

Significance
Stormorken syndrome is a rare autosomal-dominant genetic condition characterized by congenital miosis, bleeding diathesis, thrombocytopenia, and proximal muscle weakness. Other manifestations include functional or anatomical asplenia, ichthyosis, headaches, and dyslexia. A milder form of Stormorken syndrome is associated with muscle weakness and congenital miosis, but without hematologic abnormalities. Here we identify the gene mutations responsible for these syndromes and show that both conditions are caused by the hyperactivation of the Ca2+ release-activated Ca2+ (CRAC) channel. These findings contrast the pathologies associated with loss or diminished function of the CRAC channel, provide new molecular insights of the function of the CRAC channel, and suggest new approaches to combat these conditions by blocking CRAC channel activity.
Keywords: human genetics, calcium signaling
Abstract
Signaling through the store-operated Ca2+ release-activated Ca2+ (CRAC) channel regulates critical cellular functions, including gene expression, cell growth and differentiation, and Ca2+ homeostasis. Loss-of-function mutations in the CRAC channel pore-forming protein ORAI1 or the Ca2+ sensing protein stromal interaction molecule 1 (STIM1) result in severe immune dysfunction and nonprogressive myopathy. Here, we identify gain-of-function mutations in the cytoplasmic domain of STIM1 (p.R304W) associated with thrombocytopenia, bleeding diathesis, miosis, and tubular myopathy in patients with Stormorken syndrome, and in ORAI1 (p.P245L), associated with a Stormorken-like syndrome of congenital miosis and tubular aggregate myopathy but without hematological abnormalities. Heterologous expression of STIM1 p.R304W results in constitutive activation of the CRAC channel in vitro, and spontaneous bleeding accompanied by reduced numbers of thrombocytes in zebrafish embryos, recapitulating key aspects of Stormorken syndrome. p.P245L in ORAI1 does not make a constitutively active CRAC channel, but suppresses the slow Ca2+-dependent inactivation of the CRAC channel, thus also functioning as a gain-of-function mutation. These data expand our understanding of the phenotypic spectrum of dysregulated CRAC channel signaling, advance our knowledge of the molecular function of the CRAC channel, and suggest new therapies aiming at attenuating store-operated Ca2+ entry in the treatment of patients with Stormorken syndrome and related pathologic conditions.
Ca2+ influx in response to the depletion of intracellular Ca2+ stores, or store-operated Ca2+ entry, constitutes one of the major routes of Ca2+ entry in all animal cells (1). Under physiological conditions, Ca2+ influx is activated in response to numerous G protein-coupled receptors and receptor tyrosine kinases signaling via inositol-1,4,5-trisphosphate as a second messenger (2). Store-operated Ca2+ entry is mediated primarily by the Ca2+ release-activated Ca2+ (CRAC) channel (3), which consists of the pore-forming subunits ORAI1–3 (or CRAC modulators 1–3) and Ca2+ sensors, STIM1 and STIM2 (4–7). STIM proteins reside in the membrane of endoplasmic reticulum (ER), whereas ORAI proteins reside in the plasma membrane. STIM1 is a single transmembrane-spanning protein (8–12) that, in resting cells, exists as a dimer that binds Ca2+ through two EF hand-containing domains located in the ER lumen (13). Depletion of Ca2+ in the ER induces a series of molecular events in the conformation and localization of STIM1, initiated by the formation of higher-order oligomers, protein unfolding, and accumulation at discrete sites in the cell where the ER membrane is in close proximity to the plasma membrane (11, 13–16). In these sites, STIM1 binds to the cytosolic C and N termini of ORAI1 (17, 18), resulting in channel activation and generation of a highly Ca2+-selective CRAC current, or ICRAC (3, 19, 20). ICRAC is responsible not only for restoring cytosolic and ER Ca2+ concentration, thus maintaining the cell in a Ca2+ signaling-competent stage (1), but also for many cellular functions such as regulation of gene expression, exocytosis, proliferation, and apoptosis (1).
Consistent with a fundamental role of the CRAC channel in cell signaling, loss-of-function mutations in STIM1 or ORAI1 lead to immune deficiency and nonprogressive myopathy (21–23). However, evidence that gain-of-function mutations in STIM1 and ORAI1 can affect human health is only recently starting to emerge. It was shown that mutations in the domain of STIM1 that binds Ca2+ (EF hand domain) in resting conditions are associated with nonsyndromic myopathy with tubular aggregates (24). Functional studies demonstrated that these mutations cause hyperactivation of the CRAC channel (24). However, it remains unknown whether myopathy with tubular aggregates is caused by the increased activity of the CRAC channel, increased activity of another Ca2+ channel using STIM1 as a sensor (25), or a function of STIM1 that is unrelated to Ca2+ signaling, as STIM1 can function independently of ORAI1 (26-28).
Stormorken syndrome [Mendelian Inheritance in Man (MIM) 185070] is a rare autosomal-dominant condition with a constellation of symptoms, including congenital miosis, bleeding diathesis, thrombocytopenia, functional (or anatomical) asplenia, and proximal muscle weakness (29). Other manifestations include ichthyosis, headaches, and dyslexia (30). Patients typically display increased creatine kinase (CK) levels and histologic evidence of myopathy with tubular aggregates (30, 31). Here, we show that Stormorken syndrome is caused by an activating mutation in STIM1. We also identify a mutation in the STIM1-interacting molecule, ORAI1, in a Stormorken-like syndrome that presented with miosis and tubular myopathy. Functional analyses reveal that both mutations enhance the activity of the CRAC channel, but by different molecular mechanisms. These data expand the phenotypic spectrum of activating mutations in the CRAC channels from myopathy with tubular aggregates to miosis, bleeding diathesis, thrombocytopenia, asplenia, ichthyosis, headaches, and dyslexia.
Results
To define the molecular basis of Stormorken syndrome, we performed whole-exome sequencing on the DNA obtained from a child with a symptom complex consistent with Stormorken syndrome (Table S1). We identified a heterozygous frame-shift insertion in Homo sapiens keratin-associated protein 1-1 (KRTAP1-1) (NM_030967: c.144_145insGC, p.S48fs) and two heterozygous nonsynonymous mutations, one in RERE arginine-glutamic acid dipeptide (RE) repeats (RERE) (NM_012102: c.G4046A, p.R1349Q) and one in STIM1 (NM_003156: c.910C > T, p.R304W), present only in the proband (patient 1, Table S1). Each of these mutations was validated by Sanger sequencing. Next, we evaluated these mutations by Sanger sequencing in a second, unrelated patient with Stormorken syndrome (patient 2; Fig. 1B and Table S1). Although the KRTAP1-1 and RERE mutations were not present in this subject, we confirmed the presence of the identical c.910C>T transition in STIM1 (Fig. 1C and Fig. S1). These results led us to consider the p.R304W in STIM1 as the molecular lesion in Stormorken syndrome.
Fig. 1.

p.R304W in STIM1 causes Stormorken syndrome. (A and B) Pedigrees of the two families affected by Stormorken syndrome. Arrows indicate the probands in each family. Asterisk indicates individuals whose whole exome was sequenced. (C) Diagram of human STIM1_R304W and sequence alignment of the Cα2 of coiled coil domain 1 (CC1) of WT human, mouse, and zebrafish STIM1 and STIM2. Cα1–3, α-helix 1–3 in CC1; EF, EF hand; ID, inhibitory domain; TM, transmembrane domain.
The presence of the same mutant allele in two unrelated but phenotypically similar patients argues for a causal role of the p.R304W mutation in Stormorken syndrome. Given the known role of STIM1 in the store-operated Ca2+ entry pathway, we measured store-operated Ca2+ influx and ICRAC in immortalized lymphocytes derived from our patient with Stormorken syndrome and a healthy individual. Lymphocytes were used instead of platelets for several reasons: (i) platelets are not suitable for electrophysiology because of their small size, (ii) platelets should have been already preactivated/exhausted, and (iii) ICRAC has been extensively studied in B lymphocytes (32) and, therefore, we could test for a direct effect of the Stormorken mutation on native CRAC channel.
Fig. 2A shows that lymphocytes derived from the patient with Stormorken syndrome displayed larger Ca2+ influx in response to store depletion induced by thapsigargin (TG). Similar results were obtained in primary skin fibroblasts obtained from the same patient (Fig. S2). Next, we recorded native ICRAC in lymphocytes from the Stormorken and healthy individuals. ICRAC was induced by 10mM ethylene glycol tetra-acetic acid (EGTA) added in the recording pipette. Fig. 2 B and C show constitutive activation of ICRAC and an overall larger size in patient cells compared with normal cells. In addition, fast Ca2+-dependent inactivation (CDI) was suppressed in cells from the patient with Stormorken syndrome (Fig. 2D). These data suggested that STIM1_R304W acted as an activating mutation in terms of CRAC channel function.
Fig. 2.

Store-operated Ca2+ entry and ICRAC is enhanced in patients with Stormorken syndrome. (A) Store-operated Ca2+ entry in lymphocytes obtained from an unaffected individual (control; black, n = 127 cells) or a patient with Stormorken syndrome (patient; red, n = 288 cells) using single-cell Ca2+ imaging. Cells were loaded with 2 µM Fura-2/AM and placed in an extracellular solution (ECS) containing 0 mM Ca2+. Stores were depleted with 2 µM TG and Ca2+ influx was stimulated by the addition of 2 mM Ca2+ in the ECS. (B) Time course of ICRAC in lymphocytes from a control subject (black, n = 11 cells) and patient with Stormorken syndrome (red, n = 16 cells) induced by 10 mM EGTA in the recording pipette and blocked by 20 µM La3+ in the ECS. (C) Current–voltage curves taken at 50 s or 400 s after “break-in” in control cells and cells from a patient with Stormorken syndrome. (D) Quantification of inactivation as determined by the ratio (R195ms) of the peak current at the beginning of a hyperpolarizing pulse (I0) to tail current at the end of the pulse (I195) in cells from a healthy individual (black, n = 11 cells) and a patient with Stormorken syndrome (red, n = 16 cells). Representative step currents generated from hyperpolarizing pulses at the indicated test potentials at 400 s following break-in in a control subject and a patient (Stormorken) cell. Duration of the pulse was 200 ms.
Next, we sought to investigate the molecular mechanism by which STIM1 p.R304W influences store-operated Ca2+ entry through the CRAC channel. To minimize contribution from the WT allele, we used a heterologous system in which HEK293 cells were transfected with STIM1_R304W and ORAI1 and whole-cell currents were measured by using electrophysiology (Fig. 3A). Cells transfected with WT STIM1 (STIM1_WT) and ORAI1 were used as controls. In both cases, depletion of intracellular Ca2+ stores was induced by 10 mM EGTA added in the recording pipette (Fig. 3A). Cells transfected with STIM1_WT showed time-dependent activation and inactivation of ICRAC (Fig. 3A, black). Transfection with STIM1_R304W resulted in stable, maximally activated, basal currents (Fig. 3A, red), displaying the typical current–voltage relationship of ICRAC (Fig. 3 B and C). The effect of STIM1_R304W on ICRAC was suppressed when STIM1_WT was coexpressed with STIM1_R304W at equal amounts (OS_ST/R304W; Fig. 3A, burgundy). The stimulatory effect of STIM1_R304W on ICRAC was also seen when store depletion was induced by 10 mM 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), which is a faster Ca2+ chelator than EGTA (Fig. S3). In sum, these results showed that, under overexpression conditions, STIM1 p.R304W caused constitutive activation of the CRAC channel. Consistently, STIM1_R304W tagged with YFP at its C terminus (STIM_R304W-YFP) showed a characteristic accumulation in preformed puncta in resting cells, in contrast to STIM1_WT-YFP that showed uniform expression in the ER (Fig. S4).
Fig. 3.

STIM1_R304W enhances CRAC channel activity in transfected cells. (A) Time course of ICRAC in HEK 293 cells transiently cotransfected with ORAI1 plus STIM1_WT (OS_WT; black, n = 5), ORAI1 plus STIM1_R304W (OS_R304W; red, n = 7), ORAI1 plus STIM1_K(384-6)Q [OS_K(384-6)Q; dark yellow, n = 5], ORAI1 plus STIM1_R304W_K(384-6)Q [OS_R304W_K(384-6)Q; blue, n = 6], ORAI1 plus STIM1_D76A (OS_D76A; green, n = 7), or ORAI1 plus a 1:1 mix of STIM1_WT and STIM1_R304W (OS_WT/R304W; burgundy, n = 8) induced by 10 mM EGTA and suppressed by 20 µM La3+. (B and C) Current–voltage curves taken at 50 (B) or 200 s (C) after break-in in cells transfected with plasmid combinations shown in A. (D) Representative step currents generated from hyperpolarizing pulses at the indicated test potentials at 200 s following break-in in cells transfected with OS_WT, OS_R304W, OS_D76A, or OS_WT/R304W in 10 mM extracellular Ca2+. Duration of the pulse was 200 ms. (E) Quantification of inactivation as determined by the ratio (R195ms) of the peak current at the beginning of the pulse (I0) to tail current at the end of the pulse (I195) in cells transfected with OS_WT (n = 6), OS_R304W (n = 7), D76A (n = 8), or OS_WT/R304W (n = 8).
To obtain further molecular insight of the mechanism by which STIM1_R304W affected ICRAC, we compared and contrasted its function to known loss- and gain-of-function mutant forms of STIM1. STIM1_K(384-6)Q is a triple mutant of STIM1 showing diminished interaction with WT ORAI1 through the CRAC activation domain (CAD) or STIM1 ORAI1 activating region (SOAR) of STIM1 (33). Consistently, transfection of this mutant together with ORAI1 led to slowly activating ICRAC of a significantly smaller size compared with ICRAC induced by STIM1_WT (Fig. 3A, dark yellow). Introduction of the R304W mutation to STIM1_K(384-6)Q [STIM1_R304W_K(384-6)Q] led to constitutively active ICRAC but of a size similar to the size of ICRAC induced by STIM1_K(384-6)Q (Fig. 3A, blue). These data suggest that STIM1_R304W requires interaction with ORAI1 for the generation of ICRAC. This is consistent with the observation that p.R304W is not located within the CAD/SOAR or within the inhibitory helix (residues 310–340) that binds to and obstructs CAD/SOAR in resting conditions (34). Interestingly, R304 was predicted to make direct contacts with E318 and Q314, both located within an inhibitory helix (35), suggesting that R304W may indirectly affect interactions between the inhibitory helix and CAD/SOAR in resting conditions.
Fig. 3A shows that p.R304W causes constitutive activation of the CRAC channel. However, constitutive activation can also occur by mutations in the EF hand of STIM1 (p.H72Q, p.D84G, p.H109R, or p.H109N), identified recently in nonsyndromic forms of tubular-aggregate myopathy (TAM; MIM 160565) (24). These data suggest that, although both types of mutations (in CC1 and EF hand) can be activating, the effect of mutations within CC1 such as p.R304W might be more severe compared with mutations affecting the EF hand. Therefore, we compared directly the effect of STIM1_R304W and STIM1_D76A (36, 37), a well-characterized EF hand mutant, on ICRAC. Although both mutants resulted in constitutively active ICRAC of similar sizes, they differed markedly in the extent of their fast CDI (Fig. 3 D and E). STIM1_R304W showed no fast CDI, whereas STIM1_D76A showed fast CDI indistinguishable from that of STIM1_WT in 10 mM of extracellular Ca2+ (Fig. 3 D and E). Thus, these results raise the possibility that suppression of the fast CDI could account for the broader phenotypic spectrum of p.R304W in patients with Stormorken syndrome.
To probe further the pathway by which mutations in STIM1 could cause some or all of the pathologic conditions associated with Stormorken syndrome, we searched for mutations associated with Stormorken-like syndromes. In 2004, a Stormorken-like phenotype of proximal muscle weakness and miosis was described in two families, related through distant ancestors (38). Similar to Stormorken syndrome, the muscle weakness in these patients was associated with elevated CK and diagnosed histologically as a myopathy with tubular aggregates (38). However, in contrast to patients with Stormorken syndrome, these individuals lacked evidence of thrombocytopenia, bleeding diathesis, and asplenia (Table S1). Sanger sequencing failed to identify the p.R304W mutation in STIM1 in two affected siblings from this kindred (Fig. 4A). We therefore performed whole-exome sequencing and identified 371 heterozygous variants in 336 genes shared by the two affected siblings. Focusing on genes known to be associated with store-operated Ca2+ entry (39), we identified a mutation in ORAI1 (c.734C>T, p.P245L; Fig. S5), present in both patients. Sanger sequencing in the remaining available members of the family confirmed that the mutation segregated with all seven affected tested but was absent from two unaffected individuals (Fig. 4A). P245 is located within the fourth transmembrane helix (M4) of ORAI1 and is conserved from flies to humans (Fig. 4B). These results suggest that the p.P245L mutation in ORAI1 is the molecular cause of this Stormorken-like phenotype in this family and represent, to our knowledge, the first example of a mutation in ORAI1 associated with a tubular myopathy.
Fig. 4.

p.245L in ORAI1 causes a Stormorken-like syndrome with aggregate tubular myopathy. (A) Pedigree of the two branches of an extended family with common ancestry in mid-19th century with Stormorken-like syndrome. Branch 1 [Left branch (below generation II)] was described by Shahrizaila et al. (38). The disorder follows an autosomal-dominant inheritance pattern. The arrows indicate the proband in each branch of this family (patient 1, left arrow; patient 2, right arrow). Asterisk indicates individuals whose whole exome was sequenced. (B) Cartoon showing the location of P245 in transmembrane segment M4, and a sequence alignment of the M4 α-helix of Drosophila ORAI1 and human ORAI1-3.
We hypothesized that this mutation may also lead to enhanced CRAC channel activity. To test this hypothesis, we cotransfected STIM1_WT and ORAI1_WT or ORAI1_P245L and measured EGTA-induced ICRAC in HEK293 cells. Fig. 5A and Fig. S6 A and B show that ORAI1_P245L produced ICRAC with similar activation kinetics and peak size, but much reduced inactivation compared with ORAI1_WT. However, fast CDI in ORAI1_P245L was indistinguishable from ORAI1_WT (Fig. S6 C and D). Induction of ICRAC by 2 mM EGTA (no effect on fast or slow CDI) or 10 mM BAPTA (suppression of fast and slow CDI) in the pipette solution indicated that ORAI1_P245L-transfected cells showed reduced inactivation compared with cells transfected with ORAI1_WT only in cells in which ICRAC was induced by EGTA (Fig. 5 B and C), indicating that P245L suppressed the slow CDI of ORAI1. These data suggest that, although the CRAC channel was not constitutively active as was seen with STIM1_R304W (Fig. 2A) and ORAI1_P245L still required association with STIM1 for activation, ORAI1_P245L-mediated currents showed prolonged activation as a result of reduced slow CDI. To test whether store-operated Ca2+ entry was enhanced in cells of a patient with the p.P245L mutation, we measured Ca2+ influx in response to store depletion by using TG in immortalized lymphocytes derived from a healthy individual and a patient. As predicted, Ca2+ influx developed slower compared with control cells and showed prolonged activation (Fig. 5D).
Fig. 5.

ORAI1_P245L suppresses slow CDI of ICRAC. (A) Time course of ICRAC in HEK293 cells transiently cotransfected with STIM1_WT plus WT ORAI1 (S_WT/O_WT; black, n = 5) or STIM1_WT plus ORAI1_P245L (S_WT/O_P245L; dark cyan, n = 8) induced by 10 mM EGTA and suppressed by 20 µM La3+. (B) Normalized whole-cell currents measured at −80 mV of cells transfected with STIM1_WT plus ORAI1_WT (black, n = 10) or STIM1_WT plus ORAI1_P245L (dark cyan, n = 14) in the presence of 2 mM EGTA in the pipette solution. (C) Normalized whole-cell currents measured at −80 mV of cells transfected with STIM1_WT plus ORAI1_WT (black, n = 8) or STIM1_WT plus ORAI1_P245L (dark cyan, n = 9) in the presence of 10 mM BAPTA in the pipette solution. (D) Store-operated Ca2+ entry is enhanced in a patient with congenital miosis and TAM. Store-operated Ca2+ entry in lymphocytes obtained from an unaffected individual (control; black, n = 146 cells) or a patient (patient; red, n = 302 cells) using single-cell Ca2+ imaging. Cells were loaded with 2 µM Fura-2/AM and placed in an ECS containing 0 mM Ca2+. Stores were depleted with 2 µM TG, and Ca2+ influx was stimulated by the addition of 2 mM Ca2+ in the ECS.
Next, we tested for a possible effect of STIM1_R304W and ORAI1_P245L on hemostasis, one of the features of Stormorken syndrome, in developing zebrafish embryos. Bleeding was visualized directly with ο-dianisidine staining of hemoglobin (40) (Fig. 6 A–G), whereas thrombocyte (a nucleated cell type equivalent to platelets in zebrafish) numbers were visualized by using the Tg(CD41:GFP) line (Fig. 6 H–P). In this line, mature thrombocytes and hematopoietic stem cells/progenitors are labeled by GFP (41). However, thrombocytes can be distinguished from hematopoietic stem cells/progenitors by expressing high levels of GFP (42). We also examined the integrity of the caudal vein in Tg(Fli1:GFP) fish (Fig. S7), in which endothelial cells are marked with GFP (41, 43). We reasoned that reduced thrombocyte activity/numbers might impair the routine surveillance and repair of vessels associated with normal hemostatic function. All three assays yielded the same results. Injection of mouse or human WT or mouse STIM1_K(384-6)Q mRNAs yielded embryos indistinguishable from sham-injected animals in terms of bleeding in the tail or in the cranium (n = 50–100 embryos per experiment, scored blind to injection mixture; Fig. 6G), thrombocyte numbers (Fig. 6P), and formation of caudal veins. In contrast, STIM1_R304W, STIM1_R304W_K(384-6)Q, or STIM1_D76A mRNA resulted in a severe bleeding phenotype wherein embryos presented with varying degrees of brain hemorrhage and bleeding along the trunk and at the ventral portion of the tail (Fig. 6G). The number of cells with high levels of GFP in Tg(CD41:GFP) embryos was also reduced markedly (Fig. 6 H–P), and the number of embryos with a hypoplastic caudal vein was increased significantly as well (Fig. S7E). In contrast, expression of ORAI1_P245L did not show significant differences in any of the three assays compared with ORAI1_WT (Fig. 6 G and P and Fig. S7E). This is consistent with the lack of hematologic defects in patients carrying the ORAI1_P245L allele. The phenotype and effect of the R304W mutation were specific and consistent with our in vitro model, as injection of blended mixtures of WT and mutant STIM1_R304W attenuated the bleeding phenotype (Fig. 6 G and P and Fig. S7E).
Fig. 6.
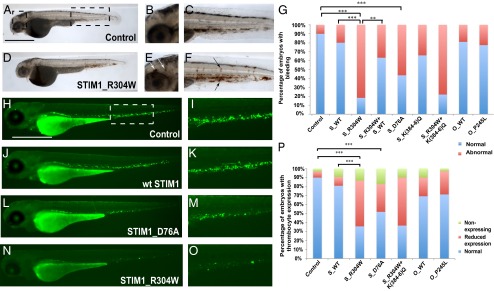
Expression of STIM1_R304W, but not ORAI1_P245L, results in bleeding and reduced expression and defective flow of thrombocyte progenitors in zebrafish embryos. (A–F) Whole-body lateral views of a control (A) and STIM1_R304W (D) injected embryos at 48 h post fertilization stained with ο-dianisidine. B and E are magnifications of the cephalic boxed area in A in all respective embryos, showing brain hemorrhages (white arrows) in the STIM1_R304W mRNA injected embryos (E). Panels C and F show magnified views of the boxed caudal area in A in the respective conditions. Areas of intrasomitic and caudal bleeding are highlighted with black arrows. (G) Percent distribution of normal embryos vs. embryos with spontaneous bleeding episodes (**P < 0.001, ***P < 0.0001). (Scale bar: 500 µm.) Whole-body lateral views of control (H), STIM1_WT- (J), STIM1_D76A- (L), and STIM1_R304W- (N) injected Tg(CD41:GFP) embryos at 72 h post fertilization. I, K, M, and O are magnifications of the ventral boxed area in H in the respective embryos, showing expression of GFP in thrombocytes. The boxed area corresponds to the site where early hematopoiesis takes place in zebrafish. (P) Percent distribution of normal embryos vs. embryos with reduced or no expression of thrombocyte progenitors (*P < 0.05, **P < 0.001, and ***P < 0.0001).
Discussion
In this study, we show that activating mutations in STIM1 and ORAI1, encoding the primary proteins regulating store-operated Ca2+ entry, result in a symptom complex characterized by myopathy with tubular aggregates, congenital miosis and, in the case of STIM1, thrombocytopenia and platelet defects. These mutations illustrate the phenotypic effects of hyperactive CRAC channel signaling and contrast the immune deficiency syndrome observed in biallelic loss-of-function mutations in these genes (21–23). Our data are supported by independent studies in mice, in which an activating mutation in the EF hand of STIM1 (p.D84G) led to premature platelet activation and bleeding, one of the symptoms in patients with Stormorken syndrome (44). We note that, in humans, the p.D84G mutation did not have the same effect, raising the possibility that compensatory mechanisms or disease modifiers could account for this difference (24). We propose that, in humans, p.R304W in STIM1 is a stronger activating allele compared with p.P245L in ORAI1 (Table S2). This idea is supported by the more severe effect of p.R304W in STIM1 than that of p.P245L in ORAI1 on ICRAC and bleeding in zebrafish embryos, and thus may explain the broader phenotypic spectrum of patients with Stormorken syndrome. However, p.P245L in ORAI1 appears to be a stronger gain-of-function allele than alleles of STIM1 bearing mutations in the EF hand of STIM1, as EF hand mutants are associated with myopathy without miosis. Our data also suggest that the mechanisms by which STIM1 and ORAI1 function are different in different tissues. It seems that the hemostatic system of platelets is more tolerant to STIM1/ORAI1 mutations, as thrombocytopenia and bleeding only manifest under conditions in which ICRAC cannot be completely turned off. Partial inactivation of the CRAC channel by preserving fast or slow CDI appears to be tolerated in platelets, as patients with EF hand mutations in STIM1 or ORAI1 mutations do not manifest thrombocytopenia or bleeding. However, this is not the case in skeletal muscle cells in which loss of either type of CRAC channel inactivation results in TAM.
Experiments in zebrafish show that overexpression of STIM1_R304W results in thrombocytopenia, bleeding, and hypoplastic caudal vein. Although we are mindful of over fitting human mutational and phenotypic data to a transient in vivo model, our combined data obtained from three separate in vivo assays do suggest that the described phenotypes approximate the Stormorken pathology, certainly within constrains of mechanistic resolution inherent to this model. All the observed defects are ameliorated when fish are coinjected with the mRNA encoding the WT allele. Although this seems paradoxical, given that patients express an equal amount of WT and mutant alleles, STIM1_R304W is expressed on top of the endogenous WT zebrafish allele. Thus, embryos injected with single mutants also express endogenous WT STIM1. This condition may approximate levels of STIM1_WT and STIM1_R304W in patients with Stormorken syndrome. In addition to recapitulating aspects of Stormorken disease, experiments in zebrafish provided mechanistic insights of the thrombocytopenia induced by STIM1 mutations. Expression of STIM1_R304W_K(384-6)Q resulted in a similar level of bleeding seen with STIM1_R304W (Fig. 6 G and P and Fig. S7E), despite the much larger ICRAC induced by STIM1_R304W (Fig. 3A, blue and red). These results suggest that it is the constitutive activation of the CRAC channel, driven by p.R304W, in resting conditions even at a submaximal level, and not necessarily the peak current, that is responsible for the bleeding phenotype in zebrafish embryos. This hypothesis is supported by the lack of an effect of ORAI1_P245L on bleeding and by the suppression of the bleeding phenotype in embryos coinjected with STIM1_R304W and STIM1_WT. This effect could be caused by the reduction of the amount of STIM1_R304W and/or formation of WT/R304W heterodimers, attenuating the constitutive activation of the CRAC channel induced by p.R304W, in resting conditions.
Our study provides molecular insights of the mechanism of CDI of the CRAC channel. We show that not all activating mutations in STIM1 can have the same effect on the fast CDI of the CRAC channel. For example, the p.R304W mutation residing in the CC1 domain suppressed CDI, which was not seen with the p.D76A mutation residing in the EF hand of STIM1. As fast CDI can be influenced by the ratio of STIM1/ORAI1 proteins in transfected cells (45–47) that could account for the observed differences, it was also shown that STIM1_R304W suppressed, but not abolished, fast CDI in native conditions. These results indicate that there is a role for this residue in modulating fast CDI. However, we do not yet understand how p.R304W suppresses fast CDI, as it is located in a region that is not involved directly in CDI (46).
Our work identifies an activating mutation in ORAI1 associated with a clinical pathologic condition in humans. Functional studies show that the p.P245L in ORAI1 does not cause constitutive activation of the CRAC channel, but rather makes a channel that cannot be completely turned off. In contrast to fast CDI, which is preserved, slow CDI is suppressed by p.P245L. The fact that fast CDI is preserved in ORAI1_P245L is consistent with the mutation being located in a region not involved in the fast CDI (46, 48). Notably, p.P245 in human ORAI1 is equivalent to p.P288 in Drosophila ORAI1, the structure of which was recently solved (49). p.P288 was shown to induce a bend in the transmembrane segment M4, suggesting an important structural role of this residue. We speculate that binding of STIM1 to the C-terminal cytosolic domain of ORAI1 induces a structural rearrangement in the transmembrane M4 segment involving P245, which is critical for the slow CDI, perhaps by promoting/stabilizing the STIM1/ORAI1 interaction. Elimination of this bend in the transmembrane M4 segment mediated by the p.P245L mutation could render ORAI1 in a state that stabilizes its interaction with STIM1, making it insensitive to slow CDI. Further work is needed to delineate the exact molecular mechanisms by which p.P245L in ORAI1 suppresses the slow CDI of the CRAC channel.
Since the first description of tubular aggregates in 1970, evidence has accumulated that these aggregates are inclusions, consisting of regular arrays of tubules derived from the sarcoplasmic reticulum (50). However, the mechanism(s) responsible for the formation of these aggregates is unknown. A relationship between disordered Ca2+ metabolism and tubular aggregates has been suggested as early as 1985 (51, 52). We propose that disordered, sustained Ca2+ entry through the CRAC channel over long periods of time results in an environment within the sarcoplasmic reticulum that is hostile to protein folding, thus initiating the formation of tubular aggregates. Compounds that target the CRAC channel may be useful therapeutic modalities for alleviating the myopathy and other associated features for patients with these mutations.
Materials and Methods
Patients.
The 9-y-old patient with Stormorken syndrome, her two healthy brothers, healthy mother, and two healthy paternal grandparents were enrolled; her father was deceased (Fig. 1A). Complete blood count, plasma CK, and platelet aggregation studies were done. A second patient with Stormorken syndrome was recruited to confirm findings (gene/mutation) in our index patient (Fig. 1B). Nine individuals from the two branches of the family described by Shahrizaila et al. (38) were recruited, two of whom were phenotypically unaffected (Fig. 4A). Genomic DNA was obtained from peripheral blood by using standard protocol from the all individuals mentioned earlier. The study was performed in accordance with the Declaration of Helsinki protocols and was approved by the Oklahoma University Health Sciences Center Institutional Review Board #2866.
Other Materials and Methods.
Details on whole exome sequencing, site-directed mutagenesis, Ca2+ imaging, electrophysiology, and zebrafish experiments are provided in SI Materials and Methods.
Statistical Analysis.
Two-tailed t tests and χ2 tests were performed to measure statistical significance between conditions.
Supplementary Material
Acknowledgments
We thank all the patients and their families for providing blood and tissue samples; Drs. George Dale, Hongguang Nie, Sanjay Bidichandani, John J. Mulvihill, Mohi Ahmad, and Gerard Elberg for comments on the manuscript; Dr. Richard Marlar and Jana Gausman for performing the platelet aggregation studies; Laura Battiest and Prof. Malcolm Taylor for EBV transformation of B lymphocytes; Drs. Shibo Li and Weihong Xu for skin fibroblast cultures and Sanger sequencing; and Prof. Leonard Zon and Dr. Ilya Shestopalov for providing the Tg(CD41:GFP) zebrafish line. This work was supported by grants from the Oklahoma Center for Adult Stem Cell Research (to L.T.) and the National Institutes of Health (NIH) (R01DK59599 and R01AR64211; to L.T.), and P20GM103456 from the NIH (to P.M.G.), an Oklahoma Medical Research Foundation Institutional Research Grant (to P.M.G.), and R01HD042601 from the NIH (to N.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312520111/-/DCSupplemental.
References
- 1.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 2.Burgess GM, et al. The second messenger linking receptor activation to internal Ca release in liver. Nature. 1984;309(5963):63–66. doi: 10.1038/309063a0. [DOI] [PubMed] [Google Scholar]
- 3.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355(6358):353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 4.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: Dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13(9):549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trebak M. STIM/Orai signalling complexes in vascular smooth muscle. J Physiol. 2012;590(Pt 17):4201–4208. doi: 10.1113/jphysiol.2012.233353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. 2009;10(1):21–27. doi: 10.1038/ni.f.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134(3):771–782. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104(22):9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174(6):815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174(6):803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis RS. Store-operated calcium channels: New perspectives on mechanism and function. Cold Spring Harb Perspect Biol. 2011;3(12) doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan JP, et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11(3):337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penner R, Matthews G, Neher E. Regulation of calcium influx by second messengers in rat mast cells. Nature. 1988;334(6182):499–504. doi: 10.1038/334499a0. [DOI] [PubMed] [Google Scholar]
- 20.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1(1):99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 22.Byun M, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207(11):2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picard C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360(19):1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Böhm J, et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet. 2013;92(2):271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng W, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32(3):439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinde AV, et al. STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci Signal. 2013;6(267):ra18. doi: 10.1126/scisignal.2003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grigoriev I, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18(3):177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth JT, Beg AM, Wu S, Putney JW, Jr, Rusan NM. Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr Biol. 2012;22(16):1487–1493. doi: 10.1016/j.cub.2012.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stormorken H, et al. A new syndrome: thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin Genet. 1985;28(5):367–374. doi: 10.1111/j.1399-0004.1985.tb02209.x. [DOI] [PubMed] [Google Scholar]
- 30.Stormorken H. [Stormorken’s syndrome] Tidsskr Nor Laegeforen. 2002;122(30):2853–2856. [PubMed] [Google Scholar]
- 31.Mizobuchi M, et al. [Muscle involvement of Stormorken’s syndrome] Rinsho Shinkeigaku. 2000;40(9):915–920. [PubMed] [Google Scholar]
- 32.Baba Y, Matsumoto M, Kurosaki T. Calcium signaling in B cells: Regulation of cytosolic Ca(2+) increase and its sensor molecules, STIM1 and STIM2. Mol Immunol. 2013 doi: 10.1016/j.molimm.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Calloway N, Holowka D, Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry. 2010;49(6):1067–1071. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korzeniowski MK, Manjarrés IM, Varnai P, Balla T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci Signal. 2010;3(148):ra82. doi: 10.1126/scisignal.2001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui B, et al. The inhibitory helix controls the intramolecular conformational switching of the C-terminus of STIM1. PLoS ONE. 2013;8(9):e74735. doi: 10.1371/journal.pone.0074735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navarro-Borelly L, et al. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586(pt 22):5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang GN, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8(9):1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 38.Shahrizaila N, Lowe J, Wills A. Familial myopathy with tubular aggregates associated with abnormal pupils. Neurology. 2004;63(6):1111–1113. doi: 10.1212/01.wnl.0000138575.14424.5f. [DOI] [PubMed] [Google Scholar]
- 39.Fujii Y, et al. Surf4 modulates STIM1-dependent calcium entry. Biochem Biophys Res Commun. 2012;422(4):615–620. doi: 10.1016/j.bbrc.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 40.Albers CA, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43(8):735–737. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin HF, et al. Analysis of thrombocyte development in CD41-GFP transgenic zebrafish. Blood. 2005;106(12):3803–3810. doi: 10.1182/blood-2005-01-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma D, Zhang J, Lin HF, Italiano J, Handin RI. The identification and characterization of zebrafish hematopoietic stem cells. Blood. 2011;118(2):289–297. doi: 10.1182/blood-2010-12-327403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns CE, et al. Isolation and characterization of runxa and runxb, zebrafish members of the runt family of transcriptional regulators. Exp Hematol. 2002;30(12):1381–1389. doi: 10.1016/s0301-472x(02)00955-4. [DOI] [PubMed] [Google Scholar]
- 44.Grosse J, et al. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J Clin Invest. 2007;117(11):3540–3550. doi: 10.1172/JCI32312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scrimgeour N, Litjens T, Ma L, Barritt GJ, Rychkov GY. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J Physiol. 2009;587(pt 12):2903–2918. doi: 10.1113/jphysiol.2009.170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci USA. 2009;106(36):15495–15500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2011;108(32):13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, et al. Crystal structure of calmodulin binding domain of orai1 in complex with Ca2+ calmodulin displays a unique binding mode. J Biol Chem. 2012;287(51):43030–43041. doi: 10.1074/jbc.M112.380964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338(6112):1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engel WK, Bishop DW, Cunningham GG. Tubular aggregates in type II muscle fibers: Ultrastructural and histochemical correlation. J Ultrastruct Res. 1970;31(5-6):507–525. doi: 10.1016/s0022-5320(70)90166-8. [DOI] [PubMed] [Google Scholar]
- 51.Pierobon-Bormioli S, et al. Familial neuromuscular disease with tubular aggregates. Muscle Nerve. 1985;8(4):291–298. doi: 10.1002/mus.880080405. [DOI] [PubMed] [Google Scholar]
- 52.Salviati G, et al. Tubular aggregates: Sarcoplasmic reticulum origin, calcium storage ability, and functional implications. Muscle Nerve. 1985;8(4):299–306. doi: 10.1002/mus.880080406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.