models (Figure 10 and 11, respectively). OxaliTEX 43 (IC50 = 0.55 ± 0.06 μM) provided a dose potency in the A2780 cell line that was nearly 3-fold greater than cisTEX (IC50 = 1.63 ± 0.2 μM). Against 2780CP cells, oxaliTEX 43 and oxaliplatin 41 (both containing DACH) maintained their potent activities, with IC50 values of 0.65 ± 0.09 and 0.30 ± 0.05 μM, respectively. In contrast, cisTEX and cisplatin provided values that reflect a 11- to 26-fold lower potency relative to oxaliTEX. It was demonstrated that 2780CP cells were two-fold cross-resistant to oxaliplatin, but were almost devoid of cross-resistance to conjugate 43 (cross-resistance factor, 1.2). This is consistent with essentially complete circumvention of resistance.
Figure 10.

Cytotoxicity profiles of oxaliTEX 43 with cisplatin sensitive A2780 and cisplatin resistant 2780CP. The complex was made up as a stock solution (for which the Pt concentration was confirmed by FAAS) and serially diluted before adding to cells, which were then incubated for five days at 37 °C in 5% CO2. Error bars represent standard deviation.
That apparent activation of wild-type p53 is sufficient to overcome multifactorial molecular mechanisms of resistance is intriguing. Normally, wild-type p53 plays a critical role in druginduced apoptosis. However, this activity becomes compromised when p53 is mutated, which leads to cisplatin/carboplatin resistance and, in the specific case of advanced ovarian cancer for which statistics are available, a four to five-fold reduction in the five year survival rate compared to the wild-type p53 cancer sub-group.50,51 Advanced cancers other than ovarian cancer (e.g., NSCLC and mesothelioma) that retain wild-type p53 also demonstrate resistance to cisplatin,51 an observation ascribed to a number of mechanisms, including the critical post-translational modifications of p53 to release p53 from its inhibitory interaction with Mdm2.61,62 Based on reports from molecularly engineered mouse models,63 it appears that activation of wild-type p53 and associated induction of apoptosis is a dominant result of DNA damage, and is sufficient to override the potential negative influence of other molecular defects that may co-exist in multifactorial resistant tumor cells.
The 2780CP tumor cells used as a model for platinum resistance in ovarian cancer have been characterized as having a multifactorial cisplatin-resistance phenotype.58 It was demonstrated that oxaliTEX restored platinum sensitivity as evidenced by induction of apoptosis (studied via flow cytometry), and upregulation of p53, phosphorylated p53, and p21 (studied via Western Blot analysis). It was also demonstrated from apoptotic investigations using Annexin V as a biomarker that the texaphyrin control, motexafin gadolinium 1, is devoid of anti-proliferative effects at concentrations that were equivalent to those employed in the studies of oxaliTEX 43.
Although circumventing molecular mechanisms of resistance can be ascribed to the design of the conjugate, the potency of oxaliTEX still relies heavily on achieving effective platinum concentrations within tumor cells. Our studies served to demonstrate that oxaliTEX (cf. Figure 11) was capable of delivering the DACH-Pt payload at similar levels in both sensitive and resistant tumor cells, a process similarly observed in cisTEX (cf. Figure 9). The similar delivery of platinum is likely due to the inherent features of the expanded porphyrin, texaphyrin, an essentially flat aromatic core that has been shown to localize selectively within tumors.64,65 That the effective delivery of platinum is due to the conjugating texaphyrin carrier and not the DACH-Pt moiety can be inferred from the knowledge that uptake and DNA adduct formation data for oxaliTEX (conjugate 43) mirror those reported by us for cisTEX (conjugate 42), which has an alternate diamine-platinum coordination environment.57
Figure 11.

Cellular uptake of platinum drugs. Levels of intracellular platinum in A2780 and 2780CP were determined by FAAS after a four hour incubation with 200 μM of the respective complex (concentrations confirmed by FAAS). p<0.05 by Student’s t-test for platinum uptake of cisplatin and oxaliplatin in 2780CP vs. A2780.
Conclusions
The results obtained to date provide support for our suggestion that texaphyrins could have a role to play in a variety of biomedical areas. These include but are not limited to use as anticancer treatments, isotope delivery vehicles, MRI contrast agents and site-localizing carriers. Their unique mode of action, involving electron capture from ascorbate and other reducing species, as well as the commensurate production of ROS, makes texaphyrins attractive scaffolds for further biological studies. Also attractive is the chemical versatility of the texaphyrins, which offer several sites for chemical modification and functionalization. It is hoped that this review, covering recent advances in the chemistry, synthesis and biological testing of new texaphyrin derivatives, will inspire additional efforts to develop more fully the biomedical potential of this class of expanded porphyrins.
Figure 1.
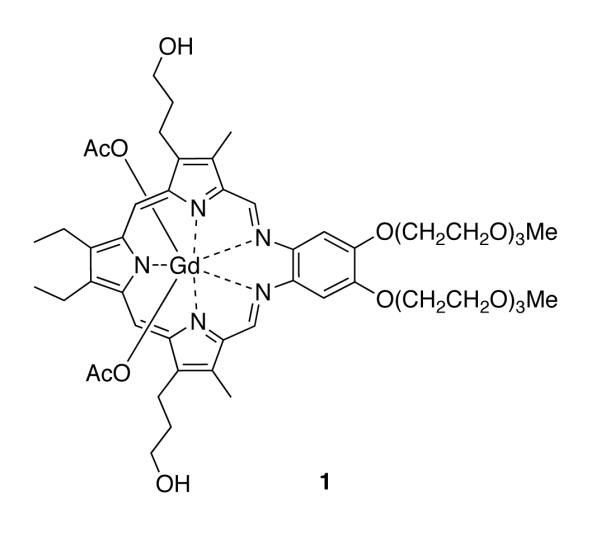
Structure of the texaphyrin species motexafin gadolinium.
Acknowledgments
Funding Sources
This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT; grant RP 120393 to J.L.S.), the U.S. National Cancer Institute (grant CA 68682 to J.L.S.; grants CA-127263 and CA-160687 to Z.H.S.), and the Robert A. Welch Foundation (Grant F-1018 to J.L.S.). Collaborative grant support from UT Austin TI-3D (Robert A. Welch Foundation Grant H-F-0032) and UT MD Anderson Cancer Center CCD (Grant 1003020-2100) is also acknowledged. JFA is supported by a postdoctoral fellowship (Grant PF-11-015-01-CDD) by the American Cancer Society. The work on texaphyrin functionalized nanoparticles was supported by grants from the Creative Research Initiative (2010-0018286 to J.C.) and the BK21 for Chemistry (to J.C.). This research was further supported by World Class University program funded by the Ministry of Education, Science and Technology through the National Research Foundation of Korea (grant no. R32-10217).
Glossary
ABBREVIATIONS
- Bcl2
B-cell lymphoma 2
- cf.
confer (compare)
- DACH
diaminocyclohexyl
- DMCA
dual-mode contrast agent
- DMSO
dimethylsulfoxide
- DNA
deoxyribonucleic acid
- at al.
et alii (and others)
- EBRT
external beam radiation therapy
- e.g.
exempli gratia (for example)
- FAAS
flameless atomic absorption spectrophotometry
- FDA
Food and Drug Administration
- GdTx-MNP
gadolinium(III) texaphyrin functionalized magnetic nanoparticles
- GSH
glutathione
- IC50
half maximal inhibitory concentration
- i.e.
id est (that is)
- Inc.
incorporated
- Mdm2
mouse double minute 2 homolog
- MRI
magnetic resonance imaging
- NADPH
nicotinamide adenine dinucleotide phosphate
- NSCL
non-small-cell lung cancer
- p53
tumor suppressor protein 53
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- T1
spin-lattice relaxtion time
- T2
spin-spin relaxation time
- TRX
thioredoxin
- TRXR
thioredoxin reductase
- XRT
X-ray therapy
Footnotes
Author Contributions
All others contributed either to the writing of this article or to the development of the original reports upon which it is based. The authors declare no competing financial interests. All authors have given approval to the final version of the manuscript.
REFERENCES
- (1).Otto SE. Pocket guide to oncology nursing. Mosby-Year Book, Inc.; St. Louis: 1995. [Google Scholar]
- (2).Gates RA, Fink RM. Oncology nursing secrets. Hanley and Belfus, Inc.; Philadelphia: 1997. [Google Scholar]
- (3).Mehta MP, Shapiro WR, Phan SC, Gervais R, Carrie C, Chabot P, Patchell RA, Glantz MJ, Recht L, Langer C, Sur RK, Roa WH, Mahe MA, Fortin A, Nieder C, Meyers CA, Smith JA, Miller RA, Renschler MF. Int. J. Radiat. Oncol. Biol. Phys. 2009;73:1069–1076. doi: 10.1016/j.ijrobp.2008.05.068. [DOI] [PubMed] [Google Scholar]
- (4).Sessler JL, Hemmi GW, Mody TD, Murai T, Burrell A. Acc. Chem. Res. 1994;27:43–50. [Google Scholar]
- (5).Mody TD, Sessler JL. In: Supramolecular Materials and Technologies. Reinhoudt DN, editor. Vol. 4. Chichester; Wiley: 1999. pp. 245–299. [Google Scholar]
- (6).Mody TD, Fu L, Sessler JL. In: Progress Inorganic Chemistry. Karlin KJ, editor. Vol. 49. Chichester; Wiley: 2001. p. 551. [Google Scholar]
- (7).Sessler JL, Mody TD, Hemmi GW, Lynch V. Inorg. Chem. 1993;32:3175–3187. [Google Scholar]
- (8).Sessler JL, Tvermoes NA, Guldi DM, Mody TD. Phys. Chem. 1999;103:787–794. [Google Scholar]
- (9).Patel H, Mick R, Finlay J, Zhu TC, Rickter E, Cengel KA, Malkowicz SB, Hahn SM, Busch TM. Clin. Cancer Res. 2008;14:4869–4876. doi: 10.1158/1078-0432.CCR-08-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sessler JL, Tvermoes NA, Guldi DM, Hug GL, Mody TD, Magda D. J. Phys. Chem. B. 2001;105:1452–1457. [Google Scholar]
- (11).Jasat A, Dolphin D. Chem. Rev. 1997;97:2267–2340. doi: 10.1021/cr950078b. [DOI] [PubMed] [Google Scholar]
- (12).Sessler JL, Murai T, Lynch V, Cyr M. J. Am. Chem. Soc. 1988;110:5586–5588. [Google Scholar]
- (13).Sessler JL, Murai T, Lynch V. Inorg. Chem. 1989;28:1333–1341. [Google Scholar]
- (14).Sessler JL, Johnson MR, Lynch V, Murai T. J. Coord. Chem. 1988;18:99–104. [Google Scholar]
- (15).Cotton FA, Wilkinson G. Advanced Inorganic Chemistry. 4th ed. John Wiley; New York: 1980. pp. 589–982. [Google Scholar]
- (16).Maiya BG, Mallouk TE, Hemmi GW, Sessler JL. Inorg. Chem. 1990;29:3738–3745. [Google Scholar]
- (17).Sessler JL, Mody TD, Ramasamy R, Sherry AD. New J. Chem. 1992;16:541–544. [Google Scholar]
- (18).Hannah S, Lynch V, Guldi DM, Gerasimchuk N, Mac-Donald CLB, Magda D, Sessler JL. J. Am. Chem. Soc. 2002;124:8416. doi: 10.1021/ja012747a. [DOI] [PubMed] [Google Scholar]
- (19).Guldi DM, Mody TD, Gerasimchuk NN, Magda D, Sessler JL. J. Am. Chem. Soc. 2000;122:8289–8298. [Google Scholar]
- (20).Acholla FV, Mertes KB. Tetrahedron Lett. 1984;25:3269–3270. [Google Scholar]
- (21).Acholla FV, Takusagawa F, Mertes KB. J. Am. Chem. Soc. 1985;107:6902–6908. [Google Scholar]
- (22).Sessler JL, Johnson MR, Lynch V. J. Org. Chem. 1987;52:4394–4397. [Google Scholar]
- (23).Sessler JL, Murai T, Lynch V, Cyr M. J. Am. Chem. Soc. 1988;110:5586–5588. [Google Scholar]
- (24).Hemmi GW. Dissertation (Ph.D.) The University of Texas at Austin: 1992. pp. 41–43. [Google Scholar]
- (25).Mody TD, Sessler JL. J. Porphy. Phthalocy. 2001;5:134–142. [Google Scholar]
- (26).Sessler JL, Dow WC, O’Connor D, Harriman A, Hemmi GW, Mody TD, Miller RA, Qing F, Springs S, Woodburn K. J. Alloys and Compounds. 1997;249:146–152. [Google Scholar]
- (27).Young SW, Sidhu MK, Qing F. Invest. Radiol. 1994;29:330–338. doi: 10.1097/00004424-199403000-00013. [DOI] [PubMed] [Google Scholar]
- (28).Rosenthal DI, Nurenberg P, Becerra CR, Frenkel EP, Carbonne DP, Lum BL, Miller R, Engel J, Young S, Miles D, Renschler MF. Clin. Cancer Res. 1999;5:739–745. [PubMed] [Google Scholar]
- (29).Viala J, Vanel D, Meingau P, Lartigau E, Carde P, Renschler MF. Radiology. 1999;3:755–759. doi: 10.1148/radiology.212.3.r99se10755. [DOI] [PubMed] [Google Scholar]
- (30).Miller RA, Woodburn K, Fan Q, Renschler MF, Sessler JL, Koutcher JA. Int. J. Radiation Oncology Biol. Phys. 1999;45:981–989. doi: 10.1016/s0360-3016(99)00274-6. [DOI] [PubMed] [Google Scholar]
- (31).Mehta MP, Shapiro WR, Glantz MJ, Patchell RA, Weitzner MA, Meyers CA, Schultz CJ, Roa WH, Leibenhout M, Ford J, Curran W, Phan S, Smith JA, Miller RA, Renschler MF. J. Clin. Oncol. 2002;20:3445–3453. doi: 10.1200/JCO.2002.07.500. [DOI] [PubMed] [Google Scholar]
- (32).Jungbauer B, editor. Pharmacyclics’ Xcytrin Gets FDA “Not Approvable” For NSCLC Patients With Brain Metastases. The Pink Sheet; Dec, 2007. [Google Scholar]
- (33).(a) Balieu S, Bouraiou AM, Carboni B, Boitrel B. Journal of Porphyrins and Phthalocyanines. 2008;12:11–18. [Google Scholar]; (b) Halime Z, Lachkar M, Roisnel T, Furet E, Halet J-F, Boitrel B. Angew. Chem. Int. Ed. 2007;46:5120–5124. doi: 10.1002/anie.200700543. [DOI] [PubMed] [Google Scholar]; (c) Halime Z, Lachkar M, Furet E, Halet J-F, Boitrel B. Inorg. Chem. 2006;45:10661–10669. doi: 10.1021/ic061422y. [DOI] [PubMed] [Google Scholar]; (d) emon CM, Brothers PJ, Boitrel B. Dalton Trans. 2011;40:6591–6609. doi: 10.1039/c0dt01711f. [DOI] [PubMed] [Google Scholar]; (e) Michaudet L, Richard P, Boitrel B. Chem.Comm. 2000;17:1589–1590. [Google Scholar]; (f) Boitrel B, Breede M, Brothers PJ, Hodgson M, Michaudet L, Rickard CEF, Al Salim N. Dalton Trans. 2003;9:1803–1807. [Google Scholar]
- (34).Kozak RW, Atcher RW, Gansow OA, Friedman AM, Hines JJ, Waldmann TA. Proc. Natl. Acad. Sci. USA. 1986;83:474–478. doi: 10.1073/pnas.83.2.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brechbiel MW, Pippin CG, McMurry TJ, Milenic D, Roselli M, Colcher D, Gansow OA. J. Chem. Soc. Chem. Commun. 1991:1169–1170. [Google Scholar]
- (36).Zalutsky MR, Pozzi OR. Quarterly Journal of Nuclear Medicine and Molecular Imaging. 2004;48:289–296. [PubMed] [Google Scholar]
- (37).Kumar K, Magerstaedt M, Gansow OA. J. Chem. Soc. Chem. Commun. 1989:145–146. [Google Scholar]
- (38).Sessler JL, Miller RA. Biochem. Pharmacol. 2000;59:733–739. doi: 10.1016/s0006-2952(99)00314-7. [DOI] [PubMed] [Google Scholar]
- (39).Preihs C, Arambula JF, Lynch VM, Siddik ZH, Sessler JL. Chem. Commun. 2010;46:7900–7902. doi: 10.1039/c0cc03528a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Arambula JF, Sessler JL, Fountain M, Wei W.-h., Magda D, Siddik ZH. Dalton Trans. 2009;48:10834–10840. doi: 10.1039/b912089k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lauffer RE. Chem. Rev. 1987;87:901–927. [Google Scholar]
- (42).Na HB, Song IC, Hyeon T. Adv. Mater. 2009;21:2133–2148. [Google Scholar]
- (43).Arbab AS, Liu W, Frank JA. Expert Rev. Med. Devices. 2006;3:427–439. doi: 10.1586/17434440.3.4.427. [DOI] [PubMed] [Google Scholar]
- (44).Jun Y.-w., Lee J-H, Cheon J. Angew. Chem., Int. Ed. 2008;47:5122–5135. doi: 10.1002/anie.200701674. [DOI] [PubMed] [Google Scholar]
- (45).Caravan P. Chem. Soc. Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- (46).Bulte DL, Kraitchman WM. NMR Biomed. 2004;17:484–499. doi: 10.1002/nbm.924. [DOI] [PubMed] [Google Scholar]
- (47).Choi J.-s., Lee J-H, Shin T-H, Song H-T, Kim EY, Cheon J. J. Am. Chem. Soc. 2010;132:11015–11017. doi: 10.1021/ja104503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yoo D, Jeong H, Preihs C, Choi J-S, Shin T-H, Sessler JL, Cheon J. Angew. Chem., Int. Ed. 2012;51:12482–12485. doi: 10.1002/anie.201206400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Siddik ZH. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- (50).Siddik ZH. In: Drug Resistance in Cancer Cells. Mehta K, Siddik ZH, editors. Springer Science; 2009. [Google Scholar]
- (51).Martinez-Rivera M, Siddik ZH. Biochem. Pharm. 2012;83:1049–1062. doi: 10.1016/j.bcp.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bosl GJ, Bajorin DF, Sheinfeld J. In: Cancer of the Testis. DeVita VTJ, Hellman S, Rosenberg SA, editors. Lippincott Williams & Wilkins; Philadelphia: 2001. [Google Scholar]
- (53).Jamieson ER, Lippard SJ. Chem. Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- (54).Kelland LR, Sharp SY, O’Neill CF, Raynaud FI, Beale PJ, Judson IR. J. Inorg. Biochem. 1999;77:111–115. [PubMed] [Google Scholar]
- (55).Fuertes MA, Alonso C, rezPé J-M. Chem. Rev. 2003;103:645–662. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- (56).Arambula JF, Sessler JL, Siddik ZH. Bioorg. Med. Chem. Lett. 2011;21:1701–1705. doi: 10.1016/j.bmcl.2011.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Arambula JF, Sessler JL, Siddik ZH. Med. Chem. Commun. 2012;3:1275–1281. doi: 10.1039/C2MD20206A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Siddik ZH, Hagopian GS, Thai G, Tomisaki S, Toyomasu T, Khokhar AR. J. Inorg. Biochem. 1999;77:65–70. doi: 10.1016/s0162-0134(99)00144-0. [DOI] [PubMed] [Google Scholar]
- (59).Hagopian GS, Mills GB, Khokhar AR, Bast RC, Jr., Siddik ZH. Clin. Cancer. Res. 1999;5:655–663. [PubMed] [Google Scholar]
- (60).He G, Kuang J, Khokhar AR, Siddik ZH. Gynecol. Oncol. 2011;122:402–409. doi: 10.1016/j.ygyno.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sionov RV, Haupt Y. Oncogene. 1999;18:6145–6157. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- (62).Shieh SY, Ikeda M, Taya Y, Prives C. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- (63).Kastan MB. Cell. 2007;128:837–840. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]
- (64).Arambula JF, Preihs C, Borthwick D, Magda D, Sessler JL. Anti-Cancer Agents in Med. Chem. 2011;11:222–232. doi: 10.2174/187152011795255894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Magda D, et al. In: Medicinal Inorganic Chemistry. Sessler JL, Doctrow S, McMurry T, Lippard SJ, editors. Oxford University Press; American Chemical Society Symposium Series 903: 2005. [Google Scholar]