

Significance
Reactions occurring at the air−sea interface have the potential to alter the chemical composition of the atmosphere. However, our knowledge of the extent to which these reactions impact the concentration of oxidants and their precursors is derived from laboratory measurements using systems that mimic the chemical, biological, and physical complexity of the surface ocean. Here, we present direct measurements of the vertical fluxes of a reactant−product pair using eddy covariance coupled with chemical ionization time-of-flight mass spectrometry to directly assess the role of the ocean surface in the exchange of reactive nitrogen and halogens. Our observations suggest that the ocean surface plays a critical role in controlling the lifetime of N2O5, a primary nocturnal reservoir for tropospheric reactive nitrogen.
Keywords: heterogeneous chemistry, halogen chemistry, atmospheric chemistry
Abstract
The lifetime of reactive nitrogen and the production rate of reactive halogens in the marine boundary layer are strongly impacted by reactions occurring at aqueous interfaces. Despite the potential importance of the air−sea interface in serving as a reactive surface, few direct field observations are available to assess its impact on reactive nitrogen deposition and halogen activation. Here, we present direct measurements of the vertical fluxes of the reactant−product pair N2O5 and ClNO2 to assess the role of the ocean surface in the exchange of reactive nitrogen and halogens. We measure nocturnal N2O5 exchange velocities (Vex = −1.66 ± 0.60 cm s−1) that are limited by atmospheric transport of N2O5 to the air−sea interface. Surprisingly, vertical fluxes of ClNO2, the product of N2O5 reactive uptake to concentrated chloride containing surfaces, display net deposition, suggesting that elevated ClNO2 mixing ratios found in the marine boundary layer are sustained primarily by N2O5 reactions with aerosol particles. Comparison of measured deposition rates and in situ observations of N2O5 reactive uptake to aerosol particles indicates that N2O5 deposition to the ocean surface accounts for between 26% and 42% of the total loss rate. The combination of large Vex, N2O5 and net deposition of ClNO2 acts to limit NOx recycling rates and the production of Cl atoms by shortening the nocturnal lifetime of N2O5. These results indicate that air−sea exchange processes account for as much as 15% of nocturnal NOx removal in polluted coastal regions and can serve to reduce ClNO2 concentrations at sunrise by over 20%.
The production rate of tropospheric ozone (O3), a criteria air pollutant, depends critically on the concentrations of nitrogen oxides (NOx ≡ NO + NO2), volatile organic compounds (VOCs), trace oxidants (e.g., OH, NO3, and Cl), and the wavelength-dependent actinic flux. Accurate model representation of O3 mixing ratios and the sensitivity of O3 to changes in NOx and VOC emissions rely heavily on a complete description of the factors that control NOx lifetimes and, in turn, the concentrations of atmospheric oxidants. Modeling studies, constrained by laboratory and field observations, suggest that nocturnal processes involving the nitrate radical (NO3) and N2O5, both products of NOx oxidation, can account for as much as 50% of the NOx removal (1). Incorporation of the heterogeneous reaction of N2O5 on chloride containing aerosol particles (2, 3) serves as both an efficient NOx recycling and halogen activation mechanism via the production of photolabile nitryl chloride (ClNO2) in both coastal (4) and continental air masses (5).
To date, study of the impact of nocturnal processes on the lifetime of NOx and the production of reactive halogen species in the marine boundary layer has concentrated on gas-phase reactions and heterogeneous and multiphase processes occurring on/within aerosol particles, with little attention paid to reactions occurring at the air−sea interface (6, 7). With nearly half of Earth’s population living within 200 km of a saltwater coastline, a significant fraction of NOx emissions are found near coastal waters (4, 8). As such, the chemical evolution of polluted air masses stemming from coastal megacities occurs to a large extent over the ocean (e.g., Beijing plume). If air−sea exchange of reactive nitrogen compounds is rapid and the reaction kinetics at the air−ocean and air−particle surface are comparable, we expect air−sea exchange processes to play an important role in setting the lifetime of compounds such as N2O5 in coastal environments. Specifically, dry deposition of N2O5 to the ocean surface could serve to help close the existing gap between models and measurements of N2O5 mixing ratios in the polluted marine boundary layer (9). In what follows, we describe direct measurements of the vertical flux of N2O5 and ClNO2 obtained via eddy covariance at a polluted coastal site to provide observation-based constraints on the role of the air−sea interface in setting the lifetime of reactive nitrogen and the production rate of reactive halogens in the marine boundary layer.
The vertical flux of trace gases across the air−sea interface is a complex function of both atmospheric and oceanic processes, where gas exchange is controlled by molecular diffusion in the interfacial regions surrounding the air−water interface (10) and the solubility and chemical reactivity of the gas in the molecular sublayer. The flux (F) of trace gas across the interface is described by Eq. E1, as a function of both the gas-phase (Cg) and liquid phase (Cl) concentrations and the dimensionless gas over liquid Henry’s law constant (KH),
where Kt, the total transfer velocity for the gas (cm s−1), encompasses all of the chemical and physical processes that govern air−sea gas exchange (11). As such, accurate, molecule-specific parameterization of Kt is critical for assessing the role of the ocean as a net source or sink for both greenhouse gases and criteria air pollutants, and the trace gases that control their abundances in the atmosphere.
With respect to N2O5 air−sea exchange, we expect the reaction mechanism to closely follow that described for reactions occurring at the air−particle interface, particularly that of sea spray aerosol. Generally, the reactive uptake of N2O5 to aqueous interfaces in the troposphere has been proposed to follow the concerted reaction mechanism (12, 13):
This mechanism is consistent with laboratory evidence that the reactive uptake of N2O5 to aqueous interfaces is dependent on: (i) liquid water content (13), (ii) nitrate (NO3−) and chloride (Cl−) concentrations (13–15), and (iii) the presence of organic surfactants and/or films (16–19). The ClNO2 product yield, Φ (ClNO2), following N2O5 hydrolysis has been shown to be a strong function of chloride concentration, where Φ (ClNO2) is 0.8 for [Cl−] = 0.5 M, increasing to 1.0 for [Cl−] > 1.0 M (2, 13, 20).
Extension of laboratory determined reaction rates (21) and equilibrium constants (2, 22) to the air−sea interface would suggest that N2O5 deposition to the ocean should be rapid (e.g., KΗ = 1.9 × 10−2, k2 = 5 × 106 s−1) and that Φ (ClNO2)ocean would be >0.8, based on an oceanic [Cl−] of 0.55 M. However, the a priori estimate for the magnitude and direction of the air−sea flux for ClNO2 (KH = 1.66) is less clear (2). Although laboratory results suggest that ClNO2 should be made at high yield at the ocean surface, it is not clear if water-side transport and subsequent chemical reactions may suppress ClNO2 release back to the atmosphere. Alternatively, reaction of the nitronium ion (NO2+) in the organic-rich sea surface microlayer (23) may also serve to reduce Φ (ClNO2)ocean, a reaction that may also proceed in organic-rich aqueous aerosol.
Results and Discussion
Eddy Covariance Measurements of N2O5 and ClNO2 Air−Sea Exchange.
Concentration and vertical flux measurements of N2O5 and ClNO2 were made at 13 m above mean lower low water (13 m above mean low water) from the end of the 330-m Scripps Institution of Oceanography (SIO) pier during January and February 2013. Briefly, N2O5 and ClNO2 mixing ratios were measured using chemical ionization time-of-flight mass spectrometry (24), using I− reagent ion chemistry (25). Spectra were saved at 10 Hz, coincident in time with measurements of 3D winds acquired with a colocated ultrasonic anemometer sampling at 20 Hz. Details on instrument calibration, inlet performance, and flux measurements can be found in Materials and Methods and SI Text.
Here, we discuss a subset of these measurements obtained on February 20, 2013 where the true wind direction ranged between 205° and 295°, resulting in a purely ocean fetch with pollution from Los Angeles entrained into the sampled air mass. Ten-meter wind speeds (u10) ranged between 6.5 and 11.1 m s−1 with a mean and SD of 9.07 ± 1.29 m s−1. The diel profile in N2O5 and ClNO2 mixing ratios is shown in Fig. 1 A and B, where N2O5 and ClNO2 mixing ratios track one another for much of the night, peaking at midnight. As expected, N2O5 mixing ratios drop sharply to zero at sunrise due to rapid photolysis of the nitrate radical (NO3), which is in thermal equilibrium with N2O5, whereas ClNO2 decays to zero with a time constant (τ = 2.71 h) consistent with its photolysis lifetime (26). The magnitude of the N2O5 and ClNO2 mixing ratios are comparable with those found in previous studies in coastal California (27).
Fig. 1.

(A) N2O5 and (B) ClNO2 mixing ratios as measured from the SIO pier in La Jolla, CA, on February 20, 2013 at 0.1, 1, and 10 Hz time resolution.
N2O5 flux measurements are shown in Fig. 2A. As expected, N2O5 displays a net downward flux, into the ocean. The magnitude of the flux tracks ambient N2O5 mixing ratios, yielding a nocturnally averaged exchange velocity (Vex, or flux divided by concentration) of −1.66 ± 0.60 (1σ) cm s−1. We note that a negative Vex indicates a downward flux from the atmosphere to the ocean. Vex,N2O5 can be interpreted within the resistance framework developed for O3 dry deposition, where Vex depends on the aerodynamic resistance, quasi-laminar boundary layer resistance, and the surface resistance that includes chemical reactions at the interface (28). To this end, we calculate the total transfer velocity (Kt) for N2O5 (Eq. E2) for comparison with the observed exchange rate,
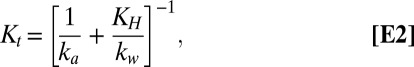 |
where ka is the air-side transfer velocity, kw is the water-side transfer velocity, and KH is the dimensionless gas over liquid Henry’s law constant. Over the past two decades, a series of parameterizations (ref. 11 and references therein) have been developed that permit calculation of both ka and kw as a function of both the molecular properties of the gas (e.g., diffusivity, reactivity, solubility) and physical forcing data (e.g., wind speed). In the case of N2O5, the hydrolysis rate (k2) is sufficiently fast (>1 × 106 s−1) that we expect N2O5 deposition to be limited only by the air-side transfer rate (ka), despite its moderate solubility (KH = 1.9 × 10−2). As such, we calculate ka (Eq. E3) following the numerical approach of Johnson (29) where the still air diffusive flux of Mackay and Yeun (30) has been added to the representation of ka found in the NOAA COARE model (31), with a numerical representation of the wind speed dependent drag coefficient (CD).
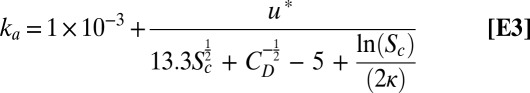 |
Here, u* is the friction velocity, Sc is the Schmidt number for N2O5, and κ is the von Karman constant (taken as 0.4). For comparison, if we neglect fast hydrolysis, N2O5 deposition is controlled by the surface resistance due to its moderate solubility and slow diffusion rate in water (Dl = 1.9 × 10−5 cm2 s−1) (32). Calculations of the total transfer velocity of N2O5 as a function of wind speed are shown in Fig. 3, with (solid black line) and without (dashed black line) surface hydrolysis. In the latter case, we calculate kw, again following the numerical approach of Johnson (29). Also included in Fig. 3 are the wind speed-dependent parameterizations for ka(N2O5) of Liss (33) and Mackay and Yeun (30) both derived from wind tunnel studies and that of Duce et al. (34), which is calculated from micrometeorological theory. The nocturnally averaged measured N2O5 exchange velocity is shown with a blue square in Fig. 3. As shown, the observed exchange rate is on average a factor of 2.17 larger than that calculated using Eq. E3; however, it is in agreement with the parameterizations of Liss (10) and Mackay and Yeun (30), to within the uncertainty and variability of the measurements. Longer-term observations of FN2O5, designed to capture wide variability in wind speed, will provide unique observation-based constraints on ka, while permitting the opportunity to assess the impact of entrainment of N2O5 and ClNO2 from the free troposphere and vertical gradients in temperature and aerosol surface area on the retrieved fluxes.
Fig. 2.

Measured vertical fluxes of N2O5 and ClNO2. Errors are determined for each 30-min flux segment as the covariance between vertical wind speed and concentration at lag times significantly longer than the delay (or lag) time. Calculated ClNO2 vertical fluxes (lines in B), as determined from the coupled time-dependent ocean−atmosphere model, constrained by the measured N2O5 vertical fluxes (A). Three different model scenarios are shown and described in detail in SI Text: C1 (a priori), model inputs taken as suggested in the literature [e.g., KH (ClNO2) = 1.66, Φ (ClNO2)ocean = 0.8, kr = 5 × 106 s−1, and δ = 1.5 × 10−6 cm]; C2, model inputs taken as 90% confidence limits of those suggested in the literature [e.g., KH (ClNO2) = 0.32, Φ (ClNO2)ocean = 0.5, kr = 2 × 105 s−1, and δ = 7.1 × 10−6 cm]; and C3, same as C2, with Φ (ClNO2)ocean = 0.
Fig. 3.

(Upper) Average N2O5 exchange velocity (Vex, cm s−1) as a function of 10-m wind speed (m s−1). Modeled exchange velocities, determined via Eq. E3 [Johnson (29)] are shown using the dimensionless gas over liquid Henry’s Law Constant, KH (N2O5) = 1.9 × 10−2 with and without incorporation of N2O5 hydrolysis (kr = 5 × 106 s−1). Parameterizations of Liss (33), Mackay and Yeun (30), and Duce (34) are also shown for comparison. (Lower) The 2012 annual median (black line), interquartile range (dark gray shaded region), and full range (light gray shaded region) for 10-m wind speed measured at the SIO pier.
Our a priori supposition for the direction of the ClNO2 flux, based on laboratory-determined reaction rates and equilibrium constants, is assumed to be out of the ocean, where hydrolyzed N2O5 produces NO2+ ions that rapidly react with Cl−, forming ClNO2 in high yield at the ocean surface. Given the N2O5 deposition rate measured above, combined with the moderate solubility of ClNO2, one would expect the magnitude of the flux to be nearly equal, yet opposite in direction, to N2O5. Surprisingly, vertical fluxes of ClNO2 show net deposition, with the exception of the time period between 0300 and 0600, where the ClNO2 flux indicates net emission from the sea surface (Fig. 2B).
Modeling Sea Surface Chemistry of Deposited N2O5.
To further our understanding of N2O5 reactions at the ocean interface, we construct a coupled atmosphere−surface ocean time-dependent box model following the framework outlined in Carpenter et al. (35). The model is constrained by the measured N2O5 vertical flux reported above and N2O5 and ClNO2 reaction rates, product branching ratios, and diffusion constants as previously determined in the literature for reactions occurring at the air−particle interface. As in Carpenter et al. (35), we define a surface aqueous layer (δ) with a depth equal to the reacto-diffusive length (Eq. E4) for the N2O5 reaction mechanism (Reactions R1−R4)
 |
where Dl is the N2O5 liquid-phase diffusion constant (Dl = 1.9 × 10−5 cm2 s−1) (32), and kr is the total reactivity of N2O5 in seawater (kr), taken as the N2O5 hydrolysis rate (k2) (21) as the lifetime of the nitronium ion (NO2+) product is estimated to be less than 10−9 s, making the hydrolysis the rate limiting step in the mechanism (2). We expect the reaction to occur in a thin film (δ < 100 nm), well within the molecular sublayer (ca. 10−3 m) where transport is driven by molecular diffusion (28). As a result, rapid volatilization of reaction products of relatively high solubility may occur following the supersaturation of dissolved gas in the thin film. For simplicity, gas transfer velocities (Kt) and the associated vertical flux for ClNO2 are calculated in the model using ka (Eq. E1) and kw (Eq. E2) parameterizations as described in Johnson (29). As in Carpenter et al. (35), mixing from the bulk, where [ClNO2] is taken to be zero, to the interfacial region is determined by the wind speed dependent expression for the transfer velocity.
To explore the apparent downward measured flux of ClNO2, we drive the coupled atmosphere−ocean model using three plausible sets of input values in the treatment of Kt. In model case 1 (C1), our a priori conditions were based on existing laboratory-based measurements, where model input parameters were taken as suggested in the literature: KH (ClNO2) = 1.66, Φ (ClNO2)ocean = 0.8, kr = 5 × 106 s−1, and δ = 1.5 × 10−6 cm. As shown in Fig. 2B, this results in a strong upward flux (sea to air) of ClNO2 that, at its maximum, is approximately half the magnitude of the N2O5 peak flux. This difference is due to the prescribed ClNO2 product yield coupled to exchange with the bulk ocean. In model case 2 (C2), we set the model input parameters at the 90% confidence limits of those suggested in the literature, in the direction of reducing the upward flux of ClNO2 [e.g., KH (ClNO2) = 0.32, Φ (ClNO2)ocean = 0.5, kr = 2 × 105 s−1, δ = 7.1 × 10−6 cm]. This results in a factor of two reduction in peak ClNO2 flux, but the direction of the flux is still positive and outside the uncertainty of the measurements for much of the night. In model case 3 (C3), we take Φ (ClNO2)ocean = 0, suggesting that perhaps NO2+ does not react with Cl− as expected but proceeds via nitration reactions (36) with enriched organic material found in the sea surface microlayer (23). It is only by setting the ClNO2 product yield to zero or invoking rapid ClNO2 aqueous-phase reaction kinetics or hydrolysis that we can force the model near the uncertainty limits of the measurements.
For the conditions sampled here, ClNO2 production rates were less than 3.0 × 10−3 ppt s−1 [calculated for the median observed khet, Φ (ClNO2)ocean = 1, and [N2O5] = 50 pptv] and the ClNO2 steady-state lifetime with respect to loss to gas-phase and aerosol reactions is estimated at greater than 30 h (4). As a result, it is unlikely that significant gradients in ClNO2 exist in the nocturnal marine boundary layer due to vertical gradients in the ClNO2 atmospheric production rate. As such, our measurements of the vertical flux of ClNO2, presented here, are most consistent with a model where the product yield of ClNO2 is near zero for reactions of NO2+ in the sea surface microlayer, and/or ClNO2 aqueous-phase reactions are significantly faster than the volatilization rate.
Relative Roles of the Particle and Ocean Surface in Regulating τ(N2O5).
To assess the relative roles of the ocean and aerosol surface in the net removal of N2O5, both reactant transport and surface reactivity must be considered. At a marine boundary layer inversion height (zi) of 810 m, taken as the nocturnally averaged zi observed during DYCOMS-II off the coast of San Diego (37), the total aerosol surface area ranges between 5% and 50% of the surface area of a slab ocean, based on aerosol surface area measurements made at the SIO pier. In gas−aerosol reactions, transport of the reactant to the particle surface is set by the gas−aerosol collision rate, which is a function of the mean molecular speed of the reactant and the total suspended aerosol surface area (Eq. E6). In contrast, reactions occurring at the ocean surface require turbulent transport of the reactants to the diffusive sublayer, where turbulence is suppressed and molecular diffusion controls the collision rate of the reactant with the ocean surface. We compare N2O5 deposition rates (kdep, Eq. E5) with N2O5 heterogeneous aerosol reaction rates (kaerosol, Eq. E6), both directly measured at the SIO pier.
 |
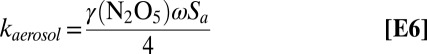 |
Here, Vex is the measured N2O5 exchange velocity, zi is the marine boundary layer inversion height, γ(N2O5) is the N2O5 reactive uptake coefficient, ω is the molecular velocity for N2O5, and Sa is the aerosol surface area concentration. Measurements of kaerosol at this location have been described previously (38), where the median kaerosol was observed to be 6.04 × 10−5 s−1 with an interquartile range of 4.5–7.4 × 10−5 s−1, where kaerosol was a strong function of particle nitrate and organic mass fractions. As described above, the mean Vex was −1.66 ± 0.60 cm s−1 measured at an average wind speed of 9 m s−1, corresponding to a range in kdep of 0.17–1.7 × 10−4 s−1 for zi between 1000 and 100 m, respectively.
The relative strengths of the ocean and the aerosol surface in the net removal of N2O5 from the atmosphere are shown in Fig. 4, where the fraction of N2O5 lost to the ocean surface is shown as a function of kdep and kaerosol. As indicated by the dashed box, the fraction of N2O5 removed by the ocean surface ranges between 19% and 79% for the conditions sampled at the SIO pier, where shallow boundary layers (1000 < zi < 100 m) and high wind speeds combined with suppressed N2O5 heterogeneous reactivity, result in large fractions of N2O5 being lost at the air−sea interface. For zi = 810 m (37) and kaerosol = 6.04 × 10−5 s−1, the fraction of N2O5 lost at the air−sea interface is 32%, ranging between 26% and 42% for the interquartile range in kaerosol. It is important to note that the measurements described here were made at wind speeds significantly larger than the annual median wind speed measured at the SIO pier (2.1 m s−1). Based on the parameterized dependence of Kt on wind speed, it is expected that the range in kdep reported in Fig. 4 may be a factor of 3 smaller at lower wind conditions, resulting in the fraction of N2O5 removed by the ocean surface to between 7% and 55% (1000 > zi > 100 m). In addition, increased aerosol surface area concentrations under low wind speeds, due to reduced dilution of aerosol from an urban source, may also serve to reduce the fraction of N2O5 removed by the ocean surface at low wind speed.
Fig. 4.

Fraction of the total N2O5 removal attributed to deposition to the ocean shown as a function of the N2O5 deposition rate (kdep) and the heterogeneous loss rate to aerosols (kaerosol). The boxed region represents the range in kdep and kaerosol measured at the SIO pier in La Jolla, CA. The median and interquartile range in the observed kaerosol is shown on the ordinate, while the range in the observed kdep [based on a marine boundary layer inversion height (zi) of 100–1000 m, at an average wind speed of 9.1 ± 1.3 m s−1] is shown on the abscissa.
NOx Removal Rates and ClNO2 Production.
At present, the majority of steady-state box model analyses as well as regional-scale chemical transport models designed to assess nocturnal NOx chemistry do not include either N2O5 or ClNO2 deposition to the ocean surface or the possibility for reaction at the air−sea interface (39, 40). Here, we use a 0D time-dependent box model to assess the impact of air−sea exchange on NOx removal rates and Cl atom production rates in the polluted marine boundary layer. The box model was run under four different N2O5 deposition rates at two different values of Φ (ClNO2)ocean, 0 and 0.8. It is important to note that the impact of N2O5 loss mechanisms on NOx removal rates and Cl atom production is coupled to nitrate radical (NO3) chemistry, where reaction of NO3 with dimethyl sulphide (DMS) can act as the primary loss process for nocturnal nitrogen oxides. In the model described here, DMS concentrations are set to 100 pptv, corresponding to a loss rate of 2.6 × 10−3 s−1 for NO3. Model details can be found in SI Text as well as in Materials and Methods.
To assess the importance of deposition processes on reactive nitrogen and halogen budgets in coastal, polluted regions, we first examine the fraction of NOx present at sunset, [NOx]sunset, that is lost to terminal sinks at night as a function of the N2O5 exchange velocity and ClNO2 product yield.
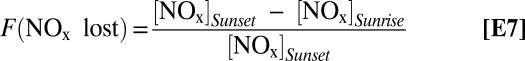 |
As shown in Fig. 5A, constraining the model with the measured Vex (−1.66 cm s−1), zi = 500 m and our best estimate of the yield of ClNO2 produced at the ocean surface [Φ (ClNO2)ocean = 0], results in an approximate 14% increase in the fraction of NOx that is lost to terminal sinks at night, compared with the model that neglects deposition (0.58–0.66). This trend shown in Fig. 5A describes the increasing fraction of NOx that is deposited to the ocean surface, where at the limit of Φ (ClNO2)ocean → 1, approximately half of deposited N2O5 is returned to the atmosphere as ClNO2 and in the limit of Φ (ClNO2)ocean → 0, all of the deposited N2O5 is terminally lost from the atmosphere. Model scenarios that neglect deposition or prescribe a high ClNO2 product yield at the air−sea interface are efficient in recycling NOx, where as much as 50% of reacted N2O5 is returned as NO2 in the early morning following the photolysis of ClNO2 (Fig. 5B). Further, these model scenarios will result in higher concentrations of ClNO2 at sunrise, compared with models that include deposition and low Φ (ClNO2)ocean.
Fig. 5.

(A) Model calculations of the fraction of available NOx ([NOx]sunset) lost during a 12-h night and (B) the mixing ratio of ClNO2 at sunrise as a function of the prescribed N2O5 exchange velocity and ClNO2 product yield [Φ (ClNO2)ocean]. Calculations were conducted using a 0D time-dependent box model constrained by the mean N2O5 reactive uptake coefficients and particle surface area (SA) concentrations measured at this site [γ(N2O5) = 0.005, SA = 500 μm2 cm3]. The model was initialized with the following conditions: Tair = 283 K, Twater = 287 K, [O3]i = 60 ppb, [NOx]i = 1.0 ppb, γ(NO3) = 0, and NO3 reactivity = 0.16 min−1.
Our observations suggest that N2O5 deposited to the ocean surface is terminally lost, thus limiting NOx recycling rates and Cl atom production that would otherwise be sustained by heterogeneous mechanisms at the air−particle interface. As shown in Fig. 5B, including deposition to the ocean surface and neglecting ClNO2 surface production results in nearly a 20% reduction in the concentration of ClNO2 at sunrise, compared with a model that does not include N2O5 deposition. For comparison, including deposition to the ocean surface at the rates measured here, with a high ClNO2 surface yield (0.8), results in nearly a 10% increase in the concentration of ClNO2 at sunrise. This analysis highlights the sensitivity of the nocturnal nitrogen and halogen budget to N2O5 deposition and the resulting chemistry in the sea surface microlayer.
Atmospheric Implications.
We present an analysis of direct measurements of N2O5 and ClNO2 air−sea exchange using eddy covariance. Our results indicate that N2O5 deposition to the ocean surface is rapid (Vex = −1.66 ± 0.60 cm s−1). We find no evidence for net ClNO2 production at the air−sea interface in this study, suggesting either that rates of aqueous-phase reactions of ClNO2 in the sea surface microlayer (SSML) are competitive with volatilization, or that the product yield for ClNO2 is small in the organic-rich SSML. Comparison with direct measurements of the N2O5 loss rate to aerosol particles at the same sampling location indicates that the ocean surface serves on average to remove 32% of N2O5 in the marine boundary layer under the conditions sampled here (assuming zi = 810 m). Our results suggest that future measurements of the exchange velocities of N2O5 under a wide range of wind conditions will provide needed constraints on parameterizations of the air-side transfer rate (ka). We hypothesize that measurements of the flux of ClNO2 will display large spatiotemporal variability and be responsive to the chemical composition and concentration of dissolved organic material (DOM) in the surface ocean due to the competition reactions of the nitronium ion with DOM and halogen ions. Our results suggest that regional modeling efforts designed to assess the impact of nocturnal nitrogen chemistry on oxidant loadings in coastal polluted environments need to properly represent air−sea interactions for adequate representation of the lifetime of both N2O5 and ClNO2.
More broadly, we show that direct measurements of trace gas vertical fluxes when combined with in situ determinations of the reactive uptake to aerosol particles provides an experimental constraint on the lifetime and reactivity of trace gases to the wide array of available surfaces in the marine boundary layer. These results indicate that under conditions of shallow boundary layer heights, uptake to the ocean surface can outpace uptake to aerosol surfaces, highlighting the vast difference in the chemical composition, morphology, phase, and pH of the aerosol and ocean surface. The results presented here highlight the future utility of combining high sensitivity and precision time-of-flight mass spectrometric measurements of reactant and product pairs with micrometeorological techniques for direct, in situ study of chemical reactions occurring at the air−sea interface. This approach will permit study of complex interfacial processes under ambient conditions, where coupled biological, chemical, and physical mechanisms often prohibit the extension of laboratory results to environmental conditions.
Materials and Methods
Sampling Location.
Concentration and vertical flux measurements of N2O5 and ClNO2 were made at 10 m from the northwest boom of the 330-m SIO Pier during February 2013. Air sampled at this location is impacted by local emissions in the La Jolla cove region as well as regional pollution attributed to both San Diego and Los Angeles. The observations presented here are for time periods where winds were sustained from the west (true wind direction between 205° and 295°) so as to ensure an ocean fetch. Backward air trajectories indicate that air sampled during these time periods was influenced by the Los Angeles plume, thus sustaining concentrations of N2O5 well above that observed in clean, marine air (41).
N2O5 and ClNO2 Concentration Measurements.
N2O5 and ClNO2 mixing ratios were measured using chemical ionization time-of-flight mass spectrometry (24), using I− reagent ion chemistry (25). N2O5 sensitivities were determined using the output of a portable N2O5 generation system, described previously (42), where N2O5 is made in situ from the dark reaction of NO2 and O3, and subsequent reaction of the NO3 product with NO2. ClNO2 sensitivities were determined by passing the output of the N2O5 source over concentrated NaCl slurry for unit conversion of N2O5 to ClNO2 (4, 25, 43). We sample N2O5 and ClNO2 through a 17-m, 3/8” o.d. Teflon perfluoroalkoxy tube. The inlet manifold is constructed of fluoropel-coated glass, closely resembling that of Ellis et al. (44), where air is drawn through a critical orifice, reducing the sample line pressure to 200 mbar. The resulting mass flow rate of 10 standard liters per minute results in a laminar flow profile, with a measured gas exchange time for the inlet of 0.7 s.
N2O5 and ClNO2 Flux Measurements.
Mass spectra, acquired at 80 kHz, were saved at 10 Hz, coincident with measurements of 3D winds acquired with a colocated ultrasonic anemometer sampling at 20 Hz (HS-50; Gill Instruments). Fluxes were determined by the eddy covariance technique (45). In this method, the vertical turbulent flux is the covariance of vertical wind speed (w) and mixing ratio from the mean (c), F = <w′c′>. Details on the application of time of flight mass spectrometry to eddy covariance flux measurement can be found elsewhere (46) and described in more detail in SI Text.
Time-Dependent Air−Sea Model.
The chemical evolution of the nocturnal boundary layer was tracked using a one-dimensional time-dependent dimensional box model, where coupled differential equations were solved using custom code written in MATLAB, analyzed with the built-in ordinary differential equation solvers. As time propagates in the model, we calculate the production and loss of NO, NO2, O3, N2O5, NO3, HNO3, and ClNO2 to gas-phase and heterogeneous reactions occurring on/within aerosol particles, as well as air−sea exchange with the ocean surface. The model is initialized with concentrations representative of those measured at the SIO pier, and reaction rates from the NASA Jet Propulsion Laboratory Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation Number 14. Detailed descriptions of the modeling efforts described here can be found in SI Text.
Supplementary Material
Acknowledgments
We thank Byron Blomquist (University of Hawaii) and Joel Thornton (University of Washington) for helpful discussions and Christian McDonald (SIO), Nicole Campbell [University of California, San Diego (UCSD)], and Kathryn Zimmerman (UCSD) for assistance in the setup for the pier observations. This research was supported by the National Science Foundation CAREER Award to T.H.B. (Grant AGS-1151430).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318694111/-/DCSupplemental.
References
- 1.Alexander B, et al. Quantifying atmospheric nitrate formation pathways based on a global model of the oxygen isotopic composition (Δ17O) of atmospheric nitrate. Atmos Chem Phys. 2009;9(14):5043–5056. [Google Scholar]
- 2.Behnke W, George C, Scheer V, Zetzsch C. Production and decay of ClNO2, from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments. J Geophys Res-Atmos. 1997;102(D3):3795–3804. [Google Scholar]
- 3.Finlayson-Pitts BJ, Ezell MJ, Pitts JN. Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2. Nature. 1989;337(6204):241–244. [Google Scholar]
- 4.Osthoff HD, et al. High levels of nitryl chloride in the polluted subtropical marine boundary layer. Nat Geosci. 2008;1(5):324–328. [Google Scholar]
- 5.Thornton JA, et al. A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature. 2010;464(7286):271–274. doi: 10.1038/nature08905. [DOI] [PubMed] [Google Scholar]
- 6.Aldener M, et al. Reactivity and loss mechanisms of NO3 and N2O5 in a polluted marine environment: Results from in situ measurements during New England Air Quality Study 2002. J Geophys Res. 2006 doi: 10.1029/2006JD007252. [DOI] [Google Scholar]
- 7.Huff DM, Joyce PL, Fochesatto GJ, Simpson WR. Deposition of dinitrogen pentoxide, N2O5, to the snowpack at high latitudes. Atmos Chem Phys. 2011;11(10):4929–4938. [Google Scholar]
- 8.Hinrichsen D. Coastal Waters of the World: Trends, Threats, and Strategies. Washington, DC: Island Press; 1998. [Google Scholar]
- 9.Wagner NL, et al. 2012. The sea breeze/land breeze circulation in Los Angeles and its influence on nitryl chloride production in this region. J Geophys Res, 10.1029/2012JD017810.
- 10.Liss PS, Slater PG. Flux of gases across air-sea interface. Nature. 1974;247(5438):181–184. [Google Scholar]
- 11.Carpenter LJ, Archer SD, Beale R. Ocean-atmosphere trace gas exchange. Chem Soc Rev. 2012;41(19):6473–6506. doi: 10.1039/c2cs35121h. [DOI] [PubMed] [Google Scholar]
- 12.Thornton JA, Braban CF, Abbatt JPD. N2O5 hydrolysis on sub-micron organic aerosols: The effect of relative humidity, particle phase, and particle size. Phys Chem Chem Phys. 2003;5(20):4593–4603. [Google Scholar]
- 13.Bertram TH, Thornton JA. Toward a general parameterization of N2O5 reactivity on aqueous particles: The competing effects of particle liquid water, nitrate and chloride. Atmos Chem Phys. 2009;9(21):8351–8363. [Google Scholar]
- 14.Mentel TF, Sohn M, Wahner A. Nitrate effect in the heterogeneous hydrolysis of dinitrogen pentoxide on aqueous aerosols. Phys Chem Chem Phys. 1999;1(24):5451–5457. [Google Scholar]
- 15.Wahner A, Mentel TF, Sohn M, Stier J. Heterogeneous reaction of N2O5 on sodium nitrate aerosol. J Geophys Res. 1998;103(D23):31103–31112. [Google Scholar]
- 16.Anttila T, Kiendler-Scharr A, Tillmann R, Mentel TF. On the reactive uptake of gaseous compounds by organic-coated aqueous aerosols: Theoretical analysis and application to the heterogeneous hydrolysis of N2O5. J Phys Chem A. 2006;110(35):10435–10443. doi: 10.1021/jp062403c. [DOI] [PubMed] [Google Scholar]
- 17.Cosman LM, Bertram AK. Reactive uptake of N2O5 on aqueous H2SO4 solutions coated with 1-component and 2-component monolayers. J Phys Chem A. 2008;112(20):4625–4635. doi: 10.1021/jp8005469. [DOI] [PubMed] [Google Scholar]
- 18.McNeill VF, Patterson J, Wolfe GM, Thornton JA. The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol. Atmos Chem Phys. 2006;6(6):1635–1644. [Google Scholar]
- 19.Escorcia EN, Sjostedt SJ, Abbatt JPD. Kinetics of N(2)O(5) hydrolysis on secondary organic aerosol and mixed ammonium bisulfate-secondary organic aerosol particles. J Phys Chem A. 2010;114(50):13113–13121. doi: 10.1021/jp107721v. [DOI] [PubMed] [Google Scholar]
- 20.Roberts JM, et al. Laboratory studies of products of N2O5 uptake on Cl− containing substrates. Geophys Res Lett. 2009 doi: 10.1029/2009GL040448. [DOI] [Google Scholar]
- 21.Griffiths PT, et al. Reactive uptake of N2O5 by aerosols containing dicarboxylic acids. Effect of particle phase, composition, and nitrate content. J Phys Chem A. 2009;113(17):5082–5090. doi: 10.1021/jp8096814. [DOI] [PubMed] [Google Scholar]
- 22.Fried A, Henry BE, Calvert JG, Mozurkewich M. The reaction probability of N2O5 with sulfuric acid aerosols at stratospheric temperatures and compositions. J Geophys Res. 1994;99(D2):3517–3532. [Google Scholar]
- 23.Hunter KA, Liss PS. Input of organic material to oceans: Air-sea interactions and organic chemical composition of sea surface. Mar Chem. 1977;5(4-6):361–379. [Google Scholar]
- 24.Bertram TH, et al. A field-deployable, chemical ionization time-of-flight mass spectrometer. Atmos Meas Tech. 2011;4(7):1471–1479. [Google Scholar]
- 25.Kercher JP, Riedel TP, Thornton JA. Chlorine activation by N2O5: Simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry. Atmos Meas Tech. 2009;2(1):193–204. [Google Scholar]
- 26.Nelson HH, Johnston HS. Kinetics of the reaction of Cl with ClNO and ClNO2 and the photochemistry of ClNO2. J Phys Chem. 1981;85(25):3891–3896. [Google Scholar]
- 27.Riedel TP, et al. Nitryl chloride and molecular chlorine in the coastal marine boundary layer. Environ Sci Technol. 2012;46(19):10463–10470. doi: 10.1021/es204632r. [DOI] [PubMed] [Google Scholar]
- 28.Fairall CW, Helmig D, Ganzeveld L, Hare J. Water-side turbulence enhancement of ozone deposition to the ocean. Atmos Chem Phys. 2007;7(2):443–451. [Google Scholar]
- 29.Johnson MT. A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas. Ocean Sci. 2010;6(4):913–932. [Google Scholar]
- 30.Mackay D, Yeun ATK. Mass transfer coefficient correlations for volatilization of organic solutes from water. Environ Sci Technol. 1983;17(4):211–217. doi: 10.1021/es00110a006. [DOI] [PubMed] [Google Scholar]
- 31.Fairall CW, Bradley EF, Hare JE, Grachev AA, Edson JB. Bulk parameterization of air-sea fluxes: Updates and verification for the COARE algorithm. J Clim. 2003;16(4):571–591. [Google Scholar]
- 32.Robinson GN, Worsnop DR, Jayne JT, Kolb CE, Davidovits P. Heterogeneous uptake of ClONO2 and N2O5 by sulfuric acid solutions. J Geophys Res. 1997;102(D3):3583–3601. [Google Scholar]
- 33.Liss PS. Processes of gas exchange across an air-water interface. Deep Sea Res. 1973;20(3):221–238. [Google Scholar]
- 34.Duce RA, et al. The atmospheric input of trace species to the world ocean. Global Biogeochem Cycles. 1991;5(3):193–259. [Google Scholar]
- 35.Carpenter LJ, et al. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat Geosci. 2013;6(2):108–111. [Google Scholar]
- 36.Heal MR, Harrison MAJ, Cape JN. Aqueous-phase nitration of phenol by N2O5 and ClNO2. Atmos Environ. 2007;41(17):3515–3520. [Google Scholar]
- 37.Faloona I, et al. Observations of entrainment in eastern Pacific marine stratocumulus using three conserved scalars. J Atmos Sci. 2005;62(9):3268–3285. [Google Scholar]
- 38.Riedel TP, et al. Direct N2O5 reactivity measurements at a polluted coastal site. Atmos Chem Phys. 2012;12(6):2959–2968. [Google Scholar]
- 39.Brown SS, Stutz J. Nighttime radical observations and chemistry. Chem Soc Rev. 2012;41(19):6405–6447. doi: 10.1039/c2cs35181a. [DOI] [PubMed] [Google Scholar]
- 40.Saiz-Lopez A, von Glasow R. Reactive halogen chemistry in the troposphere. Chem Soc Rev. 2012;41(19):6448–6472. doi: 10.1039/c2cs35208g. [DOI] [PubMed] [Google Scholar]
- 41.Draxler RR, Rolph GD. 2013. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model access via NOAA ARL READY Web site ( www.arl.noaa.gov/ready/hysplit4.html), NOAA Air Resources Laboratory. Silver Spring, MD.
- 42.Bertram TH, Thornton JA, Riedel TP. An experimental technique for the direct measurement of N2O5 reactivity on ambient particles. Atmos Meas Tech. 2009;2(1):231–242. [Google Scholar]
- 43.Thaler RD, Mielke LH, Osthoff HD. Quantification of nitryl chloride at part per trillion mixing ratios by thermal dissociation cavity ring-down spectroscopy. Anal Chem. 2011;83(7):2761–2766. doi: 10.1021/ac200055z. [DOI] [PubMed] [Google Scholar]
- 44.Ellis RA, et al. Characterizing a Quantum Cascade Tunable Infrared Laser Differential Absorption Spectrometer (QC-TILDAS) for measurements of atmospheric ammonia. Atmos Meas Tech. 2010;3(2):397–406. [Google Scholar]
- 45.Baldocchi DD, Hicks BB, Meyers TP. Measuring biosphere-atmosphere exchanges of biologically related gases with micrometeorological methods. Ecology. 1988;69(5):1331–1340. [Google Scholar]
- 46.Farmer DK, et al. Eddy covariance measurements with high-resolution time-of-flight aerosol mass spectrometry: A new approach to chemically resolved aerosol fluxes. Atmos Meas Tech. 2011;4(6):1275–1289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.