

Abstract
Recent epidemiologic studies implying differences in cancer recurrence based on anesthetic regimens raise the possibility that the mu opioid receptor (MOR) can influence cancer progression. Based on our previous observations that overexpression of MOR in human non-small cell lung cancer (NSCLC) cells increased tumor growth and metastasis, this study examined whether MOR regulates growth factor receptor signaling and epithelial mesenchymal transition (EMT) in human NSCLC cells. We utilized specific siRNA, shRNA, chemical inhibitors and overexpression vectors in human H358 NSCLC cells that were either untreated or treated with various concentrations of DAMGO, morphine, fentanyl, EGF or IGF. Cell function assays, immunoblot and immunoprecipitation assays were then performed. Our results indicate MOR regulates opioid and growth factor-induced EGF receptor signaling (Src, Gab-1, PI3K, Akt and STAT3 activation) which is crucial for consequent human NSCLC cell proliferation and migration. In addition, human NSCLC cells treated with opioids, growth factors or MOR overexpression exhibited an increase in snail, slug and vimentin and decrease ZO-1 and claudin-1 protein levels, results consistent with an EMT phenotype. Further, these effects were reversed with silencing (shRNA) or chemical inhibition of MOR, Src, Gab-1, PI3K, Akt and STAT3 (p<0.05). Our data suggest a possible direct effect of MOR on opioid and growth factor-signaling and consequent proliferation, migration and EMT transition during lung cancer progression. Such an effect provides a plausible explanation for the epidemiologic findings.
Introduction
The role of anesthesia and analgesia in the recurrence and metastatic rate of malignancies has recently received considerable attention [1], [2], [3], [4]. Retrospective studies have demonstrated a diminished incidence of cancer recurrence following regional anesthesia with lower doses of opioids following surgery for breast, prostate, colon cancer and melanoma, although other studies have failed to detect significant differences [5], [6], [7], [8]. Some hypotheses to explain these differences in recurrence rates include immune suppressive effects and direct effects on tumor cell growth [9], [10], [11]. Our research has focused on the mu opioid receptor (MOR) and its role in directly regulating cellular changes leading to tumor growth and metastasis [4], [12], [13].
Effective therapeutic strategies for lung cancer, the leading cause of cancer-associated mortality worldwide, are extremely limited exemplifying the need for early diagnosis and novel therapeutic interventions [14], [15]. We have previously reported that the MOR is upregulated in several types of human non-small cell lung cancer (NSCLC) [12]. Further, we have shown that overexpression of MOR in human NSCLC increases primary tumor growth and metastasis in xenograft models [13]. However, the exact cellular changes regulated by MOR in NSCLC are incompletely defined [4]. For cancer cells to grow and metastasize, there needs to be a loss of cell-cell adhesion (characterized by a reduction of epithelial cell adhesion proteins including the tight junction proteins, ZO-1 and claudin-1) followed by acquisition of mesenchymal characteristics including a loss of baso-apical polarization, cytoskeletal remodeling and increased cell motility (characterized by increases in specific cytoskeletal proteins (i.e. vimentin) and transcription factors (i.e. Slug and Snail) [16], [17], [18], [19]. This orchestrated oncogenic process is referred to as epithelial mesenchymal transition (EMT) [16], [17], [18], [19], [20], [21], [22].
Growth factor receptors, including the epidermal growth factor receptor (EGFR), are often overexpressed and/or mutated in NSCLC and regulate oncogenic processes including tumor cell proliferation, migration and EMT transition [23], [24], [25], [26], [27]. Several therapies targeting the EGFR in NSCLC exist including tyrosine kinase inhibitors (gefitinib, erlotinib) and monoclonal antibodies (cetuximab)[28], [29], [30], [31]. However, the overall survival rate for NSCLC remains low [32], [33], [34]. Recently, Fujioka et al., have demonstrated that morphine can stimulate EGFR signaling pathways including the serine/threonine kinases Akt and MAP kinase in NSCLC suggesting a role for MOR inhibition as a potential therapeutic strategy for NSCLC [35].
Based on the recent interest of the effects of anesthesia and analgesia regimens on the recurrence and metastatic potential of various cancers [1], [2], [3], [4], our previous published data indicating the MOR is upregulated in lung tissue from patients with NSCLC [12], overexpression of MOR promotes tumor growth and metastasis in human NSCLC xenograft models [13] as well as data from Fujioka et al., demonstrating MOR regulation of EGF-induced signaling events in NSCLC [35], this study investigated the functional effects of MOR in the fundamental oncogenic processes of opioid and growth factor-induced human lung cell migration, proliferation and epithelial mesenchymal transition (EMT)[16], [17], [18], [19], [20]. Since there is currently very little information on opioid and/or MOR regulation of EMT and the molecular mechanisms integrating cancer cell proliferation, migration and EMT, this study investigated the detailed molecular mechanisms for these events which can have potential clinical utility.
Methods
Cell Culture and Reagents
The human NSCLC cell H358 was obtained from ATCC (Walkersville, MD) and cultured in Roswell Park Memorial Institute complete medium (Cambrex, East Rutherford, NJ) at 37°C in a humidified atmosphere of 5% CO2, 95% air, with passages 6–10 used for experimentation. Unless otherwise specified, reagents were obtained from Sigma (St. Louis, MO). Reagents for SDS-PAGE electrophoresis were purchased from Bio-Rad (Richmond, CA) and Immobilon-P transfer membrane was purchased from Millipore (Millipore Corp., Bedford, MA). Rabbit anti-MOR antibody was purchased from GeneTex (San Antonio, TX). Rabbit anti-EGFR, rabbit anti-phosphotyrosine-EGFR (pY845, pY992, pY1045, pY1068), rabbit anti-Grb-2, rabbit anti-Gab-1, rabbit anti-phosphotyrosine-Gab-1 (pY307, pY627), rabbit anti-Src, rabbit anti-phosphotyrosine-Src (pY416), rabbit anti-p85 PI3 kinase, rabbit anti-p55 PI3 kinase, rabbit anti-phosphotyrosine-p85/p55 PI3 kinase (pY458, pY199), rabbit anti-STAT3, rabbit anti-phosphotyrosine-STAT3 (pY705), rabbit anti-vimentin, rabbit anti-ZO-1, rabbit anti-claudin-1, rabbit anti-Snail and rabbit anti-Slug antibodies were purchased from Cell Signaling Technologies (Danvers, MA). Mouse anti-β-actin antibody was purchased from Sigma (St. Louis, MO). Secondary horseradish peroxidase-labeled antibodies were purchased from Amersham Biosciences (Piscataway, NJ). N-methylnaltrexone bromide or methylnaltrexone was purchased from Mallinckrodt Specialty Chemicals (Phillipsburg, NJ). The PI3 kinase inhibitor LY294002, Akt Inhibitor X, the Src family kinase inhibitor PP2 and the STAT3 inhibitor Stattic were purchased from EMD Biosciences (Billerica, MA).
Immunoblotting
Immunoblotting was performed as we have previously described. Cellular materials from treated or untreated human NSCLC cells were incubated with lysis buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 20 mM MgCl2, 1% Triton X-100, 0.1% SDS, 0.4 mM Na3VO4, 40 mM NaF, 50 µM okadaic acid, 0.2 mM phenylmethylsulfonyl fluoride, 1∶250 dilution of Calbiochem protease inhibitor mixture 3). The samples were then run on SDS-PAGE in 4–15% polyacrylamide gels, transferred onto Immobilon™ membranes, and developed with specific primary and secondary antibodies. Visualization of immunoreactive bands was achieved using enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ). In some instances, immunoreactive bands were quantitated using computer-assisted densitometry.
Small Interfering RNA Transfection in Human NSCLC Cells
Stable Control and either MOR, Gab-1, or Src siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) were transfected into H358 cells as we have previously described [12]. Cells (∼40% confluent) were serum-starved for 1 hour followed by incubated with siRNA for 6 hours in serum-free media. Serum-containing media was then added (10% serum final concentration) for 42 hours. Inhibition of protein expression was confirmed by immunoblot analysis with specific antibodies.
Stable Control and MOR Small Hairpin RNA Transfection in Human NSCLC Cells
Stable Control and MOR shRNA (Santa Cruz Biotechnology, Santa Cruz, CA) were stably transfected into H358 cells as we have previously described [12]. Cells (∼40% confluent) were serum-starved for 1 hour followed by incubated with shRNA for 6 hours in serum-free media. Serum-containing media was then added (10% serum final concentration) for 42 hours and puromycin selection reagent was added. Inhibition of protein expression was confirmed by immunoblot analysis with anti-MOR antibody (GeneTex, San Antonio, TX).
Stable Vector Control and MOR1 Overexpression in Human NSCLC Cells
Myc-DDK-tagged ORF clone of Homo sapiens opioid receptor, mu 1 (OPRM1), transcript variant MOR-1 (OriGene Technologies Inc, MD) was amplified using Platinum Taq DNA polymerase high fidelity enzyme (Invitrogen, CA) and subsequently cloned into a pCR8/GW/Topo entry vector (Invitrogen, CA) according to manufacturer's instructions. Plasmid DNA was extracted from selected clones by QIAquick Plasmid Mini kit (Qiagen, CA). ORF integrity and fragment orientation were confirmed by sequencing. The MOR1-Myc fusion product was then transferred to pcDNA3.2/v5 DEST vector (Invitrogen, CA) by LR reaction. The resulting construct (pcDNA3.2-MOR1-Myc) was transfected into H358 cells using FuGENE HD™ as the transfection reagent (Roche Applied Sciences) according to the protocol provided by Roche as we have previously described. Cells (∼40% confluent) were serum-starved for 1 hour followed by incubation with pcDNA3.2-MOR1-Myc for 6 hours in serum-free media. Serum-containing media was then added (10% serum final concentration) for 42 hours and neomycin selection reagent was added. Overexpression was confirmed by immunoblot analysis with anti-MOR antibody (GeneTex, San Antonio, TX).
Human NSCLC Cell Proliferation Assay
Measurement of in vitro NSCLC cell growth was performed as we have previously described. Control or siRNA pretreated H358 cells (5×103 cells/well) were incubated with 0.2 ml of serum-free media containing either vehicle (control), methylnaltrexone (MNTX, 100 nM), the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM) for 72 h at 37°C in 5%CO2/95% air in 96-well culture plates. The in vitro cell proliferation assay was analyzed by measuring increases in cell number using the CellTiter96™ MTS assay (Promega, Madison, WI) and read at 492 nm. Each assay was set up in triplicate and repeated at least five times.
Human NSCLC Cell Migration Assay
Measurement of in vitro NSCLC cell migration was performed as we have previously described. Twenty-four transwell units with 8 µM pore size (Millipore, Billerica, MA) were used for monitoring in vitro cell migration as we have previously described [12]. Control or siRNA pretreated H358 cells (5×103 cells/well) were incubated with 0.2 ml of serum-free media containing either vehicle (control), methylnaltrexone (MNTX, 100 nM), the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM) were plated on the upper chamber and media with serum was added to the lower chamber. Cells were allowed to migrate through the pores for 18 hours. Cells from the upper and lower chamber were quantitated using the CellTiter96™ MTS assay (Promega, San Luis Obispo, CA) and read at 492 nm. % migration was defined as the # of cells in the lower chamber divided by the number of cells in both the upper and lower chamber. Each assay was set up in triplicate and repeated at least five times.
Statistical Analysis
Results are expressed as mean ± standard deviation of three independent experiments. For data analysis, experimental samples were compared to controls by unpaired Student's t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA) and post hoc multiple comparisons tests were used. Differences between groups were considered statistically significant when P value was less than 0.05. All statistical analyses were performed using the GraphPad Prism program (GraphPad Software Inc., USA).
Results
Our results in Figure 1 indicate that inhibiting MOR with the peripheral MOR antagonist MNTX attenuates EGF-induced proliferation (Figure 1-A) and migration (Figure 1-B) of human H358 NSCLC cells in a dose-dependent manner. These data suggest a link between MOR and the EGFR in H358 cells. To mechanistically evaluate the role of MOR on EGF-induced EGFR dynamics, we treated H358 cells with EGF at various times and immunoprecipitated the EGFR to determine potential MOR association. Figure 2-A demonstrates that EGF induces a complex formation between the EGFR and MOR which peaks at 5 to 15 minutes after EFG challenge. Based on our results that a MOR/EGFR complex can occur with EGF stimulation of H358 cells, we next examined whether MOR can regulate EGFR phosphorylation. Utilizing a panel of anti-phospho-EGFR antibodies, Figure 2-B demonstrates that pretreatment of H358 human NSCLC cells with the peripheral MOR antagonist MNTX failed to attenuate EGF-induced EGFR tyrosine phosphorylation.
Figure 1. The peripheral mu opioid receptor antagonist, methylnaltrexone (MNTX), inhibits epidermal growth factor (EGF)-induced proliferation and migration of human lung cancer cells in a dose-dependent manner.
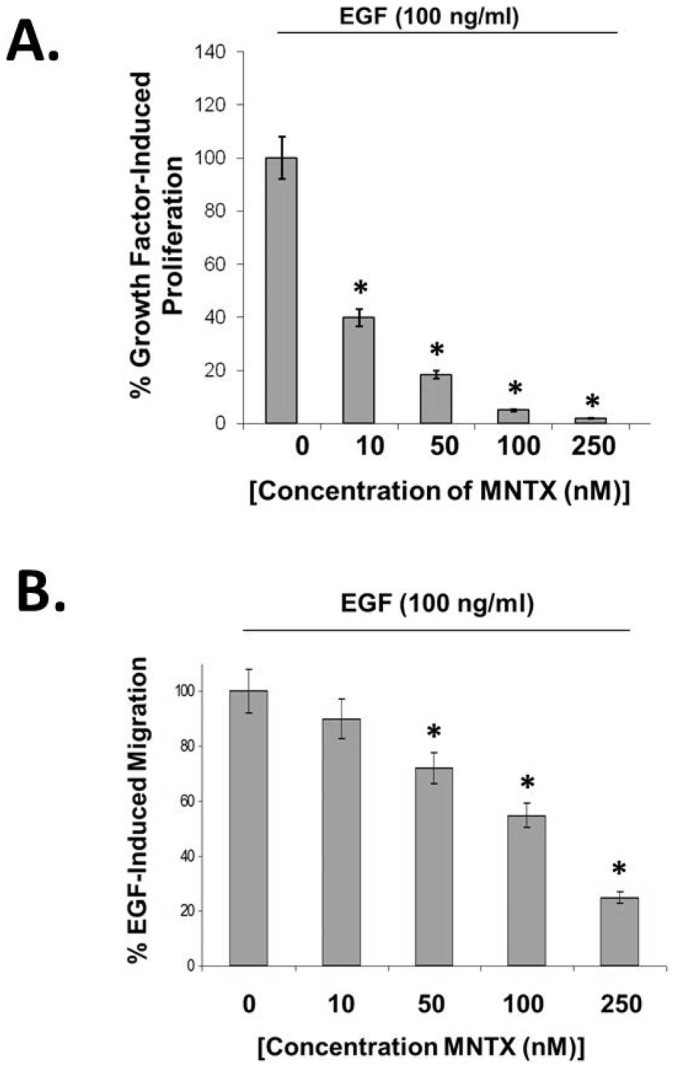
Panel A: Human H358 non-small cell lung cancer (NSCLC) cells were analyzed for methylnaltrexone (MNTX) inhibition of EGF-mediated proliferation using a MTS proliferation assay. Cells were growth in the presence of 100 ng/ml EGF and/or 0–250 nM MNTX for 72 hours. MNTX There is a statistically significant difference (p<0.05, indicated by an asterisks) between control and MNTX (10, 50, 100, 250 nM) treatment with n = 3 per condition and error bars = standard deviation. See the Methods section for experimental details. Panel B: Human H358 non-small cell lung cancer (NSCLC) cells were analyzed for methylnaltrexone (MNTX) inhibition of EGF-mediated migration using a transwell assay (8 uM pore size). Cells were allowed to migrate in the presence of 100 ng/ml EGF and/or 0–250 nM MNTX for 18 hours. There is a statistically significant difference (p<0.05, indicated by an asterisks) between control and MNTX (50, 100, 250 nM) treatment with n = 3 per condition and error bars = standard deviation. See the Methods section for experimental details.
Figure 2. The mu opioid receptor (MOR) is recruited to the EGF receptor with EGF stimulation but does not regulate EGF receptor phosphorylation.

Panel A: Human H358 non-small cell lung cancer (NSCLC) cells were treated with no (control) or 100 ng/ml EGF for 5, 15, or 30 minutes. Cell lysates were obtained and immunoprecipitated with anti-EGFR antibody. Immunoblots were performed on total cell lysates (left) and immunoprecipitated material (right) using anti-MOR (a) and anti-EGFR (b) antibodies. The mu opioid receptor is recruited to the EGFR with EGF stimulation. Panel B: Human H358 non-small cell lung cancer (NSCLC) cells were either untreated (control) or treated with 100 nM MNTX alone, 10 ng/ml EGF for 5, 15 or 30 minutes, or 100 nM MNTX and 10 ng/ml EGF for 5, 15, or 30 minutes. Cell lysates were obtained and immunoblotted using anti-pY845 EGFR (a), anti-pY992 EGFR (b), anti-pY1045 EGFR (c), anti-pY1068 EGFR (d), anti-EGFR (e) and anti-actin (e) antibodies. MNTX does not inhibit EGF-induced EGFR tyrosine phosphorylation.
We next examined whether MOR can regulate EGFR downstream signaling molecules. We first examined the adaptor protein, Growth factor receptor-bound protein 2 (Grb-2), which contains an SH2 domain and two SH3 domains and can directly bind the EGFR [36]. The results of Figure 3-A demonstrate, using immunoprecipitation of EGFR and immunoblotting with anti-Grb-2 antibody, that EGF induces EGFR/Grb-2 complex formation which is attenuated by pretreatment of H358 cells with MNTX. Recruitment of Grb-2 to the EGFR is important for plasma membrane recruitment and consequent tyrosine phosphorylation of the scaffolding protein, Grb2-associated-binding protein 1 (Gab-1) [37]. Figure 3-B indicates that EGF stimulation of H358 cells induces tyrosine phosphorylation of Gab-1 (Tyr307 and Tyr627) which peaks at ∼5 minutes and is attenuated by MOR inhibition with MNTX.
Figure 3. The peripheral mu opioid receptor antagonist, methylnaltrexone (MNTX), inhibits EGF-induced recruitment/activation of the linker protein, Grb-2, and the scaffolding protein, Gab-1, in human lung cancer cells.

Panel A: Human H358 non-small cell lung cancer (NSCLC) cells were either untreated (control) or treated with 100 nM MNTX (1 hour pre-incubation), 10 ng/ml EGF for 15 minutes, or 100 nM MNTX and 10 ng/ml EGF. Cell lysates were obtained and immunoprecipitated with anti-EGFR antibody. Immunoblots were performed on total cell lysates and immunoprecipitated material using anti-Grb-2 (a,c) and anti-EGFR (b,d) antibody. MNTX inhibits EGF-induced recruitment of Grb-2 to the EGFR. Panel B: Human H358 non-small cell lung cancer (NSCLC) cells were either untreated (control) or treated with 100 nM MNTX alone, 10 ng/ml EGF for 5, 15 or 30 minutes, or 100 nM MNTX and 10 ng/ml EGF for 5, 15, or 30 minutes. Cell lysates were obtained and immunoblotted using anti-pY627 Gab-1 (a), anti-pY307 Gab-1 (b) and anti-actin (c) antibodies. MNTX attenuates EGF-induced Gab-1 tyrosine phosphorylation.
Since tyrosine phosphorylation of Gab-1 by various tyrosine kinases including Src promotes binding to signaling molecules including Phosphatidylinositol 3-kinases (PI3Ks) which generate PIP3 and activate downstream effectors including Akt and STAT3 [37], [38], [39], [40], [41], [42], we next examined whether MOR inhibition can also influence Gab-1 binding/effector molecules. Figure 4-A shows that, like Gab-1, EGF challenge of H358 cells induces tyrosine phosphorylation of Src (Tyr416), the regulatory PI3K alpha subunits p85 and p55 (Tyr458/Tyr199), and the transcription factor STAT3 (Tyr705) which peak at ∼5 minutes and are attenuated by MOR inhibition with MNTX in a statistically significant manner (Figure 4-B).
Figure 4. The peripheral mu opioid receptor antagonist, methylnaltrexone (MNTX), inhibits EGF-induced phosphorylation of Src, PI3 kinase and STAT3 in human lung cancer cells.

Panel A: Human H358 non-small cell lung cancer (NSCLC) cells were either untreated (control) or treated with 100 nM MNTX alone, 10 ng/ml EGF for 5, 15 or 30 minutes, or 100 nM MNTX and 10 ng/ml EGF for 5, 15, or 30 minutes. Cell lysates were obtained and immunoblotted using anti-phospho-Src (pY416) (a), anti-phospho-p85/p55 PI3 kinase (pY458/pY199) (b,c), anti-phospho-STAT3 (pY705) (d) and anti-actin (e) antibodies. Panel B: Graphical quantitation of immunoreactivity of experiments performed described in Figure 3-B and Panel A with normalization to total specific protein and n = 3 independent experiments per condition. An asterisk (*) indicates a statistically significant difference (p<0.05) from control. The error bars = standard deviation.
We next examined the mechanism by which EGF and DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin), a synthetic opioid peptide with specificity for MOR) induce proliferation and migration. Figure 5 illustrates that siRNA and/or chemical inhibition of MOR, Gab-1, Src, PI3K, Akt and STAT3 markedly decrease EGF and DAMGO-induced proliferation (Figure 5-A) and migration (Figure 5-B).
Figure 5. Inhibiting the mu opioid receptor (MOR), Gab-1, Src, PI3 kinase, Akt or STAT3 attenuates EGF and DAMGO-induced proliferation and migration in human lung cancer cells.

Panel A: Human H358 non-small cell lung cancer (NSCLC) cells were analyzed for EGF and DAMGO-mediated proliferation using a MTS proliferation assay. H358 human NSCLC cells were either untreated (control) or treated with 100 ng/ml epidermal growth factor (EGF) or 100 nM DAMGO for 72 hours with or without pretreatment of cells with the peripheral MOR antagonist, methylnaltrexone (MNTX, 100 nM), MOR siRNA, Gab-1 siRNA, Src siRNA, the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM). There is a statistically significant difference (p<0.05, indicated by an asterisks) between control and treatment groups with n = 3 independent experiments per condition and error bars = standard deviation. See the Methods section for experimental details. Panel B: Human H358 non-small cell lung cancer (NSCLC) cells were analyzed for EGF and DAMGO-mediated migration using a transwell assay (8 uM pore size). H358 human NSCLC cells were either untreated (control) or treated with 100 ng/ml epidermal growth factor (EGF) or 100 nM DAMGO for 18 hours with or without pretreatment of cells with the peripheral MOR antagonist, methylnaltrexone (MNTX, 100 nM), MOR siRNA, Gab-1 siRNA, Src siRNA, the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM). There is a statistically significant difference (p<0.05, indicated by an asterisks) between control and treatment groups with n = 3 independent experiments per condition and error bars = standard deviation. See the Methods section for experimental details.
Since very little is known about MOR regulation of epithelial mesenchymal transformation (EMT) and the molecular mechanisms integrating cancer cell proliferation, migration and EMT, we next examined whether MOR1 overexpression regulates lung cancer EMT. In Figure 6-A,B, we observed that MOR overexpression in human H358 NSCLC cells induced a change in EMT marker expression which is consistent with an epithelial mesenchymal transition [19]. Since the MOR is the main receptor for certain opioids [4], [43], we next evaluated whether opioids can induce EMT in human H358 NSCLC cells. Figure 6-C indicates that DAMGO, morphine and fentanyl all induce EMT in NSCLC in a dose-dependent manner.
Figure 6. Overexpression or silencing of the mu opioid receptor (MOR) or addition of opioids in human lung cancer cells regulates epithelial mesenchymal transition (EMT).

Panel A: Control (non-transfected)(C), stable vector control (VC) and MOR1 overexpressing (O/E) H358 cell lines were generated, cell lysates obtained and immunoblotted with EMT markers anti-vimentin (a), anti-Snail (b), anti-Slug (c), anti-claudin-1 (d), anti-ZO-1 (e), anti-MOR (f) and anti-actin (g) antibodies. An increase in vimentin, Snail and Slug expression and a decrease in claudin-1 and ZO-1 expression suggest an epithelial mesenchymal transition. Panel B: Graphical quantitation of immunoreactivity of experiments performed described in Panel A with n = 3 independent experiments per condition. An asterisk (*) indicates a statistically significant difference (p<0.05) from control with error bars = standard deviation. Panel C: H358 human NSCLC cells were either untreated, treated with 100 ng/ml epidermal growth factor (EGF), 100 nM DAMGO, morphine or fentanyl or 100 ng/ml insulin growth factor (IGF) for 96 hours, cell lysates obtained and immunoblotted with EMT markers anti-vimentin (a), anti-Snail (b), anti-Slug (c), anti-claudin-1 (d), anti-ZO-1 (e), anti-MOR (f) and anti-actin (g) antibodies. A decrease in vimentin, Snail and Slug expression and an increase in claudin-1 and ZO-1 expression suggest inhibition of epithelial mesenchymal transition. Panel D: Control shRNA or MOR shRNA H358 cell lines were generated, cell lysates obtained and immunoblotted with EMT markers anti-vimentin (a), anti-Snail (b), anti-Slug (c), anti-claudin-1 (d), anti-ZO-1 (e), anti-MOR (f) and anti-actin (g) antibodies. An increase in vimentin, Snail and Slug expression and a decrease in claudin-1 and ZO-1 expression suggest an epithelial mesenchymal transition.
Considering these H358 human NSCLC cells express basal levels of MOR and respond to opioid treatment, we examined whether silencing (shRNA) of MOR would affect opioid and growth factor-induced EMT. Figure 6-D indicates MOR silencing inhibits opioid and EGF/IGF-induced changes in EMT marker expression. Figure 7-A indicates that H358 cells grow in colonies with well demarcated borders and strong cell-cell adhesions. In contrast, morphine or IGF-treated H358 cells show a loss of cell-cell adhesions and a change from cuboidal to an elongated phenotype with several cellular projections visible. These changes are consistent with an epithelial mesenchymal transition (EMT).
Figure 7. Morphologic and epithelial mesenchymal transition (EMT) marker changes in human lung cancer cells with opioids, growth factors and signal transduction pathway inhibitors.

Panel A: H358 human NSCLC cells were either untreated (control), treated with 100 nm morphine or 100 ng/ml insulin growth factor (IGF) for 96 hours and brightfield images were obtained (20×). H358 cells grow in colonies with well demarcated borders and strong cell-cell adhesions. In contrast, morphine or IGF-treated H358 cells show a loss of cell-cell adhesions and a change from cuboidal to an elongated phenotype with several cellular projections visible. These changes are consistent with an epithelial mesenchymal transition (EMT). Panel B: H358 human NSCLC cells were either untreated, treated with 100 ng/ml epidermal growth factor (EGF), 100 nM DAMGO, morphine or fentanyl or 100 ng/ml insulin growth factor (IGF) for 96 hours with or without pretreatment with the peripheral MOR antagonist, methylnaltrexone (MNTX, 100 nM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM). Cell lysates were then obtained and immunoblotted with EMT markers anti-vimentin (a,c,e) or anti-claudin-1 (b,d,f) antibodies. An increase in vimentin expression and a decrease in claudin-1 expression suggest an epithelial mesenchymal transition.
We next examined potential signal transduction proteins that could potentially regulate opioid and growth factor-induced EMT. Figure 7-B indicates pretreatment with the peripheral MOR antagonist, methylnaltrexone (MNTX), the Src family kinase inhibitor PP2 or the STAT3 inhibitor Stattic reverse the opioid and growth factor-induced changes in vimentin and claudin-1 expression. In addition, Figure 8 indicates siRNA and/or chemical inhibition of MOR, Gab-1, Src, PI3K, Akt and STAT3 dramatically inhibits EMT (as determined by inhibition of opioid and growth factor-induced increase in vimentin expression and decrease in claudin-1 expression). Both MNTX and the MOR inhibitor naloxone attenuated opioid and growth factor-induced EMT indication a general effect of MOR antagonists on this process (Figures 7 and 8 and supporting data, Figure S1). Taken together, our data suggest MOR plays a central role in the processes of proliferation, migration and epithelial mesenchymal transition alone and in conjunction with opioids and growth factors.
Figure 8. Inhibiting the mu opioid receptor (MOR), Src, Gab-1, PI3 kinase or STAT3 attenuates opioid and growth factor-induced epithelial mesenchymal transition (EMT) in human lung cancer cells.

Panel A: Graphical representation of the % control vimentin expression. H358 human NSCLC cells were either untreated, treated with 100 ng/ml epidermal growth factor (EGF), 100 nM DAMGO, morphine or fentanyl or 100 ng/ml insulin growth factor (IGF) for 96 hours with or without pretreatment of cells with the peripheral MOR antagonist, methylnaltrexone (MNTX, 100 nM), MOR siRNA, Gab-1 siRNA, Src siRNA, the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM). Cell lysates were then obtained and immunoblotted with the EMT marker anti-vimentin antibody. Experiments were repeated in triplicate and immunoreactive bands were analyzed using computer-assisted densitometry. There is a statistically significant difference (p<0.05 indicated by asterisks (*)) between control and treatment groups with error bars = standard deviation. An increase in vimentin expression is suggestive of an epithelial mesenchymal transition. Panel B: Graphical representation of the % control claudin-1 expression. H358 human NSCLC cells were either untreated, treated with 100 ng/ml epidermal growth factor (EGF), 100 nM DAMGO, morphine or fentanyl or 100 ng/ml insulin growth factor (IGF) for 96 hours with or without pretreatment of cells with the peripheral MOR antagonist, methylnaltrexone (MNTX, 100 nM), MOR siRNA, Gab-1 siRNA, Src siRNA, the PI3 kinase inhibitor LY294002 (10 uM), Akt Inhibitor X (5 uM), the Src family kinase inhibitor PP2 (100 nM) or the STAT3 inhibitor Stattic (10 uM). Cell lysates were then obtained and immunoblotted with the EMT marker anti-claudin-1 antibody. Three independent experiments per condition were performed and immunoreactive bands were analyzed using computer-assisted densitometry. There is a statistically significant difference (p<0.05 indicated by asterisks (*)) between control and treatment groups with error bars = standard deviation. A decrease in claudin-1 expression is suggestive of an epithelial mesenchymal transition.
Discussion
NSCLC, which accounts for ∼80% of all lung cancers, is a disease with high mortality and few treatment options [44], [45]. We have previously reported that the MOR is upregulated in lung tissue from patients with NSCLC [12] and that overexpression of MOR promotes tumor growth and metastasis in human NSCLC xenograft models [13]. Further, data from Fujioka et al., indicate that the MOR regulates EGF-induced signaling events in NSCLC [35]. Considering there is very limited data on opioid regulation of epithelial mesenchymal transition (EMT, a crucial process for cancer progression) and the molecular mechanisms integrating cancer cell proliferation, migration and EMT, this study examined the role of opioids and MOR in these processes [17], [18], [19], [20]. Here we present evidence that activation of growth factor receptors promotes complex formation with the mu opioid receptor (MOR) and Grb-2 as well as Src activation. These events induce recruitment of the scaffolding protein, Gab1, to the plasma membrane and consequent recruitment/activation of PI3 kinase, Akt and STAT3 signaling which are required for human lung cancer proliferation, migration and EMT. Inhibition of MOR (siRNA, shRNA and/or MOR antagonists) blocks morphine and DAMGO binding to the mu opioid receptor. In addition, inhibiting MOR attenuates growth factor-induced phosphorylation/activation of Src, Gab-1, PI3 kinase, Akt and STAT3 with consequent inhibition of human lung cancer proliferation, migration and EMT (see Figure 9).
Figure 9. Schematic diagram illustrating mu opioid receptor (MOR) regulation of growth factor receptor signaling and human lung cancer proliferation, migration and epithelial mesenchymal transition (EMT).

Activation of growth factor receptors promotes complex formation with the mu opioid receptor (MOR) and Grb-2 as well as Src activation (1). These events induce recruitment of the scaffolding protein, Gab1, to the plasma membrane (2) and consequent recruitment/activation of PI3 kinase, Akt and STAT3 signaling (3) which are required for human lung cancer proliferation, migration and epithelial mesenchymal transition (EMT) (4). Inhibition of MOR (siRNA, shRNA and/or the peripheral MOR antagonist, methylnaltrexone (MNTX)) blocks morphine and DAMGO binding to the mu opioid receptor (5). In addition, inhibiting MOR attenuates growth factor-induced phosphorylation/activation of Src, Gab-1, PI3 kinase, Akt and STAT3 with consequent inhibition of human lung cancer proliferation, migration and EMT (6).
Our results are consistent with Fujioka et al., who demonstrated that morphine and EGF challenge of human lung cancer cells (H2009, NSCLC) induced Akt and MAPK/ERK activation as well as cell proliferation and invasion [35]. These effects were blocked with the MOR antagonist, naloxone [35]. Further, these authors and others have reported that agonists of MOR can transactivate the EGRF [35], [46], [47], [48]. While the current study did not look at opioid stimulation of EGFR phosphorylation, our results indicate that blocking MOR with the peripheral MOR antagonist MNTX did not affect EGF-induced EGFR tyrosine phosphorylation. Our results further demonstrate that MNTX can block EMT transformation by another growth factor implicated in lung cancer progression, insulin growth factor (IGF) [49], [50], [51]. Whether MOR can regulate IGF receptor phosphorylation/signaling in human NSCLC is currently being investigated in our laboratory.
The current study presents the novel findings that the scaffolding protein, Grb2-associated-binding protein 1 (Gab-1), regulates opioid and growth factor-mediated human lung cancer cell proliferation, migration and EMT transformation. The adaptor protein, Growth factor receptor-bound protein 2 (Grb-2), can directly bind tyrosine phosphorylated sites on the EGRF via its SH2 domain which promotes plasma membrane recruitment of Gab-1 and binding of proline-rich motifs on Gab-1 to SH3 domains on Grb-2 [37]. Further, the proline-rich motifs on Gab-1 can bind to the non-receptor tyrosine kinase Src [37], [52]. We have previously demonstrated that MNTX is a potent inhibitor of Src activation [43], [53]. Our results that MNTX can also inhibit Grb-2/Gab-1 recruitment/activation could explain how MNTX regulates Src activity.
Tyrosine phosphorylation of Gab-1 by Src and other tyrosine kinases promotes recruitment of several signaling molecules including PI3K, PLCgamma, RasGAP and SHP2 [37], [52], [54]. Our current results indicate that MOR antagonism by MNTX blocks activation (tyrosine phosphorylation) of the PI3K regulatory subunits, p85 and p55. Further, inhibition of PI3K activity attenuates opioid- and growth factor-induced human lung cancer cell proliferation, migration and EMT. Whether PLCgamma, RasGAP and/or SHP2 also play a role in MOR regulation of NSCLC progression is currently being investigated in our laboratory.
The transcription factor, Signal transducer and activator of transcription 3 (STAT3), is an important regulator of NSCLC progression [41], [55], [56], [57]. Tyrosine phosphorylation of STAT3 induces dimerization and nuclear translocation [57]. Recently, it has been shown that PI3K can also regulate STAT3 activity [38], [39]. In this study, we have demonstrated that STAT3 regulates opioid and growth factor-induced proliferation, migration and EMT. Further, MNTX inhibits STAT3 tyrosine phosphorylation implying a role for MOR in STAT3 activation. This is in partial agreement with Debruyer et al., 2010, indicating that delta opioid receptor (DOR) agonists can differentially affect migration of HCT-8/E11 human colon cancer cells [58].
Another downstream effector of PI3K is the serine/threonine kinase Akt [42], [59]. Akt contains an SH2 domain that can bind to PIP2 and PIP3 as well as serine, threonine and tyrosine phosphorylation sites [60], [61]. We have previously reported that MNTX inhibits opioid and VEGF-induced Akt activation in human endothelial cells and that overexpression of MOR in human NSCLC activates Akt [4], [13], [62], [63]. In this study, using Akt inhibitor X (a selective inhibitor of Akt phosphorylation and activity), we demonstrate that Akt regulates opioid and EGF-induced human NSCLC cell proliferation, migration and EMT transformation.
EMT is a complex process in which there is a loss of cell-cell adhesion (characterized by a reduction of epithelial cell adhesion proteins including the tight junction proteins, ZO-1 and claudin-1) followed by acquisition of mesenchymal characteristics including a loss of baso-apical polarization, cytoskeletal remodeling and increased cell motility (characterized by increases in specific cytoskeletal proteins (i.e. vimentin) and transcription factors (i.e. Slug and Snail)[16], [17], [18], [19], [20], [22]. We have previously reported that overexpression of MOR1 (the most abundant MOR transcript that consists of exons 1, 2, 3 and 4)[64] in human H358 NSCLC cells induced elongated cellular projections suggestive of a migratory “mesenchymal” phenotype [19]. This prompted us to examine MOR regulation of EMT in human NSCLC cells. While we used a panel of EMT markers, several others exist including Twist, N-cadherin, E-cadherin, ZEB1/2, beta-catenin, fibronectin and desmoplakin [16], [17], [18], [19], [20], [22]. In addition to cancer, EMT has been implicated in several fibrotic diseases by providing a source for myofibroblasts [22], [65]. Whether MOR is involved in the regulation of fibrotic diseases via EMT is currently unknown.
Exogenous opioids are utilized clinically for their analgesic effects, cough and diarrhea suppression and pain relief [66], [67]. Opioid relief of pain is indicated in various conditions such as acute pain after surgery, injury or trauma as well as chronic pain from advanced cancer. Recently, Garcia-Recio et al., 2013, demonstrated that the pain-associated tachykinin, Substance P, and its GPCRs can transactivate HER2/EGFR signaling in human breast cancer cell lines [68]. This study provides a novel mechanism by which pain and inflammation can promote tumor progression. Substance P receptors can heterodimerize with mu opioid receptors and these receptors can transactivate each other [69], [70], [71], [72], [73], [74], [75]. Therefore, it is possible that pain and inflammation could, through Substance P transactivation of MOR, promote EMT in cancer cells. This potential link could be ameliorated with MOR antagonists. Further research is warranted.
Given our previous published data indicating the mu opioid receptor (MOR) is increased in human lung cancer [12], that lung cancer cells do not form visible tumors in MOR knockout mice [12] and that MOR overexpression promotes NSCLC primary tumor growth and metastasis [4], [43], we hypothesized that MOR regulation of EMT might be a plausible explanation for the differences in recurrence rates observed in the epidemiologic studies. Taken together, our data suggests the MOR plays a crucial role in the fundamental cellular EMT changes that occur during lung cancer progression, and provides a plausible explanation for the epidemiologic findings.
Supporting Information
The MOR antagonist, naltrexone, inhibits epithelial mesenchymal transition (EMT) in human lung cancer cells. Panel A: H358 human NSCLC cells were either untreated, treated with 100 nM morphine, DAMGO, fentanyl or 100 ng/ml EGF for 96 hours with or without pretreatment of cells with the MOR antagonist, naltrexone (100 nM). Cell lysates were obtained and immunoblotted with EMT markers anti-vimentin (1), anti-Slug (2) and anti-actin (g) antibodies. A decrease in vimentin and Slug expression suggest an inhibition of epithelial mesenchymal transition.
(TIF)
Funding Statement
Support was provided from institutional and/or departmental sources and National Institutes of Health grant CTSA UL1 TR000430. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bovill JG (2010) Surgery for cancer: does anesthesia matter? Anesth Analg 110: 1524–1526. [DOI] [PubMed] [Google Scholar]
- 2. Snyder GL, Greenberg S (2010) Effect of anaesthetic technique and other perioperative factors on cancer recurrence. British journal of anaesthesia 105: 106–115. [DOI] [PubMed] [Google Scholar]
- 3. Yeager MP, Rosenkranz KM (2010) Cancer recurrence after surgery: a role for regional anesthesia? Regional anesthesia and pain medicine 35: 483–484. [DOI] [PubMed] [Google Scholar]
- 4. Lennon FE, Moss J, Singleton PA (2012) The mu-opioid receptor in cancer progression: is there a direct effect? Anesthesiology 116: 940–945. [DOI] [PubMed] [Google Scholar]
- 5. Biki B, Mascha E, Moriarty DC, Fitzpatrick JM, Sessler DI, et al. (2008) Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: a retrospective analysis. Anesthesiology 109: 180–187. [DOI] [PubMed] [Google Scholar]
- 6. Exadaktylos AK, Buggy DJ, Moriarty DC, Mascha E, Sessler DI (2006) Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology 105: 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Christopherson R, James KE, Tableman M, Marshall P, Johnson FE (2008) Long-term survival after colon cancer surgery: a variation associated with choice of anesthesia. Anesth Analg 107: 325–332. [DOI] [PubMed] [Google Scholar]
- 8. Myles PS, Peyton P, Silbert B, Hunt J, Rigg JR, et al. (2011) Perioperative epidural analgesia for major abdominal surgery for cancer and recurrence-free survival: randomised trial. BMJ 342: d1491. [DOI] [PubMed] [Google Scholar]
- 9. Tavare AN, Perry NJ, Benzonana LL, Takata M, Ma D (2011) Cancer recurrence after surgery: Direct and indirect effects of anesthetic agents*. International journal of cancer Journal international du cancer [DOI] [PubMed] [Google Scholar]
- 10. Afsharimani B, Cabot PJ, Parat MO (2011) Morphine use in cancer surgery. Frontiers in pharmacology 2: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Afsharimani B, Cabot P, Parat MO (2011) Morphine and tumor growth and metastasis. Cancer metastasis reviews 30: 225–238. [DOI] [PubMed] [Google Scholar]
- 12. Mathew B, Lennon FE, Siegler J, Mirzapoiazova T, Mambetsariev N, et al. (2011) The novel role of the mu opioid receptor in lung cancer progression: a laboratory investigation. Anesthesia and analgesia 112: 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lennon FE, Mirzapoiazova T, Mambetsariev B, Salgia R, Moss J, et al. (2012) Overexpression of the mu-opioid receptor in human non-small cell lung cancer promotes Akt and mTOR activation, tumor growth, and metastasis. Anesthesiology 116: 857–867. [DOI] [PubMed] [Google Scholar]
- 14. Spiro SG, Tanner NT, Silvestri GA, Janes SM, Lim E, et al. (2010) Lung cancer: progress in diagnosis, staging and therapy. Respirology 15: 44–50. [DOI] [PubMed] [Google Scholar]
- 15. Stinchcombe TE, Bogart J, Veeramachaneni NK, Kratzke R, Govindan R (2011) Annual review of advances in non-small cell lung cancer research: a report for the year 2010. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 6: 1443–1450. [DOI] [PubMed] [Google Scholar]
- 16. Denlinger CE, Ikonomidis JS, Reed CE, Spinale FG (2010) Epithelial to mesenchymal transition: the doorway to metastasis in human lung cancers. The Journal of thoracic and cardiovascular surgery 140: 505–513. [DOI] [PubMed] [Google Scholar]
- 17. Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, et al. (2010) Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer science 101: 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. The Journal of clinical investigation 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Voulgari A, Pintzas A (2009) Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochimica et biophysica acta 1796: 75–90. [DOI] [PubMed] [Google Scholar]
- 20. Cannito S, Novo E, Valfre di Bonzo L, Busletta C, Colombatto S, et al. (2009) Epithelial-Mesenchymal Transition: from Molecular Mechanisms, Redox Regulation to Implications in Human Health and Disease. Antioxid Redox Signal [DOI] [PubMed] [Google Scholar]
- 21. Chow G, Tauler J, Mulshine JL (2010) Cytokines and growth factors stimulate hyaluronan production: role of hyaluronan in epithelial to mesenchymal-like transition in non-small cell lung cancer. J Biomed Biotechnol 485468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139: 871–890. [DOI] [PubMed] [Google Scholar]
- 23. Charpidou A, Blatza D, Anagnostou V, Syrigos KN (2008) Review. EGFR mutations in non-small cell lung cancer–clinical implications. In vivo 22: 529–536. [PubMed] [Google Scholar]
- 24. Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, et al. (2011) EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer 71: 241–243. [DOI] [PubMed] [Google Scholar]
- 25. Holz C, Niehr F, Boyko M, Hristozova T, Distel L, et al. (2011) Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 101: 158–164. [DOI] [PubMed] [Google Scholar]
- 26. Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, et al. (2010) Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer research 30: 2513–2517. [PubMed] [Google Scholar]
- 27. Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron-Smith D, et al. (2009) Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. British journal of cancer 100: 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carlson JJ (2009) Erlotinib in non-small-cell lung cancer: a review of the clinical and economic evidence. Expert review of pharmacoeconomics & outcomes research 9: 409–416. [DOI] [PubMed] [Google Scholar]
- 29. Choi EJ, Ryu YK, Kim SY, Wu HG, Kim JS, et al. (2010) Targeting epidermal growth factor receptor-associated signaling pathways in non-small cell lung cancer cells: implication in radiation response. Molecular cancer research : MCR 8: 1027–1036. [DOI] [PubMed] [Google Scholar]
- 30. Ganjoo KN, Wakelee H (2007) Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biologics : targets & therapy 1: 335–346. [PMC free article] [PubMed] [Google Scholar]
- 31. Gridelli C, Morabito A, Gebbia V, Mencoboni M, Carrozza F, et al. (2010) Cetuximab and gemcitabine in elderly or adult PS2 patients with advanced non-small-cell lung cancer: The cetuximab in advanced lung cancer (CALC1-E and CALC1-PS2) randomized phase II trials. Lung Cancer 67: 86–92. [DOI] [PubMed] [Google Scholar]
- 32. De Greve J, Decoster L, Van Meerbeek J, Vermeij J, Teugels E, et al. (2008) [Targeted therapies in the treatment of non-small cell lung cancer]. Bull Cancer 95: 358–364. [DOI] [PubMed] [Google Scholar]
- 33. Ng R, Loreto M, Lee R, Leighl NB (2008) Brief report: retrospective review of efficacy of erlotinib or gefitinib compared to docetaxel as subsequent line therapy in advanced non-small cell lung cancer (NSCLC) following failure of platinum-based chemotherapy. Lung Cancer 61: 262–265. [DOI] [PubMed] [Google Scholar]
- 34. Subramanian J, Madadi AR, Dandona M, Williams K, Morgensztern D, et al. (2010) Review of ongoing clinical trials in non-small cell lung cancer: a status report for 2009 from the ClinicalTrials.gov website. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 5: 1116–1119. [DOI] [PubMed] [Google Scholar]
- 35. Fujioka N, Nguyen J, Chen C, Li Y, Pasrija T, et al. (2011) Morphine-induced epidermal growth factor pathway activation in non-small cell lung cancer. Anesthesia and analgesia 113: 1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giubellino A, Burke TR Jr, Bottaro DP (2008) Grb2 signaling in cell motility and cancer. Expert opinion on therapeutic targets 12: 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, et al. (2006) Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. The Journal of biological chemistry 281: 19925–19938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vogt PK, Hart JR (2011) PI3K and STAT3: a new alliance. Cancer discovery 1: 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hart JR, Liao L, Yates JR 3rd, Vogt PK (2011) Essential role of Stat3 in PI3K-induced oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America 108: 13247–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan PC, Chen YL, Cheng CH, Yu KC, Cary LA, et al. (2003) Src phosphorylates Grb2-associated binder 1 upon hepatocyte growth factor stimulation. The Journal of biological chemistry 278: 44075–44082. [DOI] [PubMed] [Google Scholar]
- 41. Akca H, Tani M, Hishida T, Matsumoto S, Yokota J (2006) Activation of the AKT and STAT3 pathways and prolonged survival by a mutant EGFR in human lung cancer cells. Lung Cancer 54: 25–33. [DOI] [PubMed] [Google Scholar]
- 42. Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim Biophys Acta 1697: 3–16. [DOI] [PubMed] [Google Scholar]
- 43. Singleton PA, Moss J (2010) Effect of perioperative opioids on cancer recurrence: a hypothesis. Future oncology 6: 1237–1242. [DOI] [PubMed] [Google Scholar]
- 44. Maione P, Rossi A, Sacco PC, Bareschino MA, Schettino C, et al. (2010) Advances in chemotherapy in advanced non-small-cell lung cancer. Expert opinion on pharmacotherapy 11: 2997–3007. [DOI] [PubMed] [Google Scholar]
- 45. Beland MD, Wasser EJ, Mayo-Smith WW, Dupuy DE (2010) Primary non-small cell lung cancer: review of frequency, location, and time of recurrence after radiofrequency ablation. Radiology 254: 301–307. [DOI] [PubMed] [Google Scholar]
- 46. Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ (2001) mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. The Journal of biological chemistry 276: 33847–33853. [DOI] [PubMed] [Google Scholar]
- 47. Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ (2003) Mu opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: implications for mitogen-activated protein kinase signaling. Molecular pharmacology 64: 1391–1401. [DOI] [PubMed] [Google Scholar]
- 48. Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, et al. (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884–888. [DOI] [PubMed] [Google Scholar]
- 49. Gridelli C, Rossi A, Bareschino MA, Schettino C, Sacco PC, et al. (2010) The potential role of insulin-like growth factor receptor inhibitors in the treatment of advanced non-small cell lung cancer. Expert opinion on investigational drugs 19: 631–639. [DOI] [PubMed] [Google Scholar]
- 50. Gualberto A, Karp DD (2009) Development of the monoclonal antibody figitumumab, targeting the insulin-like growth factor-1 receptor, for the treatment of patients with non-small-cell lung cancer. Clinical lung cancer 10: 273–280. [DOI] [PubMed] [Google Scholar]
- 51. Yin M, Guan X, Liao Z, Wei Q (2009) Insulin-like growth factor-1 receptor-targeted therapy for non-small cell lung cancer: a mini review. American journal of translational research 1: 101–114. [PMC free article] [PubMed] [Google Scholar]
- 52. Nishida K, Hirano T (2003) The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer science 94: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singleton PA, Mambetsariev N, Lennon FE, Mathew B, Siegler JH, et al. (2010) Methylnaltrexone Potentiates the Anti-Angiogenic Effects of mTOR Inhibitors. J Angiogenes Res 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, et al. (2000) Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. The Journal of cell biology 149: 1419–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Devarajan E, Huang S (2009) STAT3 as a central regulator of tumor metastases. Curr Mol Med 9: 626–633. [DOI] [PubMed] [Google Scholar]
- 56. Yin ZJ, Jin FG, Liu TG, Fu EQ, Xie YH, et al. (2010) Overexpression of STAT3 Potentiates Growth, Survival, and Radioresistance of Non-Small-Cell Lung Cancer (NSCLC) cells. The Journal of surgical research [DOI] [PubMed] [Google Scholar]
- 57. Lim CP, Cao X (2006) Structure, function, and regulation of STAT proteins. Molecular bioSystems 2: 536–550. [DOI] [PubMed] [Google Scholar]
- 58. Debruyne D, Leroy A, O DEW, Vakaet L, Mareel M, et al. (2010) Direct effects of delta opioid receptor agonists on invasion-associated activities of HCT-8/E11 colon cancer cells. Anticancer research 30: 9–17. [PubMed] [Google Scholar]
- 59. Lee MW, Kim DS, Lee JH, Lee BS, Lee SH, et al. (2011) The roles of AKT1 and AKT2 in non-small cell lung cancer cell survival, growth and migration. Cancer science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kandel ES, Hay N (1999) The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res 253: 210–229. [DOI] [PubMed] [Google Scholar]
- 61. Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, et al. (2002) Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene 21: 6587–6597. [DOI] [PubMed] [Google Scholar]
- 62. Singleton PA, Mambetsariev N, Lennon FE, Mathew B, Siegler JH, et al. (2010) Methylnaltrexone potentiates the anti-angiogenic effects of mTOR inhibitors. Journal of angiogenesis research 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Singleton PA, Garcia JG, Moss J (2008) Synergistic effects of methylnaltrexone with 5-fluorouracil and bevacizumab on inhibition of vascular endothelial growth factor-induced angiogenesis. Molecular cancer therapeutics 7: 1669–1679. [DOI] [PubMed] [Google Scholar]
- 64. Xu J, Xu M, Hurd YL, Pasternak GW, Pan YX (2009) Isolation and characterization of new exon 11-associated N-terminal splice variants of the human mu opioid receptor gene. Journal of neurochemistry 108: 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee K, Nelson CM (2012) New insights into the regulation of epithelial-mesenchymal transition and tissue fibrosis. International review of cell and molecular biology 294: 171–221. [DOI] [PubMed] [Google Scholar]
- 66. Frank JW, Bair MJ, Becker WC, Krebs EE, Liebschutz JM, et al. (2014) Update in Pain Medicine for Primary Care Providers: A Narrative Review, 2010–2012. Pain medicine [DOI] [PubMed] [Google Scholar]
- 67. Mercadante S (2014) Managing difficult pain conditions in the cancer patient. Current pain and headache reports 18: 395. [DOI] [PubMed] [Google Scholar]
- 68. Garcia-Recio S, Fuster G, Fernandez-Nogueira P, Pastor-Arroyo EM, Park SY, et al. (2013) Substance P autocrine signaling contributes to persistent HER2 activation that drives malignant progression and drug resistance in breast cancer. Cancer research 73: 6424–6434. [DOI] [PubMed] [Google Scholar]
- 69. Aicher SA, Punnoose A, Goldberg A (2000) mu-Opioid receptors often colocalize with the substance P receptor (NK1) in the trigeminal dorsal horn. The Journal of neuroscience : the official journal of the Society for Neuroscience 20: 4345–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pfeiffer M, Kirscht S, Stumm R, Koch T, Wu D, et al. (2003) Heterodimerization of substance P and mu-opioid receptors regulates receptor trafficking and resensitization. The Journal of biological chemistry 278: 51630–51637. [DOI] [PubMed] [Google Scholar]
- 71. Illing S, Mann A, Schulz S (2013) Heterologous Regulation of Agonist-Independent mu-Opioid Receptor Phosphorylation by Protein Kinase C. British journal of pharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu YJ, Arttamangkul S, Evans CJ, Williams JT, von Zastrow M (2009) Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of mu-opioid receptors in CNS neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 29: 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Perez S, Tierney A, Deniau JM, Kemel ML (2007) Tachykinin regulation of cholinergic transmission in the limbic/prefrontal territory of the rat dorsal striatum: implication of new neurokinine 1-sensitive receptor binding site and interaction with enkephalin/mu opioid receptor transmission. Journal of neurochemistry 103: 2153–2163. [DOI] [PubMed] [Google Scholar]
- 74. Bigliardi-Qi M, Gaveriaux-Ruff C, Pfaltz K, Bady P, Baumann T, et al. (2007) Deletion of mu- and kappa-opioid receptors in mice changes epidermal hypertrophy, density of peripheral nerve endings, and itch behavior. The Journal of investigative dermatology 127: 1479–1488. [DOI] [PubMed] [Google Scholar]
- 75. Wan Q, Douglas SD, Wang X, Kolson DL, O'Donnell LA, et al. (2006) Morphine upregulates functional expression of neurokinin-1 receptor in neurons. Journal of neuroscience research 84: 1588–1596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The MOR antagonist, naltrexone, inhibits epithelial mesenchymal transition (EMT) in human lung cancer cells. Panel A: H358 human NSCLC cells were either untreated, treated with 100 nM morphine, DAMGO, fentanyl or 100 ng/ml EGF for 96 hours with or without pretreatment of cells with the MOR antagonist, naltrexone (100 nM). Cell lysates were obtained and immunoblotted with EMT markers anti-vimentin (1), anti-Slug (2) and anti-actin (g) antibodies. A decrease in vimentin and Slug expression suggest an inhibition of epithelial mesenchymal transition.
(TIF)