

Significance
New chemical entities (NCEs) with desired pharmacological and biological activity spectra fuel drug discovery and provide tools for chemical biologists. Computer-assisted molecular design generates novel chemotypes with predictable polypharmacologies. We present the successful application of fully automated de novo drug design coupled with a pioneering approach for target panel prediction to obtain readily synthesizable bioactive compounds. This innovative concept enabled the identification of relevant macromolecular targets of computationally designed NCEs and led to the discovery of previously unknown off-targets of approved drugs.
Keywords: drug design, target prediction, polypharmacology, machine learning, chemical similarity
Abstract
De novo molecular design and in silico prediction of polypharmacological profiles are emerging research topics that will profoundly affect the future of drug discovery and chemical biology. The goal is to identify the macromolecular targets of new chemical agents. Although several computational tools for predicting such targets are publicly available, none of these methods was explicitly designed to predict target engagement by de novo-designed molecules. Here we present the development and practical application of a unique technique, self-organizing map–based prediction of drug equivalence relationships (SPiDER), that merges the concepts of self-organizing maps, consensus scoring, and statistical analysis to successfully identify targets for both known drugs and computer-generated molecular scaffolds. We discovered a potential off-target liability of fenofibrate-related compounds, and in a comprehensive prospective application, we identified a multitarget-modulating profile of de novo designed molecules. These results demonstrate that SPiDER may be used to identify innovative compounds in chemical biology and in the early stages of drug discovery, and help investigate the potential side effects of drugs and their repurposing options.
Computer-assisted de novo molecular design has evolved as a popular source of ideas to combat the perceived lack of new chemical entities (NCEs) in chemical biology and drug discovery (1). We demonstrate that automated de novo design delivers readily synthesizable NCEs with desirable activity profiles. Although receptor-based design operates on a model of a macromolecular binding site, ligand-based methods are either explicitly or implicitly based on the chemical similarity principle (2) without requiring a receptor model (3). Instead, the latter typically uses some measure of chemical or pharmacophore feature similarity to a reference ligand as a fitness function, which aims to generate NCEs as template mimetics via scaffold hopping (4–8). We report the development, implementation, and successful prospective application of an innovative computational technique for the target profiling of de novo-designed molecules. The approach combines the concepts of self-organizing maps (SOMs) (9) for macromolecular target prediction (10), consensus scoring (11), and a statistical evaluation that provides confidence estimates for the predictions.
Predicting polypharmacological activities is a topic relevant to chemical biology and drug discovery, not only to take advantage of inherent drug promiscuity but also to decrease lead compound attrition caused by unfavorable off-target modulation (12). Drug activities on multiple macromolecular targets are responsible for drug side effects (13), but they can also be rationally used to increase drug efficacy (14), repurpose known drugs (15), and design multitarget NCEs (7). For example, Mestres and colleagues recently predicted the inhibition of Pim kinases using a tool compound originating from poly(ADP-ribose) polymerase (PARP) biology (16). Lounkine et al. reported a large-scale off-target prediction for marketed drugs (17). In fact, high target promiscuity appears common among drug-like molecules (18). De novo designed molecules add novelty to the chemical universe and thus risk lying outside the domain of applicability of target prediction tools that exclusively rely on the chemical similarity of molecular substructures. Here, we provide a theoretical framework to address this issue, and we demonstrate the applicability of this framework to prospective de novo design studies. We then describe the development of a ligand-based target prediction algorithm [SOM-based prediction of drug equivalence relationships (SPiDER)] and its experimental validation by finding off-targets of known drugs without strong ligand structural similarity. Finally, we demonstrate the model's applicability for identifying pharmacologically relevant macromolecular targets of de novo-designed NCEs, which we synthesized and tested in vitro.
Results and Discussion
Computer-Based Design of New Chemical Entities.
As part of an early discovery program, we generated innovative molecules using our ligand-based de novo design software DOGS (inSili.com LLC) (19). The process by which DOGS generates a compound uses virtual organic synthesis. Compound generation is steered by optimizing the topological similarity between newly generated candidate structures and a template molecule. In this study, amprenavir (Fig. 1), a potent inhibitor of HIV 1 protease (HIVP) (20), served as the template. The virtual synthesis was fueled by a set of 25,144 building blocks and 83 organic reactions (Fig. 2A). Overall, the automated compound design process generated 2,338 molecules, of which 856 were unique structures with 388 unique Murcko scaffolds (21). From this set of diverse candidates, we selected compound 1 (Fig. 1) based on its chemotype novelty (no entry in the Chemical Abstract Service database, www.scifinder.org). Automated ligand docking into the catalytic center of HIVP [Protein Data Bank (PDB) code 1HPV] (22, 23) suggested a reasonable binding pose for compound 1 (GoldScore = 86; Fig. S1). By following the computer-generated synthesis scheme, we obtained 1 through the Mitsunobu esterification of enantiomerically pure building blocks (Fig. 1). We evaluated the inhibitory potency of 1 against HIVP but did not observe any relevant activity (10% inhibition at a compound concentration of 100 µM). We then synthesized and tested derivative 2, which lacks the tert-butoxycarbonyl group of compound 1 and presents a more advantageous docking pose than 1 by potentially interacting with both catalytic Asp25 residues in the dimeric structure (GoldScore = 82; Fig. S1). However, compound 2 was also virtually inactive against HIVP (27% inhibition at 100 µM).
Fig. 1.

The de novo design software DOGS generated compound 1 as a potential HIVP inhibitor using amprenavir as the template and suggested a feasible one-step synthetic pathway. Compound 2 was synthesized as a derivative of lead design 1; (i) HCl 4 M/1,4-dioxane 0 °C → rt.
Fig. 2.

(A) Scheme of the de novo design (DOGS) and SPiDER target prediction. (B) SOM depiction of the distribution of aspartic protease inhibitors (Left) and bradykinin receptor ligands (Right). The compound density in each of the 13 × 10 = 130 clusters (circles) is represented by the black color intensity. The concentric layers in each cluster denote quantile ranges of the compound distribution according to the distance from the centroid. On the CATS-based map (B), amprenavir is located in cluster (12/4), and the de novo-designed compounds 1 and 2 are located in cluster (12/5). On the MOE-based map (C), amprenavir and 1 are located in cluster (2/10), and compound 2 is in cluster (8/10). The toroidal SOMs were trained using the in-house command line tool molmap (56). The SOM size was chosen to have ∼100 molecules per cluster, which proved expedient in preliminary experiments. The initial learning step size was 1, and the initial neighborhood update radius was 7 to allow for full signal propagation through the map topology. Training was performed using linear parameter decay and random sampling over 500,000 training cycles (9).
Target Prediction Using Publicly Available Software Tools.
Because the applicability of compounds 1 and 2 as anti-HIVP chemotypes appeared limited, we investigated the possibility of exploiting the readily synthesizable NCEs 1 and 2 by leapfrogging to another drug target. Initially, we relied on publicly available target prediction tools. HIVP was the top predicted target for 1 according to the similarity ensemble approach (SEA) (24), fully corroborating the original DOGS design intended to mimic amprenavir (Table S1). The second most confident SEA prediction for 1 was β-secretase-1 (BACE-1), which was also suggested by the semantic link association prediction (SLAP) (25) for amprenavir (Table S2). In addition, the prediction of activity spectra for substances (PASS) (26) predicted that compound 1 would exhibit HIVP and BACE-1 inhibition (Table S3). Finally, the software SuperPred (27), which suggests targets via a pairwise comparison of query molecules to known drugs, identified HIVP inhibitors, including amprenavir, as the drugs most similar to query compound 1 (Table S4). For compound 2, SuperPred and SEA again advocated HIVP as the drug target (Tables S1 and S4). These results suggested that DOGS retained the essential structural features of amprenavir in the design of compound 1 and in its derivative 2, which clearly favored HIVP and BACE-1 as the expected targets. In vitro testing revealed that compound 1 was also inactive against BACE-1, thus rendering these target predictions incorrect. We reasoned that structures 1 and 2 may lie outside the domain of applicability of the existing fingerprint- and substructure-based target prediction methods, and therefore, we pursued the development of a novel target prediction method (SPiDER) as a complementary approach with a stronger focus on the prediction of targets for NCEs.
SPiDER Approach.
Chemically abstract (“fuzzy”) molecular representations, such as pharmacophoric feature descriptors, can be used to find subtle functional relationships between compounds, thereby allowing a molecule to leapfrog onto an unrelated target (28, 29). When used in similarity searches, such fuzzy molecular representations have often demonstrated greater scaffold-hopping potential than atomistic approaches (10, 30). Consequently, we implemented SPiDER as a software tool that builds on fuzzy molecular representations for use with de novo-designed NCEs. We relied on the established concept of SOMs to capture the local domains of model applicability (Fig. 2A). SOMs were originally developed as a neural network-inspired heuristic to reduce dimensionality (9), and they have become a workhorse of molecular informatics (31, 32). Using the unsupervised SOM algorithm, we clustered 12,661 manually annotated, pharmaceutically relevant drugs and lead compounds [collection of bioactive reference analogs (COBRA); inSili.com LLC] (33). The resulting 2D map tessellates this reference space into clusters of drug molecules representing local neighborhoods (10, 34–36). A query compound is assigned to exactly one target cluster on this map. For target inference via SPiDER, only the known drugs from this local domain are considered.
One aspect of the SPiDER method is the estimation of the statistical significance of each target prediction. The pairwise Euclidean distances between the query and the local reference drugs are calculated and evaluated according to a background distance distribution. Averaging the false-positive error probabilities for the reference drugs annotated to bind to different targets emulates a false-discovery rate in multiple hypothesis testing and provides a motivated confidence score for the prediction. SPiDER performs the target prediction twice, using the following two different molecular representations for two SOM projections: (i) the chemically advanced template search [chemically advanced template search (CATS)] topological pharmacophore descriptor (SOM1) (10, 37) and (ii) the Molecular Operating Environment (MOE) physicochemical properties and indices (SOM2) (Chemical Computing Group). Jury consensus predictions are obtained as the average of the two confidence scores. Finally, a background score distribution allows for the P value calculation of the jury scores to indicate the significance of an acquired prediction (Fig. 2A).
We performed a stratified 10-fold cross-validation (38) to estimate the scope of the SPiDER model. To measure performance, we analyzed the percentage of molecules with all annotated biochemically confirmed targets scored at P < 5%. On average, 10.9 predictions per query compound were statistically significant (Table S5), which is in agreement with other studies that have reported 3–10 targets per drug depending on the target class (39). The CATS-based (SOM1) prediction alone yielded 41 ± 0.7%, and the MOE-based (SOM2) method yielded 41.3 ± 0.5% complete target profile predictions. To investigate the complementarity of the selected molecular representations, we compared the performance of the individual prediction methods per target. The two molecular representations performed differently for most targets with only weak correlation (Pearson r2 = 0.44; Fig. S2A). This finding supports the implementation of a prediction method that combines these multiple models and potentially benefits from their different scopes.
We studied different mathematical functions to merge the prediction scores from SOM1 and SOM2. We found that combination functions sensitive to low scores are the most accurate and yield target profile predictions that are ∼65% complete (Table S5). The observed accuracy level is in agreement with the values reported by Hopkins and colleagues, who found that a fingerprint-based model correctly predicted at least one target for 64% of their data (12). We calculated the ROC AUC values to include the global ranking of true positives as a second evaluation criterion. Both the geometric average and the minimum value suffer because predictions made by only one model are neglected, resulting in the loss of low-confidence true-positive predictions (Fig. S2B). The arithmetic average performed equivalently in early retrieval and exploited single model predictions, thereby yielding a high ROC AUC value of 0.92 (Table S5).
Off-Target Prediction for Known Drugs via SPiDER.
To prospectively probe the applicability of SPiDER, we predicted off-targets for the library of pharmacologically active compounds (LOPAC) collection of compounds (Sigma Aldrich) by mimicking the target prediction for NCEs. For this experiment, we only considered those predictions relying on COBRA reference drugs with a structural Tanimoto similarity <0.2 (Daylight Fingerprints) to the respective LOPAC query. This procedure attempted to find nontrivial, unexpected target relationships between reference drugs that are structurally dissimilar to the respective query molecule (expressed by a low structural Tanimoto similarity) and thus unrecognizable using substructure-based approaches. We investigated LOPAC queries for which SPiDER reported the most confident target predictions with the lowest Tanimoto similarity to the associated reference compounds. Among such high-confidence predictions (P < 0.001), we found monoamine oxygenase (MAO) as the top off-target prediction for the serotonin reuptake inhibitor fluoxetine. In full support of the SPiDER prediction, fluoxetine is in fact a MAO inhibitor both in vitro and in vivo (40). Similarly, we experimentally tested the top off-target prediction for fenofibrate (P < 0.001, Tanimoto similarity = 0.16 to the nearest reference compound; Fig. S2C) and confirmed the Nav1.5 ion channel (IC50 = 69 µM ± 1.2 log units; Fig. 3B). Fenofibrate presents natriuretic and cardiac remodeling effects (41, 42) and blocks basolateral KCNQ1 K+ channels (43). To the best of our knowledge, its binding to Na+ channels has not been previously reported. Nav1.5 is the principal Na+ channel isoform in cardiomyocytes, and variants of the Nav1.5-encoding gene have been linked to congenital and acquired long QT syndromes (44). Although fenofibrate demonstrated only a weak affinity to Nav1.5 in the assay, the data suggest that related chemotypes may present similar traits and liabilities.
Fig. 3.

Effects of compound 1, compound 2, and amprenavir (Amp.) on (A) bradykinin (B1) receptor (control agonist: 3 nM LysdesArg9[Leu8]-BK; EC50 = 0.2 nM), (B) NK1 receptor {control agonist: 1 nM [Sar9,Met(O2)11]-SP, EC50 = 0.4 nM}, and (C) TRPV1 (control agonist: 1 µM capsaicin, EC50 = 7 nM; n = 2, mean and SEM).
Target Identification for NCEs via SPiDER.
Having validated the SPiDER model for its ability to correctly infer off-targets despite a lack of structural similarity to the reference drugs, we predicted potential targets of de novo-designed compounds 1 and 2. Although HIVP and BACE-1 were also predicted, SPiDER ranked other targets with higher confidence (Table 1). Similar target profiles were predicted for 1 and 2 that occasionally overlapped the predictions for amprenavir. In contrast with all publicly available prediction models, the top consensus SPiDER prediction for 1 and 2 was the bradykinin B1 receptor, a G protein-coupled receptor involved in the mechanisms of inflammatory pain (45) and coronary vasomotor function (46). Being confidently predicted and practically exclusive to our approach, we tested compounds 1 and 2 for antagonistic activity toward the B1 receptor. Although compound 1 presented only modest antagonism (EC50 ∼ 100 µM; Fig. 4A), compound 2 exhibited high affinity and potent concentration-dependent B1 antagonistic activity (KB = 3.6 µM; EC50 = 17 µM; Fig. 3A). The B1 receptor was correctly predicted by SPiDER based on the structures of reference compounds 3 [B1 Ki ∼ 0.1 nM (47)], 4 [B1 Ki ∼ 0.5 nM (48)], and 5 [B1 Ki ∼ 1.2 µM (49)] (Fig. 4).
Table 1.
Drug targets predicted by SPiDER for amprenavir, compound 1, and compound 2 with P < 0.05
| Amprenavir | Compound 1 | Compound 2 |
| NK1–3 (0.021) | Cholecystokinin A/B receptor (0.017) | Opioid δ, κ, µ receptor (0.014) |
| 20S proteasome (0.023) | Bradykinin receptor B1 (0.018) | Dipeptidyl peptidase IV (0.025) |
| Nav1.7 channel (0.027) | TRPV1 and 4 (0.020) | CCR1, 3, 5 receptor (0.026) |
| Aspartic endopeptidasea,b,c (0.027) | Vasopressin receptor 1A, 1B, 2 (0.020) | FSH/GnRH receptor (0.027) |
| Protein tyrosine phosphatase 1B (0.028) | FSH and GnRH receptor (0.028) | TRPV1, 4 and 8 (0.028) |
| Cholecystokinin A/B receptors (0.029) | Cysteine endopeptidased,e (0.028) | Serine endopeptidase and proteasef,g,h (0.031) |
| α4β1 integrin (0.029) | Serine endopeptidasef,g (0.033) | Bradykinin receptor B1 (0.031) |
| Oxytocin receptor (0.029) | Aspartic endopeptidasec (0.033) | Aspartic endopeptidasec (0.034) |
| Serine endopeptidasef (0.029) | NK1–3 (0.038) | NK1 and NK2 (0.035) |
| Tyrosine kinasei (0.033) | Oxytocin receptor (0.042) | Urokinase plasminogen activator surface receptor (0.036) |
| Cysteine endopeptidased,j (0.034) | Na+v1.7 channel (0.048) | Histone deacetylase (0.040) |
| Bombesin receptor 1/2 (0.039) | Capsid assembly inhibitor (0.042) | |
| Serine threonine kinasek (0.043) | ||
| α4β1 integrin (0.045) | ||
| RasFTase (0.048) |
P values are in parentheses.
Includes cathepsin D, HIV protease, Pol polyprotein, and SIV protease.
Includes endothiapepsin and saccharopepsin (proteinase A).
Includes plasmepsins, renin, and secretase (Abeta secretion).
Includes calpain, cathepsins B, K, L, and S, cruzain, falcipain, picornavirus 3C-like protease, and virus 3C protease.
Includes caspase 1.
Includes elastase, factor Xa, and thrombin.
Includes β-tryptase, chymase, factor IXa, trypsin, tryptase, and urokinase.
Includes factor VIIa.
Includes c-Src, LCK, and VEGFR1.
Includes papain.
Includes CDK, NF-κB, MAPK14, NF-protein kinase B and C, and rho kinase.
Fig. 4.

Reference B1 receptor antagonists. Compound 3 is the CATS reference for both 1 and 2. Compound 4 is the MOE reference for 1, and 5 is the MOE reference for 2.
SPiDER further suggested that neurokinin 1 (NK1) and vanilloid 1 (TRPV1) were receptors targeted by compounds 1 and 2 with favorable P values (Table 1). Again, these represent clinically relevant drug targets exclusively predicted by SPiDER for both NCEs under investigation. In full agreement with the computational analysis, we observed concentration-dependent effects for both compounds. NK1 receptor antagonism (KB = 15 µM, EC50 = 100 µM ± 0.7 log units; Fig. 3B) and agonism on the TRPV1 ion channel (EC50 = 76 µM ± 0.7 log units; Fig. 3C) were measured for compound 2. To probe whether any of these activities were inherited from the de novo design template, we investigated the effect of amprenavir on the B1, NK1, and TRPV1 receptors. As correctly recognized by SPiDER, amprenavir did not exhibit activity against the B1 or TRPV1 receptor up to 100 µM. Its weak NK1 receptor antagonism (EC50 > 100 µM) also agrees with the prediction (P = 0.021). Taken together, we successfully and efficiently used SPiDER to determine subtle functional bioactivity traits for both de novo-designed molecular frameworks and their drug template. For further assessment of SPiDER for NCE target prediction, we provide a public version of the model on our webserver (http://modlab-cadd.ethz.ch/software/).
Conclusions.
Structural genomics and systems pharmacology greatly benefit from innovations in molecular informatics (50, 51). We implemented and experimentally validated a unique method for ligand-based target prediction that extends the capabilities of related approaches. Our software combines multiple representations of small molecules in a conservative jury approach, thereby reducing false-positive predictions in the high confidence range. The binding association of de novo designs 1 and 2 to the B1 receptor indicates the explorative nature of the SPiDER approach. Because SPiDER determined all of the correct targets for two-thirds of the test data in retrospective studies, the method is expected to perform at least equivalently to other computational target prediction tools (12, 24, 26, 27). The results of our study demonstrate how the approach can be included in de novo drug design to obtain target-binding confidence in the proposed NCEs or to redirect stalled drug discovery projects.
Materials and Methods
De Novo Design.
DOGS (19) was used for the de novo molecular design with amprenavir as the template, 25,144 molecular building blocks, and 83 coupling reactions (52) (inSili.com LLC). The similarities between the designs and the template were computed using the iterative similarity optimal assignment kernel (ISOAK) method on reduced molecular graph representations (5). Nine independent runs were performed with 200 randomly selected start fragments (ISOAK α values ranging from 0.1 to 0.9). Scaffold occurrences in the set of designed molecules were determined by comparing the canonical simplified molecular input line entry system (SMILES) representations of extracted scaffolds on the KNIME platform v2.6.0 (53) using RDKit nodes (www.rdkit.org) for scaffold extraction and KNIME-native nodes to determine their frequency.
Reference Compounds.
A manually curated collection of bioactive reference compounds (COBRA, v12.6, inSili.com LLC) (33) containing 12,661 unique pharmacologically active molecules annotated with one or more of 251 biomolecular targets and specific subtype activities was used for training, retrospective, and prospective evaluations of the SPiDER model.
Data Preparation and Molecular Representation.
Compounds were provided as SMILES and processed in KNIME v2.6.0 (53) with the MOE “wash” function (2011.10; Chemical Computing Group) using the options “disconnect salts,” “remove ion pairs,” “deprotonate strong acids,” “remove minor component,” “protonate strong bases,” and “add hydrogen.” The 210-dimensional CATS vectors were calculated with an in-house software tool with a maximal correlation distance of 10 bonds and pharmacophoic feature type-sensitive descriptor scaling (10). The following 186 descriptors from MOE were calculated using the MOE “QSAR descriptors” function (MOE 2011.10): simple descriptors, chi indices, BCUT descriptors, PEOE descriptors, Q_charge descriptors, Lipinski rule of fives, Kier indices, GCUT descriptors, SlogP descriptors, VSA descriptors, SMR descriptors, and “other 2D descriptors.” The raw descriptor values were standardized according to the training data distribution to prevent bias due to different value ranges.
SPiDER Prediction.
The respective query molecule was projected onto the two SOMs trained with the corresponding molecular representation of the COBRA compound set according to the winner-takes-all rule (54) using in-house Java code. The projection defined the relevant cluster of reference compounds (the query's local domain). To score a target C, only the set CΘ of coclustered reference compounds labeled to bind this target C was considered. Euclidean distances  from each reference ligand x in CΘ to the query molecule y were calculated in descriptor space and transformed into P values P(D ≤ dxy). The empirical probability distribution P(D) of pairwise distances between all reference compounds that are annotated to not bind to the same target was precomputed to yield a distribution of distances between the intertarget compounds as false-positive error estimates for assuming that two molecules at a certain distance have a common biomolecular target. The P values were averaged for all ligands in CΘ, as motivated by the false discovery rate calculations for the multiple hypothesis testing of individual pairwise comparisons (54). The score S for target C is computed as
from each reference ligand x in CΘ to the query molecule y were calculated in descriptor space and transformed into P values P(D ≤ dxy). The empirical probability distribution P(D) of pairwise distances between all reference compounds that are annotated to not bind to the same target was precomputed to yield a distribution of distances between the intertarget compounds as false-positive error estimates for assuming that two molecules at a certain distance have a common biomolecular target. The P values were averaged for all ligands in CΘ, as motivated by the false discovery rate calculations for the multiple hypothesis testing of individual pairwise comparisons (54). The score S for target C is computed as 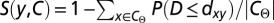 , in which a value of S close to 1 indicates that the coclustered reference ligands are close to the query compound in descriptor space. Individual scores were obtained for both molecular representations (SSOM1, SSOM2) (Fig. 2A) and were combined as an arithmetic average:
, in which a value of S close to 1 indicates that the coclustered reference ligands are close to the query compound in descriptor space. Individual scores were obtained for both molecular representations (SSOM1, SSOM2) (Fig. 2A) and were combined as an arithmetic average: 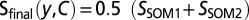 . Predicting the targets for all reference compounds generated a distribution of Sfinal, from which P values for new predictions were computed based on the likelihood of a certain confidence in the prediction of known drug targets. Note that these P values are relatively high due to the likelihood of a more realistic null hypothesis in our background distribution compared with other approaches that report the P value as a measure of significance, such as the random shuffling in sequence alignment (55) or the random assembly of ligand sets in the target prediction by SEA (24).
. Predicting the targets for all reference compounds generated a distribution of Sfinal, from which P values for new predictions were computed based on the likelihood of a certain confidence in the prediction of known drug targets. Note that these P values are relatively high due to the likelihood of a more realistic null hypothesis in our background distribution compared with other approaches that report the P value as a measure of significance, such as the random shuffling in sequence alignment (55) or the random assembly of ligand sets in the target prediction by SEA (24).
Retrospective Evaluation.
Stratified 10-fold cross-validation was performed (38). For each fold, the model fitting included standardizing the descriptors according to the training fold, retraining the SOMs, and estimating the background distance distributions. A prediction was considered successful if all annotated targets of a test compound were predicted with a confidence score of P < 5%. Scores for all compounds from the cross-validations were merged to calculate the ROC curves and to integrate them numerically with the trapezoid method for AUC calculation using in-house Java code.
Synthesis of (2R,3R)-(R)-1-Tosylpyrrolidin-3-yl 3-((tert-Butoxycarbonyl)Amino)-2-Hydroxy-4-Phenylbutanoate (Compound 1).
(2R,3R)-3-(Boc-amino)-2-hydroxy-4-phenylbutyric acid (1.0 molar equivalent) and (R)-1-tosylpyrrolidin-3-ol (2.0 molar equivalent) were dissolved in dry tetrahydrofuran (18 mL/mmol carboxylic acid). Triphenylphosphine (0.8 molar equivalent) and diethyl azodicarboxylate (0.8 molar equivalent) were added to the solution. The mixture was heated under microwaves (150 W) for 30 min. The crude mixture was washed with water and purified via preparative HPLC using a gradient of 50–75% (acetonitrile: H2O + 0.1% trifluoroacetic acid in each solvent) run over 16 min. White solid, mp = 106–107 °C, 42% (Fig. S3A).
Synthesis of (2R,3R)-(R)-1-Tosylpyrrolidin-3-yl 3-Amino-2-Hydroxy-4-Phenylbutanoate (Compound 2).
Compound 1 (0.04 mmol) was cooled to 0 °C under nitrogen. A 4-N solution of HCl in dioxane (1 mL) was added, and the suspension was stirred at room temperature for 15 min. The solvent was evaporated under a nitrogen stream, and the crude product was purified via preparative HPLC (ACN/H2O + 0.1% formic acid – 30–95% ACN gradient run over 16 min) to afford 2. White oil, 55% (Fig. S3B).
Supplementary Material
Acknowledgments
We thank Dr. Nikolay Todoroff for support in setting up the trial version of the SPiDER tool. This work was supported by the Swiss Federal Institute of Technology Zurich, Deutsche Forschungsgemeinschaft Grant FOR1406TP4, and the OPO Foundation, Zurich. The Chemical Computing Group Inc. (Montreal, Canada) provided a research license for the Molecular Operating Environment. The inSili.com LLC (Zurich, Switzerland) contributed research licenses for the software tools DOGS, molmap, and the COBRA database.
Footnotes
Conflict of interest statement: P.S. and G.S. are shareholders of inSili.com LLC, Zurich. G.S. is a shareholder of AlloCyte Pharmaceuticals Ltd., Basel, and a scientific consultant of the pharmaceutical industry.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320001111/-/DCSupplemental.
References
- 1.Bennani YL. Drug discovery in the next decade: Innovation needed ASAP. Drug Discov Today. 2012;17(Suppl):S31–S44. doi: 10.1016/j.drudis.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Johnson MA, Maggiora GM. Concepts and Applications of Molecular Similarity. New York: Wiley; 1990. [Google Scholar]
- 3.Schneider G. De Novo Molecular Design. Weinheim, Germany: Wiley-VCH; 2013. [Google Scholar]
- 4.Hartenfeller M, Schneider G. De novo drug design. Methods Mol Biol. 2011;672:299–323. doi: 10.1007/978-1-60761-839-3_12. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues T, et al. De novo design and optimization of Aurora A kinase inhibitors. Chem Sci. 2013;4(3):1229–1233. [Google Scholar]
- 6.Spänkuch B, et al. Drugs by numbers: Reaction-driven de novo design of potent and selective anticancer leads. Angew Chem Int Ed Engl. 2013;52(17):4676–4681. doi: 10.1002/anie.201206897. [DOI] [PubMed] [Google Scholar]
- 7.Besnard J, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492(7428):215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues T, et al. Steering target selectivity and potency by fragment-based de novo drug design. Angew Chem Int Ed Engl. 2013;52(38):10006–10009. doi: 10.1002/anie.201304847. [DOI] [PubMed] [Google Scholar]
- 9.Kohonen T. Self-Organizing Maps. New York: Springer; 2001. [Google Scholar]
- 10.Reutlinger M, et al. Chemically advanced template search (CATS) for scaffold-hopping and prospective target prediction for 'orphan' molecules. Molec Inf. 2013;32(2):133–138. doi: 10.1002/minf.201200141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 12.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24(7):805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 13.Tatonetti NP, Liu T, Altman RB. Predicting drug side-effects by chemical systems biology. Genome Biol. 2009;10(9):238. doi: 10.1186/gb-2009-10-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16(1):127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Hopkins AL. Network pharmacology. Nat Biotechnol. 2007;25(10):1110–1111. doi: 10.1038/nbt1007-1110. [DOI] [PubMed] [Google Scholar]
- 16.Antolín AA, Jalencas X, Yélamos J, Mestres J. Identification of pim kinases as novel targets for PJ34 with confounding effects in PARP biology. ACS Chem Biol. 2012;7(12):1962–1967. doi: 10.1021/cb300317y. [DOI] [PubMed] [Google Scholar]
- 17.Lounkine E, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486(7403):361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 19.Hartenfeller M, et al. DOGS: Reaction-driven de novo design of bioactive compounds. PLOS Comput Biol. 2012;8(2):e1002380. doi: 10.1371/journal.pcbi.1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter DJT, Hanlon MH, Carter LH, 3rd, Danger DP, Furfine ES. Effectors of HIV-1 protease peptidolytic activity. Biochemistry. 2001;40(37):11131–11139. doi: 10.1021/bi010418p. [DOI] [PubMed] [Google Scholar]
- 21.Bemis GW, Murcko MA. The properties of known drugs. 1. Molecular frameworks. J Med Chem. 1996;39(15):2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 22.Rose PW, et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013;41(Database issue):D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim EE, et al. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117(3):1181–1182. [Google Scholar]
- 24.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25(2):197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 25.Chen B, Ding Y, Wild DJ. Assessing drug target association using semantic linked data. PLOS Comput Biol. 2012;8(7):e1002574. doi: 10.1371/journal.pcbi.1002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagunin A, Stepanchikova A, Filimonov D, Poroikov V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics. 2000;16(8):747–748. doi: 10.1093/bioinformatics/16.8.747. [DOI] [PubMed] [Google Scholar]
- 27.Dunkel M, Günther S, Ahmed J, Wittig B, Preissner R. SuperPred: Drug classification and target prediction. Nucleic Acids Res. 2008;36(Web Server issue):W55–W59. doi: 10.1093/nar/gkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGregor MJ, Luo Z, Jiang X. Virtual screening in drug discovery. In: Huang Z, editor. Drug Discovery Research: New Frontiers in the Post-Genomic Era. New York: Wiley; 2007. pp. 63–88. [Google Scholar]
- 29.Klenner A, Hartenfeller M, Schneider P, Schneider G. 'Fuzziness' in pharmacophore-based virtual screening and de novo design. Drug Discov Today Technol. 2010;7(4):e237–e244. doi: 10.1016/j.ddtec.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Schneider G. De novo design: Hop(p)ing against hope. Drug Discov Today. 2013;10(4):e453–e460. doi: 10.1016/j.ddtec.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Schneider P, Tanrikulu Y, Schneider G. Self-organizing maps in drug discovery: Compound library design, scaffold-hopping, repurposing. Curr Med Chem. 2009;16(3):258–266. doi: 10.2174/092986709787002655. [DOI] [PubMed] [Google Scholar]
- 32.Zupan J, Gasteiger J. Neural Networks in Chemistry and Drug Design. Weinheim, Germany: Wiley-VCH; 1999. [Google Scholar]
- 33.Schneider P, Schneider G. Collection of bioactive reference compounds for focused library design. QSAR Comb Sci. 2003;22(7):713–718. [Google Scholar]
- 34.Schneider P, Schneider G. Navigation in chemical space: Ligand-based design of focused compound libraries. In: Kubinyi H, Müller G, editors. Chemogenomics in Drug Discovery. Weinheim, Germany: Wiley-VCH; 2004. pp. 341–376. [Google Scholar]
- 35.Schneider G, Tanrikulu Y, Schneider P. Self-organizing molecular fingerprints: A ligand-based view on drug-like chemical space and off-target prediction. Future Med Chem. 2009;1(1):213–218. doi: 10.4155/fmc.09.11. [DOI] [PubMed] [Google Scholar]
- 36.Rupp M, Schneider P, Schneider G. Distance phenomena in high-dimensional chemical descriptor spaces: Consequences for similarity-based approaches. J Comput Chem. 2009;30(14):2285–2296. doi: 10.1002/jcc.21218. [DOI] [PubMed] [Google Scholar]
- 37.Schneider G, Neidhart W, Giller T, Schmid G. 'Scaffold-hopping' by topological pharmacophore search: A contribution to virtua screening. Angew Chem Int Edu. 1999;38(19):2894–2896. [PubMed] [Google Scholar]
- 38.Tropsha A. Best practices for QSAR model development, validation, and exploitation. Molec Inf. 2010;29(6-7):476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 39.Antolín AA, Mestres J. Knowledge base for nuclear receptor drug discovery. In: Botana LM, Loza MI, editors. Therapeutic Targets: Modulation, Inhibition, and Activation. New York: Wiley; 2012. pp. 309–326. [Google Scholar]
- 40.Holt A, Baker GB. Inhibition of rat brain monoamine oxidase enzymes by fluoxetine and norfluoxetine. Naunyn Schmiedebergs Arch Pharmacol. 1996;354(1):17–24. doi: 10.1007/BF00168701. [DOI] [PubMed] [Google Scholar]
- 41.Shatara RK, Quest DW, Wilson TW. Fenofibrate lowers blood pressure in two genetic models of hypertension. Can J Physiol Pharmacol. 2000;78(5):367–371. [PubMed] [Google Scholar]
- 42.Lebrasseur NK, et al. Effects of fenofibrate on cardiac remodeling in aldosterone-induced hypertension. Hypertension. 2007;50(3):489–496. doi: 10.1161/HYPERTENSIONAHA.107.092403. [DOI] [PubMed] [Google Scholar]
- 43.Bajwa PJ, et al. Fenofibrate inhibits intestinal Cl- secretion by blocking basolateral KCNQ1 K+ channels. Am J Physiol Gastrointest Liver Physiol. 2007;293(6):G1288–G1299. doi: 10.1152/ajpgi.00234.2007. [DOI] [PubMed] [Google Scholar]
- 44.Abriel H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007;76(3):381–389. doi: 10.1016/j.cardiores.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 45.Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429(1-3):161–176. doi: 10.1016/s0014-2999(01)01318-8. [DOI] [PubMed] [Google Scholar]
- 46.Aptecar E, et al. Coronary vasomotor response to the selective B1-kinin-receptor agonist Des-Arg9-bradykinin in humans. J Heart Lung Transplant. 2006;25(2):187–194. doi: 10.1016/j.healun.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 47.Ferrari B, et al. 1997. Novel N-(arylsuphonyl)amino acid derivatives having bradykinin receptor affinity, Patent WO 9725315.
- 48.Hart T, Ritchie TJ. 2002. Sulfonamide derivatives, Patent WO 02092556.
- 49.Feng DM, et al. 2,3-Diaminopyridine as a platform for designing structurally unique nonpeptide bradykinin B1 receptor antagonists. Bioorg Med Chem Lett. 2005;15(9):2385–2388. doi: 10.1016/j.bmcl.2005.02.077. [DOI] [PubMed] [Google Scholar]
- 50.Taboureau O, Baell JB, Fernández-Recio J, Villoutreix BO. Established and emerging trends in computational drug discovery in the structural genomics era. Chem Biol. 2012;19(1):29–41. doi: 10.1016/j.chembiol.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Futamura Y, et al. Morphobase, an encyclopedic cell morphology database, and its use for drug target identification. Chem Biol. 2012;19(12):1620–1630. doi: 10.1016/j.chembiol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Hartenfeller M, et al. A collection of robust organic synthesis reactions for in silico molecule design. J Chem Inf Model. 2011;51(12):3093–3098. doi: 10.1021/ci200379p. [DOI] [PubMed] [Google Scholar]
- 53.Berthold MR, et al. KNIME: The Konstanz information miner. In: Preisach C, Burkhardt H, Schmidt-Thieme L, Decker R, editors. Data Analysis, Machine Learning and Applications. Berlin: Springer; 2008. pp. 319–326. [Google Scholar]
- 54.Storey JD. A direct approach to false discovery rates. J R Stat Soc B. 2002;64(3):479–498. [Google Scholar]
- 55.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Schneider G, Wrede P. Artificial neural networks for computer-based molecular design. Prog Biophys Mol Biol. 1998;70(3):175–222. doi: 10.1016/s0079-6107(98)00026-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.