

Abstract
Dendritic cells (DCs) control the balance between effector and regulatory T cells in vivo. Hence, the study of DCs might identify mechanisms of disease pathogenesis and guide new therapeutic approaches for immune-mediated disorders. We found that IL-27 signaling in murine DCs limits the generation of effector TH1 and TH17 cells and the development of experimental autoimmune encephalomyelitis (EAE). The effects of IL-27 were mediated, at least partially, through the induction of the immunoregulatory molecule ENTPD1 (CD39) in DCs. IL-27-induced ENTPD1 decreased extracellular ATP levels, down-regulating nucleotide-dependent NLRP3 inflammasome activation. Finally, therapeutic vaccination with IL-27-conditioned DCs suppressed established relapsing-remitting EAE. Thus, IL-27 signaling in DCs limits pathogenic T cell responses and the development of autoimmunity.
The dysregulated activity of effector TH1 and TH17 cells results in the development of tissue inflammation and autoimmunity. Myelin-specific TH1 and TH17 cells, for example, contribute to disease pathogenesis in multiple sclerosis (MS) 1 and its animal model experimental autoimmune encephalomyelitis (EAE) 2. During EAE, dendritic cells (DCs) control the activation and differentiation of myelin-specific effector and regulatory T (Treg) cells 3, 4. Moreover, DCs isolated from MS patients produce high amounts of TH1 and TH17 polarizing cytokines 5, suggesting that DCs promote the generation of the pathogenic T cell response in MS. Indeed, DCs control several pathogenic mechanisms associated with the development of central nervous system (CNS) autoimmunity. DCs promote T cell entry into the central nervous system (CNS) 6, the activation and differentiation of pathogenic T cells within the CNS 3, and the spreading of the autoimmune response to new CNS epitopes 7. Thus, it is important to study the pathways that regulate DC activity during the course of autoimmunity, to identify mechanisms of disease pathogenesis and also to develop new approaches for therapeutic intervention.
IL-27 is a cytokine structurally related to IL-12, composed of IL-27-p28 and the Epstein-Barr Virus-induced gene 3 (Ebi3) 8. IL-27 signals through a receptor composed of the common IL-6 receptor chain gp130, used by several other IL-6 and IL-12 family members, and a unique IL-27 receptor α chain (IL-27RA) homologous to the IL-12Rβ2 chain of the IL-12 receptor 8. IL-27 is produced by DCs in response to Toll-like receptor (TLR) activation through a mechanism that involves the autocrine effects of interferon-β (IFN-β) 9. Indeed, the therapeutic effects of IFN-β administration on relapsing-remitting MS have been linked to the induction of IL-27 production by DCs 10.
Based on its structural homology to IL-12 and its ability to trigger IFN-γ production, IL-27 was initially thought to be a pro-inflammatory cytokine 11. However, it was later discovered that IL-27 suppresses TH1, TH2 and TH17 responses and limits CNS inflammation in several experimental models 8. In the EAE model, IL-27 administration inhibits disease development 12. Conversely, the lack of a functional IL-27 receptor results in exaggerated TH17 responses and the worsening of EAE 12, 13. IL-27 has been shown to act directly on T cells to inhibit the development of pathogenic TH17 cells and promote the differentiation of IL-10 producing type 1 regulatory T cells (Tr1 cells) 12–17. Thus, the arrest of EAE by IL-27 is thought to reflect its direct effects on T cells. However, several cell types besides T cells express the IL-27 receptor, but the relevance of IL-27 signaling in these cells for the control of EAE and autoimmunity is unknown.
DCs express a functional IL-27 receptor 18, but although IL-27 has been reported to interfere with the activation of DCs in vitro 18–20, the physiological relevance of IL-27 signaling in DCs and its effects on the control of the T cell response and autoimmunity are unknown. In this work, we studied the effects of IL-27 signaling in DCs on the differentiation of effector and Treg cells and CNS autoimmunity. We found that IL-27 signaling in DCs up-regulates ENTPD1 (CD39) expression and limits the development of EAE. ENTPD1 expressed by conventional DCs (cDCs) reduces the extracellular levels of ATP, decreasing ATP-triggered activation of the NLRP3 inflammasome. Moreover, we found that vaccination with IL-27-conditioned DCs suppresses established chronic relapsing-remitting EAE. Taken together, these data demonstrate that IL-27 signaling in DCs controls pathogenic T cell responses in an ENTPD1-dependent manner, and identify IL-27 conditioned DCs as a possible therapeutic approach for immune-mediated disorders.
Results
cDCs express higher IL-27RA than plasmacytoid DCs
To investigate the role of IL-27 signaling in DCs on the regulation of autoimmunity, we first analyzed the expression of IL-27RA in plasmacytoid DCs (pDCs, F4/80−CD11b−CD11cloB220+MHC-IIloLy6c+) and cDCs (cDCs, F4/80−CD11b+CD11c+ B220− MHC-II+Ly6c−) isolated from naive mice by flow cytometry (Supplementary Fig. 1). IL-27RA was predominantly expressed in cDCs with only low or absent expression on pDCs (Fig. 1a). Similar results were obtained when IL-27RA expression was analyzed by quantitative PCR (qPCR) and immunoblot in sorted pDCs and cDCs (Figs. 1b,c). Thus, the expression pattern of IL-27RA suggests that IL-27 controls the activity of cDCs.
Figure 1. Il27ra expression in DCs.
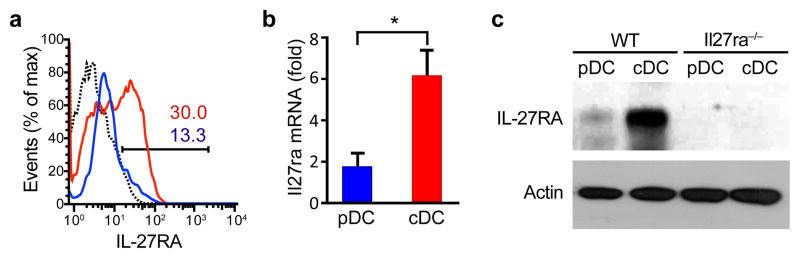
(a–c) IL-27RA expression was analyzed by flow cytometry (a), q-PCR (b) and immunoblot (c) in sorted cDCs and pDCs. The numbers within the histogram show the percentage of positive cells. *P < 0.05 (Student’s t-test). Data are representative of more than three independent experiments with similar results.
IL-27 modulates the function of cDCs
Following antigen uptake in the presence of DC-maturating stimuli, DCs up-regulate the expression of the major histocompatibility complex class II (MHC class II) and co-stimulatory molecules 21. To investigate the effects of IL-27 on DC activation, we pre-treated splenic cDCs from naive mice with vehicle or IL-27 and studied their response to activation with Escherichia coli lipopolysaccharide (ecLPS). LPS pre-treatment with IL-27 led to a significant decrease in the expression of MHC-II, CD40, CD80 and CD86 (Fig. 2a and Suppl. Fig. 2).
Figure 2. IL-27 modulates the APC function of cDCs.

(a) Flow cytometry of IL-27- (20 ng/ml) treated cDC in the presence or absence of ecLPS (100 ng/ml). Mean fluorescence intensity (MFI) +/− SD is shown. (b) Cytokine secretion by IL-27-treated cDCs, determined in culture supernatants by ELISA. (c) q-PCR analysis of Il27 expression in IL-27-treated cDCs. (d–f) Naive 2D2+ CD4+ T cells were stimulated with MOG35–55 and cDCs pre-treated with IL-27 and ecLPS, and proliferation (d), cytokine secretion to culture supernatants (e) and frequency of CD4+ IFN-γ+, IL-17+, IL-10+ and FoxP3+ (f) were analyzed. (g–h) Naive 2D2+ CD4+ T cells were stimulated with MOG35–55 and cDCs pre-treated with IL-27 and ecLPS in the presence of exogenous cytokines to promote the differentiation of TH1, TH2, Tr1 or FoxP3+ T cells, and cytokine secretion (g) and the frequency of CD4+ FoxP3+ T cells (h) were analyzed. Data are representative (h, left panel) or are the mean ± SEM (a–h) of three independent experiments. *P < 0.05; **P < 0.01 (One-way ANOVA).
DCs control T cell differentiation via the secretion of polarizing cytokines 21. Pre-treatment of splenic cDCs with IL-27 led to a significant reduction in the production of IL-12, or IL-6 and IL-23, which promote the differentiation of TH1 and TH17 cells, respectively (Fig. 2b). Pre-treatment of cDCs with IL-27 also up-regulated Il27 expression, suggesting a positive feedback loop for IL-27 production (Fig. 2c). Indeed, we detected an increased production of IFN-β, which is reported to act in an autocrine manner to trigger IL-27 production (Fig. 2b) 9. We also detected an increased production of IL-10 and transforming growth factor-β1 (TGF-β1) in IL-27 treated DCs (Fig. 2b). Taken together, these data suggest that IL-27 decreases the production of cytokines that promote the differentiation of effector TH1 and TH17 cells, while it up-regulates the production of anti-inflammatory cytokines by cDCs.
The effects of IL-27 on the expression of MHC classII, co-stimulatory molecules and cytokines suggested that IL-27 affects the ability of DCs to activate and polarize T cells into specific subsets. Thus, we examined the ability of cDCs pre-treated with IL-27 and extensively washed to activate naive 2D2+ CD4+ T cells in the presence of their cognate target antigen, the region between amino acids 35–55 of the myelin oligodendrocyte protein (MOG (35–55)). Pre-treatment of cDCs with IL-27 led to a significant decrease in the proliferative response of naive 2D2+ T cells to MOG (35–55) (Fig. 2d). Moreover, IL-27 treated cDCs had a decreased ability to induce IFN-γ and IL-17 production by T cells as measured by ELISA and intracellular cytokine staining (Figs. 2e,f). Conversely, pre-treatment of cDCs with IL-27 boosted their ability to promote the differentiation of IL-10+ and FoxP3+ CD4+ T cells (Figs. 2e,f). Similar effects were observed when bone marrow-derived DCs were treated with IL-27 (data not shown).
IL-27 is known to act directly on T cells to suppress their differentiation into effector T cells 12, 15–17. We found that IL-27 treated cDC showed a reduced ability to trigger the production of IFN-γ and IL-17 by T cells in the presence of exogenously added TH1 and TH17 polarizing cytokines (Fig. 2g). Conversely, IL-27 treatment of cDC increased IL-10 production and the expression of FoxP3 in T cells when Tr1 or Treg cell (FoxP3) polarizing cytokines were added to the co-culture (Figs. 2g,h), suggesting that IL-27 signaling in DCs modulates T cell differentiation in vivo even in the context of inflammation or other physiological conditions that generate a polarizing cytokine milieu (Figs. 2g–h). Taken together, these data demonstrate that IL-27 signaling controls the antigen-presenting cell (APC) function of cDCs.
IL-27RA in DCs limits EAE development
IL-27 plays an important role in the control of CNS inflammation during EAE 12, 13, 15. In agreement with previous reports 13, we found a significant worsening of EAE in IL-27RA-deficient (Il27ra−/−) mice, characterized by an increase in the frequency of CNS infiltrating IFN-γ+ and IL-17+ CD4+ T cells and a reduction in IL-10+ CD4+ T cells (Suppl. Figs. 3a,b). IL-27RA-deficient mice also showed an increased recall response to MOG (35–55) and increased frequencies of CD4+CD44+CD40Lhi IFNγ+, IL-17+ and IFNγ+ IL-17+ CD4+ T cells in lymph nodes and spleen, concomitant with a reduction in FoxP3+ and IL-10+ CD4+ T cells (Suppl. Figs. 3c,d).
The published effects of IL-27 on encephalitogenic and Treg cells 14, 15, 17 suggest that the worsening of EAE in IL-27RA-deficient mice results from the lack of IL-27 signaling in T cells. However, Il27ra−/− mice carry a non-cell specific deletion of IL-27RA, thus it is possible that IL-27 acts on additional cells besides T cells to limit the development of EAE. To investigate the role of IL-27 signaling in DCs during EAE we isolated cDCs from WT and Il27ra−/− mice 21 days after disease induction. We found that cDCs from Il27ra−/− mice showed an increased ability to activate naive 2D2+ T cells in the presence of MOG (35–55) (Suppl. Figs. 3e), suggesting that defective IL-27 signaling in DCs contributes to the worsening of EAE in Il27ra−/− mice.
In vivo, DCs are influenced by their interactions with T cells and the cytokine milieu. Thus, the increased APC function of cDCs isolated from Il27ra−/− mice might reflect the exposure of the DCs to a more inflammatory cytokine milieu, and not direct effects of IL-27 on DCs. To study the role of IL-27 signaling in DCs during EAE we used a chimera-based approach to generate mice lacking IL-27RA expression in DCs (Suppl. Fig. 4a). Briefly, we reconstituted lethally irradiated WT mice with bone marrow (BM) cells from mice expressing the diphtheria toxin receptor (DTR) under the control of the CD11c (itgax) promoter (CD11c-DTR mice). Following reconstitution, the CD11c+ DCs in these mice can be depleted by the administration of diphtheria toxin (DTx) 22. DTx cannot be chronically administered to CD11c-DTR mice because of adverse side effects, however no adverse effects are associated to the chronic administration of DTx to CD11c-DTR→WT chimeras 4. Thus, 2 months after reconstitution with CD11c-DTR BM, we depleted the DTR+ DCs in the CD11c-DTR→WT chimeras by the chronic administration of DTx, and the DC compartment was reconstituted with DC precursors from WT or Il27ra−/− mice (Suppl. Fig. 4b). DTx was administered to these mice once every other day until the completion of the experiment; no antibodies against DTx were detected following 2 months of DTx administration (Suppl. Fig. 4c). We refer to the CD11c-DTR→WT chimeras reconstituted with WT or IL-27RA-KO DC precursors as DC (WT) or DC (IL-27RA-KO) mice, respectively.
DC (IL-27RA-KO) mice showed a significant decrease in IL-27RA expression in cDCs, but not in other antigen presenting cell populations (Fig. 3a and Suppl. Fig. 4d). No differences were detected in the frequency and absolute number of DCs between DC (WT) and DC (IL-27RA-KO) mice (Suppl. Fig. 4e). EAE induction with MOG (35–55) resulted in a faster development of EAE in DC (IL-27RA-KO) mice, which also reached significantly higher disease scores (P<0.01, Fig. 3b). The worsening of EAE in DC (IL-27RA-KO) mice was associated with an increased frequency of TH1 and TH17 cells in the CNS, and a significant decrease in IL-10+ T cells (Fig. 3c). Moreover, the analysis of the CD4+ CD44+CD40Lhi splenic T-cell compartment revealed a higher recall proliferative response to MOG (35–55) in DC (IL-27RA-KO) mice, and a higher frequency of IFN-γ+ and IL-17+ CD4+ cells, concomitant with a reduction in FoxP3+ Tregs cells and IL-10+ producing CD4+ cells (Figs. 3d,e). Similar results were obtained when EAE was induced by the transfer of TH1 or TH17 MOG (35–55)-reactive T cells (Suppl. Figs. 4f–h).
Figure 3. IL-27RA signaling in cDCs controls T cell differentiation and EAE development.

(a) Flow cytometry of IL-27RA expression in splenic cDCs sorted from naive DC (WT) or DC (IL-27RA-KO) mice. (b) Development of EAE in DC (WT) and DC (IL-27RA-KO) mice, clinical score (left panel) and linear-regression curves of disease for each group (dashed lines indicate 95% confidence interval). (c) CNS-infiltrating CD4+ T cells analyzed for the expression of IFN-γ, IL-17, IL-10 and FoxP3 by flow cytometry. (d) Recall response to MOG (35–55) in splenocytes from DC (WT) and DC (IL-27RA-KO) mice isolated 21 days after EAE induction. (e) Frequency of CD4+CD44+CD40Lhi splenic IFN-γ+, IL-17+, IFN-γ+ IL-17+ (DP), IL-10+ and FoxP3+ CD4+ T cells in DC (WT) and DC (IL-27RA-KO) mice 21 days after EAE induction. (f) Expression of Il27ra, Il6, Il12a, Il23a, Il27, Ifnb, Il10 and Tgfb1 in cDCs sorted from DC (WT) and DC (IL-27RA-KO) mice 21 days after immunization. (g) Quantitative nCounter expression profiling of cDCs isolated from DC (WT) and DC (IL-27RA-KO) mice 21 days after immunization. (h–i) Naive CFSE labeled 2D2+ CD4+ T cells were stimulated with MOG35–55 and cDCs sorted from DC (WT) and DC (IL-27RA-KO) mice 21 days after immunization, and proliferation (h) and cytokines secretion (i) were analyzed. The frequency of proliferated cells is shown in the histogram and the proliferation index is shown in the right (h). Numbers within histograms show the percentage of positive cells. Shown is a representative experiment (of three) with n ≥ 5 mice/group. *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test) compared with DC (WT).
To study the effects of IL-27 signaling in DCs during EAE we isolated cDCs from WT or IL-27RA-KO 21 days after EAE induction. cDCs from DC (IL-27RA-KO) mice showed an increased expression of the pro-inflammatory cytokines IL-6, IL-12, IL-23, concomitant with a decrease in IL-10 and IL-27 expression (Fig. 3f). These results were confirmed by additional nCounter analyses, which detected a pronounced pro-inflammatory transcription profile in cDCs isolated from DC (IL-27RA-KO) mice during EAE (Fig. 3g). Moreover, cDCs from DC (IL-27RA-KO) mice showed an increased ability to activate 2D2+ CD4+ T cell proliferation, promoting the production of increased amounts of IFN-γ and IL-17 and decreased quantities of IL-10 and TGF-β1 (Fig 3h,i). Taken together, these data show that IL-27 acts on cDCs in vivo to limit the development of encephalitogenic T cells and EAE.
Transcriptional effects of IL-27 on DCs
To investigate the mechanisms mediating the effects of IL-27 on DCs, we analyzed the expression profile of primary splenic cDCs isolated from naive WT mice and treated in vitro with IL-27 using Affymetrix arrays. The study of the expression data with the Ingenuity Pathway Analysis (IPA) identified a significant reduction in the expression of pro-inflammatory genes associated with NF-κB and TLR signaling pathways (p=2.16 10−21 and p=4.55 10−27, respectively, Suppl. Figs. 5a,b). Conversely, IL-27 treatment led to a significant increase in Tnip3 and Tnfaip3 expression, genes known to inhibit NF-κB activation 23 (Suppl. Fig. 5c). We also found a significant up-regulation of the anti-inflammatory molecules Ido1, Ido2, Il27, Il10 and Entpd1 (Suppl. Fig. 5c).
We used NetGenerator to analyze the transcriptional response of DCs to IL-27. NetGenerator integrates in a model the expression profiles with prior knowledge on the genes under investigation and their connections 24. The resulting model suggested that IL-27 controls the expression of the anti-inflammatory molecules described above in a STAT1 and STAT3 transcription factor-dependent manner (Suppl. Fig. 5d). Taken together, these data show that IL-27 limits the inflammatory response of cDCs and triggers the expression of tolerogenic molecules.
ENTPD1 mediates the inhibitory effects of IL-27 on DCs
Our transcriptional profiling studies identified several candidate molecules likely to mediate the effects of IL-27 signaling in cDCs on T cell activation. To identify the mechanisms mediating the effects of IL-27 on the APC function of cDCs, we used blocking antibodies to IL-27, IL-10, IFN-β or TGF-β, or the IDO-specific inhibitor 1-methyl-tryptophan (1-D-MT). Splenic cDCs were pre-treated with IL-27, extensively washed and used to activate naive CD4+ T cells with anti-CD3 in the presence of the blocking antibodies to cytokines or 1-D-MT. The suppressive effect of cDC-pretreatment with IL-27 was not blocked by the antibodies or 1-D-MT suggesting that neither IDO nor IL-27, IL-10, IFN-β or TGF-β were mediating the suppressive effects of IL-27-pretreated cDC (Figs. 4a,b).
Figure 4. ENTPD1 is required for the inhibitory effects of IL-27 on DCs.

(a–c) Naive CD4+ T cells were stimulated with anti-CD3 and ecLPS- or ecLPS+IL-27-treated cDCs in the presence of either isotype control (IC), anti-IL-27, anti-IL-10, anti-IFN-β or anti-TGF-β blocking antibodies (a) or in the presence of the IDO inhibitor 1-D-MT (b) and proliferation was analyzed. (c) T cells were stimulated with anti-CD3 and ecLPS- or ecLPS+IL-27-treated cDCs from ENTPD1-deficient mice (DC (CD39-KO)) and T-cell proliferation was analyzed. (d) Expression of Entpd1 in cDCs sorted from naive DC (WT) and DC (IL-27RA-KO) mice analyzed by qPCR (left panel) and flow cytometry. The numbers within the histogram show the percentage of positive cells. (e–f) Splenic cDCs were exposed to IL-27 (20 ng/ml) and after indicated time periods, cells were harvested and analyzed by immunoblot (e) and flow cytometry (f) for phosphorylated or unphosphorylated STAT 1 and STAT3. (g) STAT1 and STAT3 binding sites in the Entpd1 promoter. Schematic representation of the Entpd1 promoter with STAT1- (green; IRF-1), STAT3- (blue; SRE-1 and SRE-2) and STAT1 and STAT3-binding sites. (h) ChIP analysis of the interaction of STAT3 in the Entpd1 promoter of cDCs treated with IL-27 and ecLPS. (i) ChIP analysis of the interaction of STAT1 in the Entpd1 promoter of cDCs treated with IL-27 and ecLPS. (j) Luciferase activity in HEK293 cells transfected with a Entpd1 (CD39) luciferase reporter, alone (Control) or with a construct encoding constitutively activated STAT1 or STAT3, separately or together (STAT1c + STAT3c). *P < 0.05; **P < 0.01 (One-way ANOVA). Data are representative of more than three independent experiments with similar results.
PD-L1 expression is reported to be up-regulated following IL-27 treatment of murine pDCs 20 or human monocyte-derived DCs 19. We did not detect a significant up-regulation of PD-L1 expression following cDC treatment with IL-27 (Suppl. Fig. 6a). Moreover, the suppressive effects of IL-27-treated cDCs on T cell activation were also observed when we used PD-L1 or IL-10 deficient cDCs (Suppl. Fig. 6b). Taken together, these data suggest that the suppressive effects of IL-27 on the APC function of cDCs are independent of PD-L1 or IL-10.
ENTPD1, the ectonucleoside triphosphate diphosphohydrolase 1, has been linked to the suppressive activity of mouse and human Treg cells 25, 26. Entpd1 is significantly up-regulated in DCs in response to IL-27 treatment in vitro (Suppl. Fig. 5c). Hence, we investigated the role of ENTPD1 on the effects of IL-27 in cDCs. ENTPD1-deficiency abolished the suppressive effects of DC-pretreatment with IL-27 on T cell activation and polarization. Pre-treatment of ENTPD1-deficient cDCs with IL-27 did not significantly decrease the proliferative response and the production of IFN-γ and IL-17, and had no significant effect on IL-10 production and FoxP3 expression by 2D2 T cells (Fig. 4c and Suppl. Figs. 6b,c). Thus, these data suggest that ENTPD1 mediates the effects of IL-27 signaling in cDCs on T cell activation.
To further characterize the role of ENTPD1 on the effects of IL-27 in DCs we studied the regulation of ENTPD1 expression by IL-27. Freshly isolated cDCs from DC (IL-27RA-KO) mice showed a significant decrease in the expression of ENTPD1 at both mRNA and protein levels, reflected as a significant decrease in ENTPD1 (CD39)+ DCs (Fig. 4d). Thus, IL-27 controls Entpd1 expression in cDCs in vitro and in vivo.
STAT1 and STAT3 mediate the response to IL-27 in T cells 17. We detected a significant phosphorylation of STAT3 in cDCs 5 min after treatment with IL-27, and a modest but significant increase in STAT1 phosphorylation (Figs. 4e,f). A bioinformatics analysis identified a putative STAT1-binding element (IRF-1), two STAT3-binding elements (SRE-1 and SRE-2) and a common STAT1-STAT3 binding element upstream of the transcription start site in the Entpd1 promoter (Fig. 4g). Chromatin immunoprecipitation assays (ChIP) detected STAT3 binding to SRE-1, SRE-2 and the STAT1-STAT3 binding element in the Entpd1 promoter in response to IL-27 (Fig. 4h). STAT1 binding to the Entpd1 promoter, however, was unresponsive to IL-27 and triggered by LPS activation instead (Fig. 4i).
To investigate the functional consequences of the interaction of STAT1 and STAT3 with the Entpd1 promoter we performed reporter assays. Co-transfection of a reporter construct containing the firefly luciferase gene under the control of the Entpd1 promoter together with constructs encoding constitutively activated STAT1 or STAT3 led to a significant increase in luciferase activity, but this effect was significantly stronger in response to constitutively activated STAT3 (Fig. 4j). These data are in agreement with the reported effects of STAT3 on ENTPD1 expression in T cells 27. Taken together, these data suggest that IL-27 controls Entpd1 expression in cDCs via STAT3.
ENTPD1 controls NLRP3 inflammasome activation
ENTPD1 is an ectonucleotidase that catalyzes the degradation of extracellular adenosine triphosphate and diphosphate (ATP and ADP, respectively) 28. Extracellular ATP triggers NLRP3 inflammasome activation 29, a process shown to control the differentiation of encephalitogenic TH1 and TH17 cells during EAE 30. Based on the requirement of ENTPD1 expression for the effects of IL-27 signaling in cDCs on T cell activation and differentiation (Fig. 4c and Suppl. Fig. 6), we investigated the effects of IL-27 on the extracellular concentration of ATP and NLRP3 inflammasome activation. IL-27 treatment led to a significant inhibition of the extracellular levels of ATP detected after activation of WT cDCs with LPS (Fig. 5a). IL-27, however, had no effect on the extracellular levels of ATP measured in supernatants of IL-27RA- or ENTPD1-deficient DCs (Fig. 5a). Moreover, IL-27RA and ENTPD1 deficiency led to a significant increase in the extracellular levels of ATP detected after activation with LPS (Fig. 5a).
Figure 5. IL-27-induced ENTPD1 controls extracellular ATP and NLRP3 inflammasome activation.

(a) Extracellular ATP levels in culture supernatants of ecLPS- or ecLPS+IL-27-treated cDCs. (b) Residual extracellular ATP in culture supernatants of ecLPS- or ecLPS+IL-27-treated cDCs after incubation with 500 μM of exogenous ATP. Please note different scale used in comparison with the data showed in (a). (c) TLC assays to assess CD39 enzyme activity in ecLPS- or ecLPS+IL-27-treated cDCs. (d) Quantification of AMP band intensity, expressed relative to ADP in CD39 deficient DCs. (e) Immunoblot blot (left) and densitometric analysis of caspase-1 and IL-1β in ecLPS- or ecLPS+IL-27-treated cDCs. (f) Quantification of IL-1β in culture supernatants of ecLPS- or ecLPS+IL-27-treated cDCs. *P < 0.05; **P < 0.01; ***P < 0.001 (One-way ANOVA). Data are representative of two independent experiments with similar results. AU, arbitrary units.
To further investigate the mechanisms linked to the reduced levels of extracellular ATP detected in tissue culture supernatants of WT cDCs treated with IL-27, we added exogenous ATP (500 μM) to cDCs treated with LPS and/or IL-27 and quantified the residual amount of extracellular ATP in the supernatants after 2 hours of incubation. We found significant lower levels of residual extracellular ATP remaining in supernatants from IL-27-treated WT cDCs (Fig. 5b). IL-27, however, did not affect the amount of residual extracellular ATP in IL-27RA- or ENTPD1-deficient cells, suggesting that the reduction in ATP in supernatants from WT cDCs treated with IL-27 results from its increased catabolism by ENTPD1 (Fig. 5b). In agreement with this interpretation, IL-27 treatment significantly increased nucleoside triphosphate diphosphohydrolase activity in WT DCs, but not in IL-27RA or ENTPD1-deficient cDCs (Fig. 5c,d). Moreover, we detected decreased nucleoside triphosphate diphosphohydrolase activity (manifested as higher residual amounts of ATP) in IL-27RA-deficient DCs, suggesting that the autocrine effects of IL-27 control the ability of cDCs to degrade extracellular ATP.
ATP activates the NLRP3 inflammasome in certain APCs such as macrophages29. NLRP3 inflammasome activation results in the generation of active caspase 1, leading to the maturation and release of IL-1β and IL-18 31. We found that treatment with IL-27 significantly suppressed NLRP3 inflammasome activation in WT cDCs, as evidenced by a reduced detection of activated caspase-1 and mature IL-1β (Fig. 5e) and the secretion of significantly lower amounts of IL-1β to the culture medium (Fig. 5f). In accordance with our data on extracellular concentrations of ATP, the inhibitory effects of IL-27 on NLRP3 inflammasome activation and IL-1β release were not detected on IL-27RA- or ENTPD1-deficient cDCs (Figs. 5e,f). Indeed, IL-27RA or ENTPD1 deficiency resulted in a significant increase in IL-1β release (Figs. 5f). Taken together, these data suggest that the up-regulation of ENTPD1 expression triggered by IL-27 limits extracellular ATP levels and consequently, NLRP3 inflammasome activation in cDCs.
ENTPD1 in DCs limits EAE development
To study the role of ENTPD1 in DCs during EAE we reconstituted CD11c-DTR→WT chimeras with ENTPD1-deficient DC precursors, to generate mice lacking ENTPD1 expression in DCs (DC (CD39-KO) mice). DC (WT) mice reconstituted with WT precursors were used as controls. We found a significant decrease in ENTPD1 protein expression in cDCs from DC (CD39-KO) mice DC (CD39-KO) (Fig. 6a), but not in other antigen presenting cell populations (Suppl. Fig. 6d). No differences were detected in the frequency and absolute number of DCs between DC (WT) and DC (CD39-KO) mice (Suppl. Fig. 6e). EAE induction led to an earlier onset of EAE in DC (CD39-KO) mice, which also reached significantly higher scores (P<0.01, Fig. 6b). The worsening of EAE in DC (CD39-KO) mice was correlated with an increased frequency of TH1 and TH17 cells in the CNS (Fig. 6c). In agreement with the worsening of EAE associated with ENTPD1-deficiency in DCs, splenic T cells showed a significant increase in the proliferative recall response to MOG (35–55) and in the frequency of CD4+CD44+CD40Lhi splenic IFN-γ+ and IL-17+ CD4+ T cells in DC (CD39-KO) mice (Figs. 6d,e). No differences were observed in the frequency of FoxP3+ Tregs cells and IL-10+ CD4+ T cells (Figs. 6d,e).
Figure 6. ENTPD1 in DCs controls T cell differentiation and EAE development.

(a) flow cytometry of ENTPD1 (CD39) expression of splenic DC sorted from naive WT (DC (WT))- or CD39-KO (DC (CD39-KO))-reconstituted mice. (b) Development of EAE in WT DC (WT) and DC (CD39-KO) mice, clinical score (left panel) and linear-regression curves of disease for each group (dashed lines indicate 95% confidence intervals). (c) CNS-infiltrating CD4+ T cells analyzed for the expression of IFN-γ, IL-17, IL-10 and FoxP3 by flow cytometry. (d) Recall response to MOG (35–55) in splenocytes from DC (WT) and DC (CD39-KO) mice isolated 21 days after EAE induction. (e) Frequency of CD4+CD44+ CD40Lhi splenic IFN-γ+, IL-17+, IFN-γ+ IL-17+ (DP), IL-10+ and FoxP3+ CD4+ T cells in DC (WT) and DC (CD39-KO) mice 21 days after EAE induction. (f) Expression of Entpd1, Il6, Il12a, Il23a, Il27, Ifnb, Il10 and Tgfb1 in cDCs sorted from DC (WT) and DC (CD39-KO) mice 21 days after immunization. (g–h) Naive CFSE labeled 2D2+ CD4+ T cells were stimulated with MOG35–55 and cDCs sorted from DC (WT) and DC (CD39-KO) mice 21 days after immunization, and proliferation (g) and cytokine secretion (h) were analyzed. The frequency of proliferated cells is shown in the histogram and the proliferation index is shown in the right (g). Numbers within histograms show the percentage of positive cells. Shown is a representative experiment (of three) with n ≥ 5 mice/group. *P < 0.05 and **P < 0.01 (Student’s t-test) compared with DC (WT).
The analysis of cDCs isolated from DC (CD39-KO) mice 21 days after EAE induction revealed a significant decrease in ENTPD1 expression, concomitant with an increased mRNA expression of the pro-inflammatory cytokines IL-6, IL-12 and IL-23 (Fig. 6f). No significant differences were found in IL-10, TGF-β, IFN-β or IL-27 mRNA expression (Fig. 6f). In vitro, cDCs from DC (CD39-KO) mice showed an increased ability to trigger the proliferation of naive 2D2+ CD4+ T cells in the presence of MOG (35–55) and induce IL-17 and IFN-γ production, at the expense of a decreased secretion of IL-10 and TGF-β (Figs. 6g,h). Thus, ENTPD1 (CD39) expression in DCs limits the encephalitogenic TH1 and TH17 T cell response and the development of EAE.
Vaccination with IL-27-conditioned DCs suppresses EAE
The tolerogenic effects of IL-27 signaling in T cells and DCs support its therapeutic potential in immune-mediated disorders. IL-27, however, has been reported to act on T cells to boost cytotoxic CD8+ T-cell responses 11, suggesting that direct IL-27 administration could potentially have detrimental side effects in immune-mediated disorders.
DC vaccination induces immunity to tumors and pathogens 32, but vaccination with tolerogenic DCs has been shown to induce antigen-specific tolerance33. Thus, based on the tolerogenic effects of IL-27 signaling in DCs, and to avoid the potential pathogenic effects of IL-27 administration, we investigated the therapeutic effects of vaccination with IL-27-conditioned DCs on CNS autoimmunity. First, we studied the effects of vaccination with IL-27-conditioned bone marrow derived-DCs on the model of relapsing-remitting EAE induced in SJL mice by immunization with the 139–151 region of the proteolipid protein (PLP (139–151)). On day 20 after EAE induction, during the remission phase of the disease, the mice were randomly allocated into 4 groups and treated as follows: group 1, saline; group 2, received DCs loaded with PLP (139–151); group 3, received DCs treated with IL-27; group 4, received DCs treated with IL-27 and loaded with PLP (139–151). An additional control group received DCs not loaded with peptide nor pretreated with cytokine. The treatment was repeated 3 additional times, once every 4 days. DCs not loaded with peptide nor pretreated with cytokine had no significant effects on disease development (Suppl. Fig. 7a). However, the administration of IL-27-conditioned DCs loaded with PLP (139–151) led to a significant reduction of EAE, and a significant reduction in the proliferative, IFN-γ and IL-17 recall response to PLP (139–151) concomitant with an increased production of IL-10 and TGF-β (Figs. 7a–c).
Figure 7. Vaccination with IL-27 conditioned DCs suppresses EAE.

EAE was induced by immunization of naive SJL mice with PLP131–159, and DCs were administered i.v. 4 times, once every 4 days, starting at day 20. (a) The course of EAE is shown as the mean EAE score ± SEM (n = 5 mice per group) for the whole observation period (left panel), and also as the linear regression curves (right panel) of the disease for each group from day 20 until the termination of the experiment. Arrows indicate treatment with DC vaccines. (b,c) Recall proliferative (b) and cytokine (c) response to PLP131–159 in splenocytes taken from DCs-treated mice 55 days after EAE induction. (d,e) Recall proliferative (d) and cytokine (e) response to PLP178–191 in splenocytes taken from DCs-treated mice 55 days after EAE induction. (f) Heatmap depicting the antibody response to myelin antigens on day 55 after EAE induction as measured on antigen microarrays. Each column represents the mean IgG serum reactivity in each treatment group, according to the colorimetric scale shown on the bottom. Data are representative of at least three independent experiments. NS, not significant. *P < 0.05, **P < 0.01 and **P < 0.001 (One-way ANOVA)versus control mice.
In this model, the chronic phase of EAE is characterized by the spreading of the T-cell response to the PLP epitope placed between residues 178 and 191 (PLP (178–191)) 7. Thus, we analyzed the recall response to PLP (178–191) epitope in DC-vaccinated mice. We found that administration of IL-27-treated DCs loaded with PLP (139–151) led to a significant suppression of the proliferative response and IFN-γ and IL-17 production triggered by PLP (178–191), concomitant with an increased production of IL-10 and TGF-β (Figs. 7d–e). Thus vaccination with IL-27 treated cDC also seem to reduce epitope-spreading in this model of EAE.
To further investigate the effects of IL-27-conditioned DCs on the immune response, we used myelin antigen arrays, which we 34 and others 35 found can detect epitope spreading in EAE and MS. The microarrays consisted of a collection of CNS-related autoantigens including tissue lysates, recombinant proteins, peptide libraries spanning the whole sequence of myelin proteins and lipids found in the central and peripheral nervous system 34 (Supplmentary table 1). The therapeutic effect of DC vaccination in SJL EAE was accompanied by a decrease in IgG serum antibodies to 31 myelin antigens (Fig. 7f).
We then studied the mechanisms involved in the suppression of EAE with IL-27 conditioned DCs using the B6 model of EAE induced with MOG (35–55). We initiated the administration of IL-27 conditioned DCs 10 days after EAE induction. Vaccination with IL-27-conditioned WT DCs loaded with MOG (35–55) led to a significant reduction of EAE (Suppl. Figs. 7b–d). A similar suppression of EAE was observed following administration of MOG (35–55) loaded, IL-27-conditioned DCs deficient in IL-10 or PD-L1, suggesting that these molecules do not play a significant role in the regulation of the encephalitogenic response by IL-27 signaling in DCs. Conversely, MOG (35–55) loaded IL-27-conditioned DCs deficient in IL-27R or CD39 did not have a significant effect on EAE. Taken together, these data demonstrate that vaccination with IL-27-treated DCs controls the encephalitogenic response and established relapsing-remitting EAE in a therapeutic paradigm in a CD39 dependent manner.
Discussion
IL-27 limits tissue inflammation and autoimmunity in different scenarios 11. Indeed, IL-27R deficiency is linked to the development of exacerbated inflammation in animal experimental models, and IL-27 polymorphisms are associated to human inflammatory disorders 11. The immunoregulatory actions of IL-27 are usually attributed to its direct effects on T cells, where it arrests TH17 cell development and promotes the differentiation of type 1 regulatory T cells through mechanisms that involve the aryl hydrocarbon receptor 26, 36. In this work we report that IL-27 signaling in DCs plays a significant role in the control of the T-cell response and CNS autoimmunity.
We found that ENTPD1 is required for the modulation of the APC function of cDCs by IL-27 in vitro and in vivo. However, IL-27 also up-regulates the expression of additional immunoregulatory molecules such as IL-27 itself, IFN-β, TGF-β and IDO. Although under the experimental conditions that we tested these molecules did not play a significant role, they might contribute to the suppressive effects of IL-27 on DCs in alternative scenarios.
IL-10, known to activate STAT3 signaling, has also been shown to induce a tolerogenic phenotype in DCs 37. Conversely, STAT3 deficiency restricted to DCs results in the spontaneous development of inflammation 38. These data support a tolerogenic role for STAT3 in DCs, and suggest that other cytokines that activate STAT3 signaling might trigger ENTPD1-dependent regulatory pathways similar to those triggered by IL-27. Moreover, microglia and tissue macrophages express ENTPD1, and their function is also regulated by cytokines that activate STAT3 signaling 39, 40. Thus, it is possible that the induction of ENTPD1 expression constitutes a common immunoregulatory mechanism triggered by STAT3-activating cytokines in cells of the innate immune system.
ENTPD1 mediates the suppressive activity of mouse and human regulatory T cells, probably through the generation of adenosine 25, 26. The mechanisms by which ENTPD1 in APCs regulates adaptive immunity are less clear, but recent data suggest that these effects involve the control of NLRP3 inflammasome activation. Extracellular ATP triggers NLRP3 inflammasome activation 28. Initial support for a role of ATP-dependent NLRP3 activation in the control of T-cell immunity was provided by the observation of augmented contact hypersensitivity in ENTPD1-deficient mice 41. Conversely, NLRP3-deficiency results in decreased contact hypersensitivity 42. Follow up studies determined that the activation of the NLRP3 inflammasome in APCs controls TH1, TH2 and TH17 responses 43, 44. Indeed, NLRP3 inflammasome activation is required for the differentiation of encephalitogenic TH1 and TH17 cells and the development of EAE 30, probably as a result of its effects on IL-1β and IL-18 secretion.
In our studies, the induction of ENTPD1 expression in DCs by IL-27 led to a significant reduction of extracellular ATP levels, concomitant with a significant reduction of NLRP3 inflammasome activation and the reduced differentiation of TH1 and TH17 cells. These data are compatible with a model in which, through the up-regulation of ENTPD1 expression, IL-27 limits ATP-dependent NLRP3 inflammasome activation in DCs and their ability to promote the differentiation of pathogenic TH1 and TH17 cells. It is possible, however, that in addition to its effects on ENTPD1 expression, IL-27-triggered signaling directly interferes with the expression of NLRP3 inflammasome components and their activation in DCs.
The immunoregulatory properties of IL-27 support its use for the treatment of human autoimmune diseases. IL-27, however, can boost cytotoxic CD8+ T-cell responses 11, suggesting that IL-27 administration could potentially worsen immune-mediated disorders. Hence, to explore the therapeutic potential of IL-27 and avoid its potential deleterious side effects, we studied the effects of vaccination with IL-27-conditioned DCs. We found that vaccination with IL-27-conditioned DCs suppresses the encephalitogenic TH1 and TH17 response and halts established chronic EAE. DC vaccination has been recently approved for the treatment of prostate cancer 45, thus vaccination with IL-27-conditioned tolerogenic DCs might provide a new avenue for the treatment of autoimmune diseases. Alternatively, we recently described the development of nanoparticles for the simultaneous activation of tolerogenic pathways and the delivery of antigens to APCs in vivo to induce antigen-specific tolerance 46. Thus, it is conceivable that nanoparticles engineered to activate IL-27 signaling and deliver myelin antigens to DCs will induce tolerogenic DCs in vivo and arrest pathogenic T-cell responses. This approach would exploit the immunoregulatory properties of IL-27 and overcome the limitations associated to the implementation of cell-based therapies in a clinical setup.
In summary, we report that IL-27 signaling in DCs limits the differentiation of pathogenic TH1 and TH17 T cells and the development of CNS autoimmunity, through a mechanism that is at least partially dependent on ENTPD1. This immunoregulatory pathway might provide new therapeutic avenues for MS and other autoimmune disorders.
Methods
Animals
IL-27RA and CD39-deficient mice have been previously described 25, 36. SJL, 2D2, C57BL/6, CD11c-Cre:DTR and IL-10 deficient mice were purchased from the Jackson Laboratories. PD-L1 deficient mice were a generous gift from Dr. Arlene Sharpe (Harvard Medical School). For conditional DC ablation [CD11c-DTR→wt], BM chimeras were inoculated i.p. every second day for 2 wk with 16ng DTx/g body weight. For BM chimera generation, recipient mice were lethally irradiated with a 9.5 Gy dose and a day later i.v. injected with 5×106 BM cells isolated from donors femora and tibiae. BM recipients were then allowed to rest for 8 wk before use. Four to five randomly assigned mice were used per experimental group per experiment. Mice were kept in a conventional, pathogen-free facility at the Harvard Institutes of Medicine. All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School.
Isolation of Splenic DCs and CNS Infiltrates
Spleens were incubated with 2 mg/ml Collagenase D for 20 min and then mashed through a 70 μm cell strainer. DCs were sorted as previously described into F4/80−CD11b− CD11clow B220+ MHC-IIlow LY6C+ pDCs and F4/80− CD11b+ CD11c+ B220− MHC-II+ LY6C− cDCs (eBioscience). Conventional DCs were cultured with IL-27 (20 ng/ml) or LPS (100 ng/ml; Escherichia coli strain 0111:B4; Sigma; ecLPS) for 48 h. Parallel cultures were maintained without stimuli and used as controls. IL-27 was purchased from eBioscience and prepared according to the manufacturer’s protocol.
CNS infiltrates were isolated as previously described 47. In brief, mice were perfused using ice-cold PBS. The brain and spinal cords were removed and incubated in PBS containing collagenase type III (2 mg/ml, Worthington) and DNase (20 units/ml, Sigma-Aldrich). The tissue was then homogenized and loaded on a 30:37:70% Percoll gradient for enrichment of CNS infiltrates.
Flow Cytometry Staining and Acquisition
The following antibodies were used for flow cytometry: peridinin chlorophyll protein–conjugated anti-LY6C (HK1.4) and allophycocyanin-conjugated anti-CD11b (M1/70) from BioLegend, phycoerythrin-conjugated anti-MHCII (M5/114.15.2), fluorescein isothiocyanate (FITC)-conjugated anti-F4/80 (BM8) and Alexa Fluora 647–anti-CD39 (24DMS1) from eBioscience, allophycocyanin-cyanine7 (APC-Cy7)-conjugated anti-CD11c (HL3), phycoerythrin-conjugated anti-B220 (RA3-6B2) from BD Pharmingen and fluorescein isothiocyanate (FITC)-conjugated anti-Il-27RA (263503) from R&D.
For analysis of intracellular Foxp3, cell preparations were stained for cell surface markers, then were fixed and made permeable with fixation-permeabilization buffers (eBioscience) and stained with peridinin chlorophyll protein-cyanine5.5 (PerCP-Cy5.5)–conjugated anti-Foxp3 (FJK-16s; eBioscience).
For intracellular cytokine staining, cells were stimulated for 6 h with phorbol 12-myristate 13-acetate (50 ng/ml) and ionomycin (500 ng/ml) in the presence of GolgiPlug (BD Pharmingen). Then, cells were stained for surface molecules, fixed and made permeable with a Cytofix/Cytoperm Plus kit (BD Bioscience) and stained with the following antibodies: phycoerythrin-cyanine7 (PE-Cy7)-conjugated anti-IFN-γ (XMG1.2; BioLegend), phycoerythrin-conjugated anti-IL-17 (eBio17B7; eBioscience) and allophycocyanin-conjugated anti-IL-10 (JES5-16E3; BD Pharmingen). Data were collected with a LSR II or flow cytometryAria flow cytometer (BD Biosciences), then were analyzed with FlowJo software (Treestar).
Proliferation assays
For in vitro experiments, cDCs treated as described above were used to stimulate naive T cells from WT or 2D2 mice at a ratio of 1:10. Polarizing conditions were used as described 48. Briefly, IL-12 (30 ng/ml) to generate TH1 cells, or IL-6 (30 ng/ml) and TGF-β1 (3 ng/ml) to generate TH17 cells, or TGF-β1 (5 ng/ml) and IL-27 (30 ng/ml) to generate Tregs and Tr1 cells respectively. Mouse IL-6, IL-12, IL-23 and TGF-β were all purchased from R&D Systems. For in vivo experiments, splenic cells were harvested from MOG immunized wild type or IL27RA-, CD39-deficient mice at day 21 and restimulated in vitro for 3 days in the presence of MOG (35–55) peptide. SJL mice were immunized with PLP (139–151) and at the end of the experiment splenic cells were harvested and restimulated in vitro for 3 days in the presence of PLP (139–151) or PLP (178–191) peptides. The cells were pulsed with 3H-thymidine (1μCi/well) for the last 24 h of the incubation period. The percentage of IL-17-, IFN-γ-, IL-10-producing T-cells and FoxP3 positive cells was assessed by flow cytometry analysis. For CFSE-based proliferation assay, 2D2+ CD4+ T cells were labeled with 1μM of carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular Probes, USA). Data were acquired on a LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Measurement of cytokines
Secreted cytokines were measured after 48 h by ELISA as described 49.
qPCR
RNA was extracted with RNAeasy columns (Qiagen, USA), cDNA was prepared following the manufacturer’s instructions (Applied Biosystems, USA) and used as template for real-time PCR. All the primers and probes in this work were provided by Applied Biosystems, and were used on the ViiA™ 7 Real-Time PCR System (Applied Biosystems). Expression was normalized to the expression of gapdh. Primers-probe mixtures were purchased from Applied Biosystems (USA): Il6 (Mm00446190_m1), Il10 (Mm0043614_m1), Il12a (Mm00434165_m1), Il23a (Mm00518984_m1), Il27 (Mm00461162_m1), Il27ra (Mm00497259_m1), Entpd1 (Mm00515447_m1), Tgfb1 (Mm01178820_m1), Ifnb1 (Mm00439552_s1), Ido1 (Mm0001218007_m1), Ido2 (Mm01218007_m1), Tnip3 (Mm01181626_m1), Tnfaip3 (Mm00437121_m1), Ramp3 (Mm00840142_m1), Esr1 (Mm00433151_m1) and Gapdh (Mm99999915_g1).
Gene expression analysis
Transcriptome analysis was done using Affymetrix microarray MoGene_1_0_st on samples taken 0, 2 and 6 h after stimulation with LPS or IL-27 and LPS. Data were normalized using the Robust Multi-array Average algorithm. Network behavior was inferred using NetGenerator, and R package in its 2.1–3 version 24. Using as input fold change of gene expression over time points. The network graph was produced using DOT language through the open software collection Graphviz (http://www.graphviz.org/). Black edges denote inferred connections without prior knowledge, green edges present an agreement, and grey dashed edges stand for prior knowledge not reproduced in the inferred network.
For Nanostring nCounter analysis of gene expression we customized a multiplexed target profiling of 146 inflammation- and immune-related transcripts and used in accordance with the manufacturer’s protocol (Nanostring, USA). This combination of genes and their differential expression in vivo in DCs allowed us to interrogate immune-related pathways using the Expander (Expression Analyzer and Displayer) pathway analysis during EAE.
Immunoblot analysis
For immunoblotting cells were lysed with RIPA buffer supplemented with protease inhibitor cocktail (Sigma, USA). Total cell lysates of DCs (40 μg) were resolved on 4–12% Bis-Tris Nupage gels (Invitrogen, USA) and transferred onto PVDF membranes (Millipore). The following primary antibodies were used: Anti-IL-27RA (MAB21091) from R&D; anti-pSTAT3 (Cat. #9134), anti-pSTAT1 (Cat. #9167), anti-STAT3 (Cat. #9132), anti-STAT1 (Cat. #9172) and anti-GAPDH (#2111) from Cell Signaling Technology, USA; anti-caspase-1 (ab17820), anti-IL-1β (ab9722) and anti-beta actin (ab20272) from Abcam, USA. Immunoblot analysis was performed as described 47 and developed using SuperSignal West Femto Maximum Sensitivity Substrate, as instructed by the manufacturer (Pierce).
Chromatin immunoprecipitation (ChIP)
Cells were cross-linked with 1% paraformaldehyde and lysed with 0.35 ml of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) containing 1X protease inhibitor cocktail (Roche Molecular Biochemicals, USA). Chromatin was sheared by sonication and supernatants were collected after centrifugation and diluted in buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1). Five μg of antibody was prebound for a minimum of 4 h to protein A and protein G Dynal magnetic beads (Invitrogen, USA) and washed three times with ice-cold PBS plus 5% BSA, and then added to the diluted chromatin and immunoprecipitated overnight. The magnetic bead-chromatin complexes were then washed 3 times in RIPA buffer (50 mM HEPES [pH 7.6], 1 mM EDTA, 0.7% Na deoxycholate, 1% NP-40, 0.5 M LiCl) followed by 2 times with TE buffer. Immunoprecipitated chromatin was then extracted with 1% SDS, 0.1 M NaHCO3 and heated at 65°C for at least 6 h to reverse the paraformaldehyde cross-linking. DNA fragments were purified with a QIAquick DNA purification Kit (Qiagen, USA) and analyzed using SYBR green real time PCR (Takara Bio Inc., USA). We used the following antibodies for ChIP: anti-STAT3 (Cat. #9132, Cell Signaling Technology, Inc., USA), anti-STAT1 (Cat. #9172, Cell Signaling Technology, Inc., USA), The following primer pairs were used: Stat3 (SRE-1): for: 5′-GCTGGGCTTTAGAGACTTGTGGGC-3′, rev: 5′-ACCCATGCAAATGGTTTGGGCA-3′; Stat3 (SRE-2): for: 5′-TGAGGGCCAGCCCACACTTCA-3′. rev: 5′-GCTCACTGGGTACCTCTTGCCA-3′; Stat1 (IRF-1): for: 5′-GGAACAAAAATATAGAGAGAACTTGGGA-3′, rev: 5′-GTAGTTTGACCTAAGTGGACATAGG-3′; Stat1/3: for: 5′-AGGCTCTTGTATCCTTGCCACCTCT-3′, rev: 5′-TGATGGTGGAGTGCTGTGTGCTG-3′.
Plasmids
The Entpd1 promoter reporter was generously provided by Dr. David J. Pinsky (University of Michigan Health Systems, Ann Harbor, MI, USA), vectors coding for STAT3C and STAT1C were provided by Dr. David Frank (Dana-Farber Cancer Institute, Boston, MA, USA).
Transfection and luciferase assays
HEK293 cells were grown in DMEM supplemented with 10% fetal bovine serum, and transfected using the FuGENE HD (Roche, USA) transfection reagent and 2 μg of each plasmids according the manufacturer’s instructions. Firefly and renilla luciferase activity was analyzed 48 h after transfection using a dual luciferase assay kit (Promega, USA).
Free ATP measurement
Conventional DCs were cultured with IL-27 or LPS as described above for 48 h. Cells were then washed 2 times with phenol red-free RPMI 1640 (Gibco, USA) and serum starved for 24 h. Cell-free medium was then analyzed for endogenous ATP using ENTILEN rLuciferase/Luciferin Reagent (Promega, USA). Bioluminescent activity was measured using an Infinite 200 Pro luminometer (Tecan, USA).
Ectonucleotidase enzymatic activity analysis
Thin Layer Chromatography (TLC) was performed as described previously 25 with slight modifications. Specifically, 1×105 DCs treated with IL-27 or LPS for 48 h, were incubated with 2 mCi/ml [C14]ADP (GE Healthcare Life Sciences, USA) in 10 mM Ca2+ and 5 mM Mg2+. Five μl aliquots, collected at 1.5, 3, and 6 min, were analyzed for the presence of [C14]ADP hydrolysis products by TLC and applied onto silica gel matrix plates (Sigma-Aldrich, USA). [C14]ADP and the radiolabeled derivatives were separated using an appropriate solvent mixture as previously described 25, 50. Adenosine uptake and deamination was blocked with 10 μM of dipyridamole. [C14]ADP, [C14]AMP and [C14]ADO were incubated in PBS served as standards.
EAE Induction and DC Vaccination
EAE was induced by s.c. immunization with 150 μg of the MOG35–55 peptide or 30 μg of PLP (139–151) peptide (ANASPEC, USA) as described 49. Adoptive transfer EAE was induced as described 48 with some modifications. 2D2 mice were immunized with 150 μg MOG35–55 in complete Freund’s adjuvant, and draining lymph nodes and spleens were collected 7 d after immunization and cultured for 48 h with MOG35–55 (20 μg/ml) and carrier free recombinant IL-12 (20 ng/ml; R&D Systems) or IL-23 (20 ng/ml; R&D Systems). Subsequently, 5×106 cells were transferred intravenously into DC (WT) or DC (IL-27RA-KO) mice. Clinical signs of EAE were assessed by investigators blind to treatment according to the following score: 0, no disease; 1, loss of tail tone; 1.5, poor righting ability; 2, hind-limb weakness; 3, hind-limb paralysis; 4, quadreparesis; and 5, moribund.
To generate bone marrow-derived DCs, bone marrow cells were isolated from the femurs of naive mice and cultured for 7 days in the presence of GM-CSF (20 ng/mL, Peprotech, USA). On day 7, the cells were purified with CD11c+ magnetic beads (Miltenyi, USA), cultured with IL-27 (20 ng/ml) for 18 h and for the last 3 h before administration loaded with 20 μg of MOG35–55 or PLP (139–151). DCs (2×106 per mouse) were then extensively washed and administered i.v. 4 times, once every 4 days. All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School.
Antigen Microarrays
The antigens listed in Supplementary Table 1 were spotted onto Epoxy slides (TeleChem, Sunnyvale, CA, USA) as described 34. The microarrays were blocked with 1% bovine serum albumin, and incubated with a 1:100 dilution of the test serum in blocking buffer. The arrays were then washed and incubated with goat anti-mouse IgG Cy3-conjugated detection antibodies (Jackson ImmunoResearch Labs, West Grove, PA, USA). Antigen reactivity was defined by the mean intensity of binding to the replicates of that antigen on the microarray. Raw data were normalized and analyzed using the GeneSpring software (Silicon Genetics, Redwood City, CA, USA).
Statistical analysis
Prism software was used for statistical analysis. Statistical analysis was performed following Nature’s recommendations for Reporting Life Sciences Research. For comparison of two groups, linear regression with 95% confidence interval, and unpaired, two-tailed Student’s t-test were used. One-way ANOVA test for paired data was used to determine the significance of the time-response curves. Values of P<0.05 were considered statistically significant. To adjust the significance level for multiple comparisons, a Bonferroni correction was applied using a corrected significance level of 0.017.
Supplementary Material
Acknowledgments
This work was supported by grants AI075285, and AI093903 from the National Institutes of Health and RG4111A1 from the National Multiple Sclerosis Society to FJQ.
Footnotes
Authors’ contribution
I.D.M, A.Y, S.M.V., E.J.B., Y.W. and L.M performed in vitro and in vivo experiments, B.P., I.S. and S.E. performed bioinformatics analysis V.K.K and S.R. provided unique reagents, I.D.M and F.J.Q. wrote the manuscript and F.J.Q. supervised the study and edited the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122:1180–1188. doi: 10.1172/JCI58649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey SL, Schreiner B, Mcmahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ TH-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 4.Yogev N, et al. Dendritic Cells Ameliorate Autoimmunity in the CNS by Controlling the Homeostasis of PD-1 Receptor+ Regulatory T Cells. Immunity. 2012;37:264–275. doi: 10.1016/j.immuni.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 5.Comabella M, Montalban X, Münz C, Lünemann JD. Targeting dendritic cells to treat multiple sclerosis. Nature Reviews Neurology. 2010;6:499–507. doi: 10.1038/nrneurol.2010.112. [DOI] [PubMed] [Google Scholar]
- 6.Greter M, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nature Medicine. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 7.Mcmahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nature Medicine. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 8.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 9.Molle C, Goldman M, Goriely S. Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J Immunol. 2010;184:1784–1792. doi: 10.4049/jimmunol.0902005. [DOI] [PubMed] [Google Scholar]
- 10.Mitsdoerffer M, Kuchroo V. New pieces in the puzzle: how does interferon-beta really work in multiple sclerosis? Ann Neurol. 2009;65:487–488. doi: 10.1002/ana.21722. [DOI] [PubMed] [Google Scholar]
- 11.Hunter CA, Kastelein R. Interleukin-27: balancing protective and pathological immunity. Immunity. 2012;37:960–969. doi: 10.1016/j.immuni.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald DC, et al. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:3268–3275. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- 13.Batten M, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 14.Awasthi A, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 15.Fitzgerald DC, et al. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- 16.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 17.Stumhofer JS, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Miyazaki Y, Shinozaki Y, Yoshida H. Augmentation of antigen-presenting and Th1-promoting functions of dendritic cells by WSX-1(IL-27R) deficiency. J Immunol. 2007;179:6421–6428. doi: 10.4049/jimmunol.179.10.6421. [DOI] [PubMed] [Google Scholar]
- 19.Karakhanova S, Bedke T, Enk AH, Mahnke K. IL-27 renders DC immunosuppressive by induction of B7-H1. J Leukoc Biol. 2011;89:837–845. doi: 10.1189/jlb.1209788. [DOI] [PubMed] [Google Scholar]
- 20.Matta BM, Raimondi G, Rosborough BR, Sumpter TL, Thomson AW. IL-27 production and STAT3-dependent upregulation of B7-H1 mediate immune regulatory functions of liver plasmacytoid dendritic cells. J Immunol. 2012;188:5227–5237. doi: 10.4049/jimmunol.1103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 22.Jung S, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012;12:774–785. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber M, et al. Inference of dynamical gene-regulatory networks based on time-resolved multi-stimuli multi-experiment data applying NetGenerator V2.0. BMC Syst Biol. 2013;7:1. doi: 10.1186/1752-0509-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gandhi R, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nature Immunology. 2010;11:846–853. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalmin F, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. 2012;36:362–373. doi: 10.1016/j.immuni.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 30.Gris D, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–981. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 32.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 33.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintana FJ, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci U S A. 2008;105:18889–18894. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson WH, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol. 2003;21:1033–1039. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- 36.Apetoh L, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–861. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 38.Melillo JA, et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184:2638–2645. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker BJ, Park KW, Qin H, Ma X, Benveniste EN. IL-27 inhibits OSM-mediated TNF-alpha and iNOS gene expression in microglia. Glia. 2010;58:1082–1093. doi: 10.1002/glia.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farber K, et al. The ectonucleotidase cd39/ENTPDase1 modulates purinergic-mediated microglial migration. Glia. 2008;56:331–341. doi: 10.1002/glia.20606. [DOI] [PubMed] [Google Scholar]
- 41.Mizumoto N, et al. CD39 is the dominant Langerhans cell-associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat Med. 2002;8:358–365. doi: 10.1038/nm0402-358. [DOI] [PubMed] [Google Scholar]
- 42.Sutterwala FS, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jahnisch H, et al. Dendritic cell-based immunotherapy for prostate cancer. Clin Dev Immunol. 2010;2010:517493. doi: 10.1155/2010/517493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starossom SC, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37:249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quintana FJ, et al. Aiolos promotes T(H)17 differentiation by directly silencing Il2 expression. Nature Immunology. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintana FJ, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 50.Sun X, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010;139:1030–1040. doi: 10.1053/j.gastro.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.