

Abstract
Iron is an essential nutrient for most bacteria. Depending on the oxygen available in the surrounding environment, iron is found in two distinct forms: ferrous (FeII) or ferric (FeIII). Bacteria utilize different transport systems for the uptake of the two different forms of iron. In oxic growth conditions, iron is found in its insoluble, ferric form, and in anoxic growth conditions iron is found in its soluble, ferrous form. Enterobacteriacea, have adapted to transporting the two forms of iron by utilizing the global, oxygen-sensing regulators, ArcA and Fnr to regulate iron transport genes in response to oxygen.
Keywords: iron transport, siderophore, oxygen, regulation
1. Introduction
Iron metabolism and oxygen sensing are closely linked in facultative anaerobic bacteria. In the Enterobacteriacea, a group of gram-negative, facultative anaerobes, iron is required in micromolar concentrations [1]. These bacteria, which include the major human pathogens Shigella, Salmonella, Yersinia and pathogenic Escherichia coli, must be able to obtain iron over a wide range of oxygen concentrations, from the anoxic state of the human colon, to microoxic niches in other body sites, to highly oxic conditions in the external environment [2–6]. Both the availability and oxidation state of iron are highly influenced by the amount of oxygen in the environment. The ability of the bacterium to sense and respond to the levels of both iron and oxygen have significant consequences in preventing either iron starvation or iron toxicity.
Iron is a requirement for the activity of proteins or co-factors involved in a variety of cellular processes. These include TCA cycle enzymes, electron transport chain, and oxygen metabolism [7]. From an evolutionary perspective, the selection of iron for essential functions was ideal. Iron is highly abundant, and before the introduction of oxygen to the environment, iron was readily available in its soluble, ferrous (FeII) form. Iron is an excellent biocatalyst, due to its ability to adopt two stable valences, ferric and ferrous, thereby giving considerable oxidation-reduction potential to iron-containing proteins [8,9]. Once oxygen was introduced into the environment, iron was found more often in its insoluble, oxidized ferric (FeIII) form. Therefore, bacteria evolved mechanisms for solubilizing and transporting ferric iron into the cell. Iron acquisition is even more difficult for bacteria inhabiting the human host, as iron in mammals is primarily found inside cells as heme, metalloproteins or stored in ferritin [10,11]. The small amounts of extracellular iron are tightly bound to carrier proteins such as transferrin, lactoferrin and hemopexin [10]. To compete with their hosts for the available pool of iron, bacterial pathogens have evolved a variety of systems for the acquisition and transport of the metal within the host as well as in the external environment [8,12,13].
While iron is a necessity of the cell, excess iron is toxic, particularly if it is not bound to protein or chelated, since it can act as a catalyst for the Haber-Weiss reaction, in which highly reactive hydroxyl radicals are generated from peroxide and superoxide (Fig. 1). In oxygen-rich environments, incomplete reduction of O2 can lead to formation of superoxide and peroxide. Superoxide can convert ferric iron to ferrous iron, and, in the Fenton reaction, Fe2+ reacts with peroxide to produce hydroxyl radicals and oxidize the iron (Fig. 1) [14, 15]. Hydroxyl radicals cause significant cell damage or death by damaging DNA, resulting in an increase in spontaneous mutations, and by damaging unsaturated lipids and proteins [8]. A variety of reactive oxygen species (ROS) are generated during aerobic metabolism, which can cause oxidative stress in the cell. In addition to hydrogen peroxide, superoxide anion and the hydroxyl radical described above, these reactive species include organic hydroperoxides, nitric oxide, and singlet oxygen. Given the linkage between oxygen and both iron availability and toxicity, it is not surprising that bacteria have linked the regulation of iron transport to the amount of available iron and to the levels of oxygen in the environment. In this review, we describe the major iron transport systems of enteric pathogens and their regulation in response to varying levels of iron and oxygen encountered by the enteric pathogens in the course of an infection.
Figure 1.
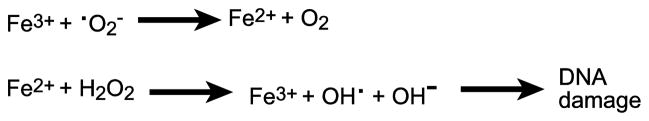
Fenton chemistry. Iron (Fe) catalyzes the production of highly reactive hydroxide radicals (•OH) from superoxide anion (•O2−) and hydrogen peroxide (H2O2).
2. Ferric Iron Transport
In oxygen rich systems, iron is primarily found in the insoluble ferric state. The Enterobacteriacea, like many other bacteria, use low molecular mass compounds, termed siderophores, to solubilize and transport iron [12]. Siderophores have extremely high affinities (Kaff > 1030) for ferric iron [16] and in the enterics, these are usually catechols, secondary hydroxamates or polyketide and non-ribosomal peptide compounds. The bacteria synthesize these compounds and secrete them into the environment, where they bind free ferric iron or sequester it from lower affinity chelates [16]. The iron-siderophore complexes are then transported into the cell using outer membrane receptors that bind the ferri-siderophores with high specificity (Fig. 2) [17]. Bacteria often express additional receptors for siderophores that they do not produce, thereby using siderophores secreted by other microorganisms to scavenge for iron. For example, E. coli produces the catechol siderophore enterobactin, but it is also able to transport the fungal hydroxamate siderophore ferrichrome [18].
Figure 2.
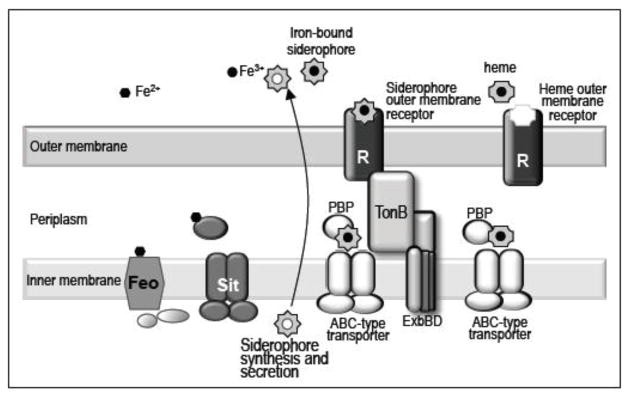
Iron transport systems. Outer membrane receptors (R) specifically bind the ferri-siderophore complexes and transport them across the outer membrane using energy provided by the TonB-ExbB-ExbD complex. Heme is also bound by a specific outer membrane receptor and transported across the outer membrane using energy provided by the TonB-ExbB-ExbD complex. Once in the periplasm, ferri-siderophore or heme is bound by a periplasmic binding protein (PBP) and delivered to a cytoplasmic ABC-type transporter which delivers it to the cytoplasm. Ferrous iron is transported into the cell by either the Sit or Feo systems, which are located in the inner membrane. It is unknown how free ferric and ferrous ions cross the outer membrane.
The energy required to transport the ferri-siderophore across the outer membrane is provided by the TonB-ExbB-ExbD complex (Fig. 2) which transduces the energy from the electrochemical charge gradient of the cytoplasmic membrane to the outer membrane receptor, allowing active transport of ferri-siderophore into the periplasm [2,3]. Once in the periplasm, a periplasmic binding protein relays the iron-siderophore complex to a cytoplasmic ABC-type transporter that delivers the ferri-siderophore into the cytoplasm (Fig. 2) [16,19]. Iron is then removed from the siderophore by reduction or by siderophore degradation [17,20,21]. Although the immediate fate of the released ferrous iron is unknown, it must be rapidly sequestered to prevent damage to the cell.
2.1 Siderophore Biosynthesis
Enterobactin
Enterobactin is a catechol siderophore produced by E. coli, Salmonella, Shigella, and other enteric bacteria. It consists of a cyclic trimer of 2, 3-dihydroxybenzoy serine (Fig. 3A) [16,22,23]. Six enzymes encoded by the entA,B,C,D,E,F genes produce enterobactin from the precursor chorismate [24,25]. Chorismate is derived from the shikimic acid pathway and is a precursor not only for enterobactin, but also for other aromatic compounds such as quinones and aromatic amino acids [26]. Chorismate is converted to isochorismate by isochorismate synthase, EntC [25]. Next, EntB, isochorismatase converts isochorismate to 2, 3-dihydro-2, 3-dihydroxybenzoate [25], which is then converted to 2, 3-dihydroxybenzoate (DHB) by EntA [25]. Finally, enterobactin synthase, a complex of EntD, EntE, EntF and the bifunctional enzyme EntB, combines three molecules of DHB and three serines to form enterobactin [25].
Figure 3.

Structure formulas of (A) Enterobactin, (B) Salmochelin S4, (C) Aerobactin, and (D) Yersiniabactin.
It is interesting to note that the isochorismate synthase, EntC, performs the same enzymatic reaction as MenF [27]. Although both enzymes synthesize isochorismate, the EntC product is channeled into enterobactin and the isochorismate produced by MenF is used for menaquinone synthesis. Thus, an entC mutant is deficient for enterobactin synthesis but produces menaquinone, while a menF mutant produces normal amounts of enterobactin during iron starvation but lacks menaquinone [28]. When E. coli is growing anaerobically, MenF activity is greatly increased and menaquinone synthesis increases under conditions where enterobactin, and thus EntC, are less important [27].
Although the Ent proteins are sufficient for enterobactin production, the peroxiredoxin AhpC enhances enterobactin biosynthesis [29], indicating a link between oxidative stress and siderophore biosynthesis. AhpC is a member of the alkyl hydroperoxide reductase system and catalyzes the reduction of organic hydroperoxides and hydrogen peroxide. Studies in E. coli have shown that AhpC, which contains two active cysteines, operates as a homodimer. In its oxidized state, C46 of one subunit forms a disulfide bond with C165 of the other subunit. AhpC is reduced by AhpF, a flavoprotein reductase. The C46 of one subunit of the reduced AhpC attacks the peroxide, thereby being oxidized to cysteine sulfenic acid, and the C165 of the other subunit reduces the cysteine sulfenic acid and regenerates the disulfide bond with the release of a water molecule [30,31]. C46 is necessary for peroxide reductase activity, while a C146S mutant retains activity [30,31].
In E. coli, an ahpC mutant had reduced growth in iron-limiting medium, and this was linked to a lower internal iron level and a reduction in the amount of DHB produced by the ahpC mutant [29]. The reduced production of DHB was suppressed by providing entC on a multi-copy plasmid indicating that the defect was in the biosynthesis of enterobactin [29]. Interestingly, not only was the reduction of DHB production suppressed by providing entC on a plasmid, it was also suppressed by providing a mixture of aromatic amino acids and para-aminobenzoate which, like enterobactin, are synthesized from chorismate. This suggested that AhpC is either involved in the delivery of chorismate to the enterobactin biosynthesis pathway or in maintaining an optimal concentration of chorismate inside E. coli cells [29]. It is unknown whether or not AhpC peroxidase activity is directly involved in its contribution to the efficiency of enterobactin production, but there is evidence that peroxide stress influences the chorismate pool. Waters et al. [32] identified a small protein, MntS, that may act as a manganese chaperone, and they showed that overproduction of MntS resulted in an increase of entC expression when E. coli was grown with a high manganese concentration. Gerstle et al. [33] showed that the RNA product of this gene, which they named RybA, can function as a small regulatory RNA. RybA, down-regulates aromatic amino acid biosynthesis under peroxide stress, which could increase the availability of chorismate to other biosynthetic pathways, including enterobactin production. These data suggest a link between peroxide stress and an increase in the chorismate pool or in EntC. An ahpC mutant, which should be more susceptible to peroxide stress, would be predicted to have an increase in chorismate available for EntC, resulting in an increase in enterobactin synthesis. However, Ma and Payne [29] showed that an E. coli ahpC mutant had reduced production of enterobactin. Further, a mutant in which the cysteine at position 165 of AhpC is changed to serine (C165S), had reduced enterobactin synthesis and failed to support wild-type growth in iron-limiting medium, even though it retained peroxidase activity, indicating that the effect of AhpC on enterobactin synthesis is not entirely mediated by peroxides [29]. Thus, it remains to be determined how AhpC participates in enterobactin biosynthesis and how it relates to peroxide stress.
2.2 Salmochelin
Enterobactin is further modified by glucosylation in Salmonella species, Shigella dysenteriae, and some E. coli, such as uropathogenic E. coli (Fig. 3B) [34,35]. These modified catechols, termed salmochelins, and their transport proteins are the products of the iroBCDEN genes. IroB glucosylates enterobactin and the resulting salmochelin is secreted by IroC [34]. The other products of this locus are required for transport and utilization of the siderophore [34].
The ability to produce salmochelin gives bacterial pathogens an advantage over their mammalian host. Lipocalin-2, a protein produced by epithelial cells during inflammation and found in granules of neutrophils, binds enterobactin, effectively sequestering the siderophore from the bacteria [36,37]. However, lipocalin-2 cannot bind the glucosylated enterobactin, giving salmochelin-producing pathogens an advantage in transporting iron during infection of a mammalian host [38].
The production of either salmochelin or enterobactin helps protect Salmonella enterica serovar Typhimurium from ROS. A mutant defective in enterobactin synthesis was more sensitive to H2O2 and to paraquat, which generates superoxide [39]. Transport of enterobactin and its ability to supply iron to the cells was not required for protection, since a siderophore transport mutant that still secreted enterobactin was resistant to both compounds. The protective effect appeared to be specific for the catechol structure, and no protection was observed with the non-catechol siderophores aerobactin and yersiniabactin (described below). Achard et al. [39] showed that this resistance to ROS is important in resisting the oxidative burst following uptake of S. enterica by the macrophage. The enterobactin synthesis mutant had lower survival than wild type in interferon gamma (INFγ)–treated macrophages at the early stage of infection.
2.3 Aerobactin
Although enterobactin is produced by many of the Enterobacteriaceae, the hydroxamate siderophore aerobactin is commonly synthesized by enteric pathogens. Aerobactin has a lower affinity for ferric iron than enterobactin, but the ability to produce aerobactin gives selective advantage to pathogens in the host. Unlike enterobactin, aerobactin is not sequestered by serum proteins in the host [40,41]. Production of aerobactin allows pathogens to scavenge ferric iron without host proteins effectively diluting the levels of siderophore they produce [40,42].
Aerobactin is a hydroxamate siderophore (Fig. 3C) that is biosynthesized from lysine and citrate by the products of the iucABCD genes. [43,44]. IucD oxygenates lysine to hydroxylysine [43,44], which is acetylated by IucB to form acetylhydroxylysine [43]. The synthetase, containing IucA and IucC subunits, catalyzes the attachment of two acetylhydroxylysine side chains to citrate, forming aerobactin [44]. Like ferri-enterobactin, the aerobactin-iron complex is transported back into the cell via a specific outer membrane receptor and a periplasmic protein dependent ABC permease complex.
2.4 Yersiniabactin
Yersiniabactin, a siderophore first isolated from Yersinia enterocolitica, is distinct from the catechol and hydroxamate siderophores and contains a phenolic group and thiazolidine and thiazoline rings (Fig. 3D) [45,46]. Yersiniabactin was subsequently identified in Yersinia pestis [47] and other enterics, including pathogenic E. coli [48–51] and Klebsiella [52].
Yersiniabactin is synthesized from salicylate, three cysteines and one malonyl moiety. Four proteins, YbtE, HMWP2, HMWP1, and YbtU, are required for the synthesis of the siderophore in an assembly-line process, in which the intermediates are tethered and passed along multiple domains of the synthase complex [53–56]. HMWP2, a non-ribosomal peptide synthase (NRPS) combines salicylate, activated by YbtE, and two cysteines to a hydroxyphenyl-thiazolinyl-thiazolinyl (HPTT)-S-enzyme intermediate. The intermediate is transferred to HMWP1, a mixed polyketide synthase/NRPS, which completes the synthesis by addition of the malonyl group and condensation and heterocyclization of the final cysteine residue. YbtU reduces the second thiazoline ring to thiazolidine [56].
Interestingly, yersiniabactin can bind copper at physiological concentrations and protects the bacteria against copper toxicity [57]. Cu(II)-yersiniabactin was isolated from the urine of patients with acute E. coli urinary tract infections, indicating that the binding occurs in vivo. Sequestration of copper by yersiniabactin prevents catechol-mediated reduction of Cu(II) to the more toxic Cu(I). Thus, siderophores not only provide iron to the bacteria but also play roles in protection against ROS and metal toxicity during infection of the host.
3. Heme Iron Transport
Heme is the most abundant potential iron source for pathogens infecting mammalian hosts. The bacteria may express receptors for free heme or for heme bound to hemoglobin or hemopexin [58]. Additionally, some species, such as Serratia marcescens and Y. pestis secrete hemophores, proteins that bind free heme or extract heme from hemoglobin and then bind to hemophore receptors on the bacterial surface [59]. The transport of heme into the cell is analogous to siderophore transport (Fig. 2). The TonB-dependent cell surface receptor transports heme across the outer membrane to a periplasmic heme-binding protein, which delivers heme to an ABC transporter for import into the cytoplasm [60,61].
4. Ferrous Iron Transport
In low oxygen environments or in the presence of reducing agents, iron is found primarily in its reduced form, FeII. Most bacteria have systems to transport ferrous iron in these environments. E. coli, Salmonella and Shigella generally use the Feo and Sit systems to transport ferrous iron (Fig. 2).
4.1 Feo
The Feo transport system is found in most bacteria, and in the Enterobacteriaceae it is encoded by the feoABC operon [62–64]. FeoB is a cytoplasmic membrane protein with multiple membrane-spanning regions predicted to form the channel for iron transport. Its structure is distinct from other characterized iron transporters, and it contains a G protein in its cytoplasmic N-terminal region [65]. GTP binding and hydrolysis are essential for FeoB transport function; a mutation in the GTP-binding site resulting in an inability to bind GTP abolishes the ability to transport ferrous iron [65]. FeoA is a small cytoplasmic protein that is required for FeoB-mediated iron transport [66,67] and in Salmonella may act through direct interactions with FeoB [66]. Kim et al. [68] showed that the binding of FeoC to FeoB in S. enterica Typhimurium protected FeoB from proteolytic degradation by the FtsH protease. This resulted in an increase in FeoB and increased iron transport through Feo in low iron, low oxygen condition. FeoC, which has not been identified in all species that have FeoB and is not highly conserved among those that do, is required for Feo iron transport in Vibrio cholerae and interacts with FeoB [67]. However, FeoC was shown not to be required in Yersinia [69].
4.2 Sit
The Sit ferrous iron transport system is encoded by sitABCD. This system is found in S. enterica Typhimurium [70], all Shigella species [71] and some pathogenic E. coli [71], but is generally absent from non-pathogens. An orthologous system named Yfe is found in Yersinia pestis [72]. In S. enterica Typhimurium, the Sit transport system has been shown to transport both iron and manganese [70,73]. Similarly, Sit transports ferrous iron in Shigella flexneri [13], but has overlapping functions with the manganese transporter, MntH, in uptake of manganese [74].
4.3 Ferrous iron transport and virulence
Both Feo and Sit have been shown to have roles in colonization or virulence of the enteric pathogens. A feoB mutant of S. enterica Typhimurium was out-competed by the wild type during colonization of the mouse intestine [75], and the Feo transport system was required for full virulence in a susceptible strain of mice [76]. The Sit transport system is required for replication of S. enterica Typhimurium inside macrophages and for full virulence in mice [76]. The Feo and Sit transport systems are not redundant in S. enterica Typhimurium in that both are required for full virulence, and combining the feo and sit mutations had an additive effect on attenuation [76]. This could be due to differences in regulation or kinetics of the systems, as well as differences in transport of manganese [76].
In S. flexneri, the Sit system appears to be critical for iron acquisition in the host. Shigella spp. are intracellular pathogens, invading and replicating within human colonic epithelial cells. These steps in pathogenesis can be reproduced in cell culture, and virulent Shigella are able to form plaques in epithelial cell monolayers. Because S. flexneri has only three iron transport systems that could be used in host cells, aerobactin synthesis and transport and the Feo and Sit ferrous iron transporters [77], it was possible to make mutants in one or two systems and test the ability of the mutant to use the remaining system for growth inside human cells. A sit mutant was outcompeted by the wild type in epithelial cell monolayers and had reduced virulence in a mouse lung model [77]. A feo mutant was able to invade and multiply in cultured cells [71,78], but a mutant with only the Feo transporter (sitA, iucB double mutant) was able to form plaques only if the cell cultures were maintained under anoxic conditions [78]. This is consistent with iron being more prevalent in ferrous form in the absence of oxygen. A sitA mntH double mutant of S. flexneri, which is defective in manganese transport, was unable to survive in activated macrophage lines and was more sensitive to hydrogen peroxide, suggesting that the Sit transport system may contribute to survival of S. flexneri in macrophages due to its ability to transport both iron and manganese [74]. The transport of manganese by the Sit transport system [74,76] could facilitate the use of the manganese superoxide dismutase, SodA, which would help protect the bacteria against oxidative stress when the function of the iron-containing enzyme SodB is restricted by lack of iron [79].
In Y. pestis, the Yfe (Sit) and Feo ferrous iron transport systems are both required for efficient growth under static, low-oxygen conditions and for disease in a bubonic plaque model [72]. The effects of mutations in these systems were additive, and the double mutant had a more severe phenotype in an experimental infection than either of the single mutants [72]. Taken together, the results of studies on the role of ferrous iron transporters in virulence of enteric pathogens suggest that ferrous iron transport is important in certain stages of infection and that, although the Feo and Sit/Yfe systems both transport ferrous iron, they are not fully redundant.
5. Iron- and oxygen-regulated expression of iron transport systems
Iron transport systems are tightly regulated in response to the level of available iron. In Enterobacteriacea, the primary regulator of iron transport genes is the ferric uptake regulator, Fur [80–82]. Fur responds to the availability of iron in its regulation of iron transport genes and in regulation of the non-coding small RNA, RyhB.
5.1 Response to iron: Fur and RyhB
The Fur protein acts as a transcriptional repressor of iron-regulated promoters, by binding as a dimer to specific sequences upstream of the transcription start site [82]. The binding site, or Fur Box, has been described as the palindrome 5′GATAATGATAATCATTATC3′ or as smaller repeated or overlapping motifs that allow the binding of additional Fur dimers [83]. When iron levels are high, Fur binds Fe2+ allowing its configuration to be appropriate for binding to its DNA target sequences, thereby repressing transcription [82]. Fur regulated genes include those for siderophore biosynthesis, siderophore-dependent ferric iron transporters, and ferrous iron transporters [83–85]. In conditions of low iron availability, the iron-regulated genes are derepressed due to displacement of Fe2+ from Fur and its subsequent release from target DNA sequences.
While the majority of iron-regulated genes are repressed by Fur, some genes were expressed at lower levels in a fur mutant than in wild type, suggesting activation by Fur [83,86]. These genes (e.g. acnA, bfr, ftnA, fumA, sdhCDAB, sodB) encode proteins that have iron or iron-sulfur clusters [83,86]. Masse and Gottesman [87,88] discovered that a small non-coding RNA, RyhB, repressed these genes, and Fur repressed RyhB (Fig. 4). Therefore, in conditions of low iron, RhyB is expressed, and it reduces the expression of a number of genes encoding iron-containing or iron-storage proteins, thereby freeing up iron for essential processes.
Figure 4.

Fur- and RyhB-mediated regulation of iron-regulated genes. In iron-replete conditions, Fur represses genes for iron transport systems, including feo and ent genes, and the small non-coding RNA, RyhB. RyhB expression results in reduced levels of proteins such as aconitase and SodB that contain iron or iron-sulfur clusters. Thus, Fur and RyhB function together to balance the demands for iron in the cell.
Fur auto-regulates its own expression by binding to a Fur box overlapping the fur promoter, repressing its transcription under iron-replete conditions [89]. The transcription of fur is also regulated by the redox regulator, OxyR, which responds to oxidative stress [90]. After activation by hydrogen peroxide, OxyR induces the transcription of a set of antioxidant genes including ahpCF (alkylhydroperoxidase), katG (hydroperoxidase I), gorA (glutathione reductase), and grxA (glutaredoxin I) [91]. Because iron can cause the production of hydroxyl radicals through the Fenton reaction, it is not surprising that OxyR induces the transcription of fur during times of oxidative stress [90]. The increased production of Fur in oxidative stress conditions results in the repression of iron transport genes and the reduction of hydroxyl radicals via the Fenton reaction.
5.2 Response to oxygen: ArcA and Fnr
While iron transport genes are regulated primarily by the availability of iron in the environment, their expression is also modulated by the amount of oxygen in the environment via the transcription factors ArcA and Fnr. This allows bacteria to respond to environments with differing oxygen levels or reducing agents, in order to utilize iron transport systems according to the form of iron available.
ArcA
ArcA is part of the ArcA/B two-component response regulator system and is the regulator of aerobic and microaerobic metabolism [92,93]. The sensor kinase, ArcB, is a membrane-anchored protein that autophosphorylates under anoxic or reducing conditions and subsequently transfers the phosphoryl group to ArcA, thereby stimulating its DNA-binding activity (Fig. 5). This results in repression or activation of genes involved in a variety of catabolic pathways in response to redox growth conditions [94–99]. ArcB activation and inhibition are linked to respiratory growth conditions by the utilization of electron carriers, ubiquinone and menaquinone (Fig. 5), which differ in their abundance under oxic and anoxic conditions [100,101]. Ubiquinone predominates in cells growing aerobically and menaquinone is more abundant in anaerobically growing cells. ArcB contains two cytosol-located redox-active cysteine residues that control the catalytic activity of the protein. Under aerobic growth, ubiquinone oxidizes the two cysteine residues resulting in intermolecular disulfide bond formation which silences the ArcB kinase activity preventing the activation of ArcA [100,102]. Recently, it has been suggested that during a switch from oxic to anoxic conditions or in microaerobic growth, menaquinones reduce the cysteine residues of ArcB causing the disulfide bonds to break, resulting in activation of ArcB kinase activity and subsequent activation of ArcA [101,102].
Figure 5.
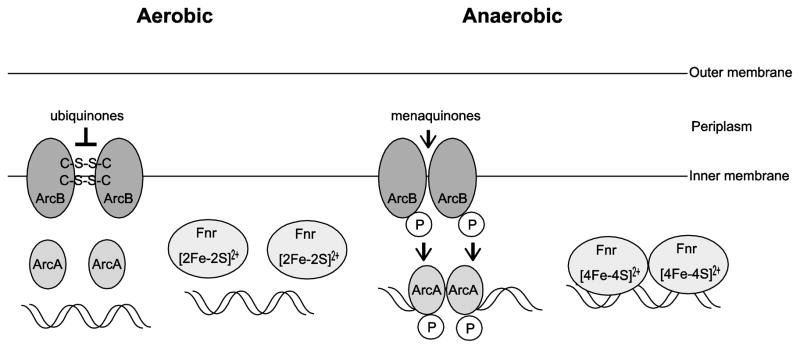
Activities of regulators ArcA and Fnr. Under oxic conditions, ubiquinones oxidize the cysteine residues of ArcB resulting in intermolecular disulfide bond formation and inactivation of ArcB kinase activity. Under microoxic conditions or upon a shift from oxic to anoxic conditions, the ubiquinone pool is replaced by meniquinones, which reduce the cysteine residues of ArcB resulting in breakage of intermolecular disulfide bonds and activation of ArcB kinase activity. ArcB kinase then phosphorylates and activates ArcA. The phosphorylated ArcA binds to target DNA sites to repress or activate transcription. During aerobic metabolism, the iron-sulfur cluster of Fnr is oxidized which prevents Fnr from forming an active homo dimer. During anaerobic metabolism, the iron-sulfur cluster is reduced resulting in the formation of the active complex and binding to target DNA sites.
Fnr
Fnr is the major regulator of anaerobic metabolism. Fnr regulates many genes with differing functions, such as respiratory enzymes, transmembrane substrate carriers, enzymes involved in anaerobic catabolism or fermentation, and genes involved in biosynthetic pathways [103]. Fnr is a cytoplasmic sensor-regulator that contains an oxygen-responsive [4Fe-4S] cluster. Fnr binds specifically to DNA as a dimer under anoxic conditions, and exposure to oxygen results in the loss of its DNA-binding activity (Fig. 5) [104–106]. Fnr is inactivated by oxygen through oxidation of the [4Fe-4S] iron-sulfur cluster in the dimer to a [2Fe-2S] cluster. This favors the the monomeric form of the protein which is unable to bind DNA (Fig. 5) [104–106].
There is often an overlap in the genes regulated by ArcA and Fnr. This overlap allows maximum expression of genes under the appropriate conditions. For example, the cydAB operon, encoding the terminal oxidase of the oxygen respiratory chain, is induced by ArcA and repressed by Fnr under microoxic conditions [107,108].
6. Regulation of iron uptake systems in response to oxygen
Both ArcA and Fnr have been shown to regulate the expression of iron transport systems (Fig. 6). Boulette and Payne [78] showed that in response to oxic environments, S. flexneri repressed the expression of the ferrous iron transporter feo, while inducing the aerobactin biosynthesis and transport genes iucABCD and iutA. In anoxic conditions, the opposite was observed, induction of feo and repression of iuc. S. flexneri employs the transcription factors ArcA and Fnr to differentially regulate its iron transporters in response to oxygen levels (Fig 6) [78]. In low oxygen, ArcA and Fnr act additively to activate feo in S. flexneri, while ArcA represses the iuc siderophore genes (Fig 6). ArcA binds to specific sequences in the S. flexneri feo and iuc promoters. The ArcA binding site in feo is upstream of the predicted transcription start site, while it is downstream in iuc [78]. The difference in the positioning of ArcA relative to the polymerase binding site may explain activation of expression of one operon (feo) but repression of the other (iuc). Similarly, in E coli, Fnr was shown to induce the feoABC promoter under anoxic conditions [63]. A putative ArcA binding site was also identified upstream of the feo operon, but the feo transcript was not derepressed in an arcA mutant, suggesting that factors other than ArcA are involved in anaerobic regulation of feo in E. coli [98]. Transcription from the Y. pestis feo promoter was not affected by mutations in either arcA or fnr and the levels of feo expression were similar when the bacteria were grown aerobically or statically [72]. However, under oxic conditions, regulation by Fur was lost and feo expression was the same in the presence or absence of Fur [72].
Figure 6.
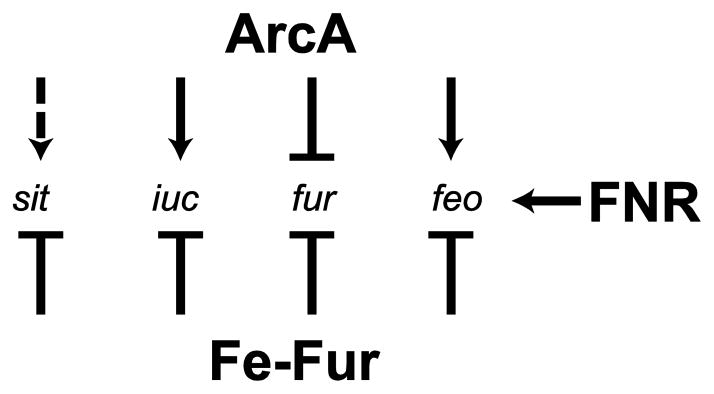
ArcA, Fnr and Fur-mediated regulation of iron transport genes in S. flexneri. The ArcA protein represses the transcription of fur and iuc genes, and induces the transcription of feo, in anoxic conditions. sit expression is low in an arcA mutant growing aerobically, suggesting that ArcA induces transcription of sit in oxic conditions. The Fnr protein induces the transcription of feo in anoxic conditions. All of the iron transport system genes and fur are repressed by Fur when iron is present. Arrowhead = induction, Bar = repression. Dashed line = effect determined by comparing gene expression aerobically and anaerobically in an arcA mutant but direct activation has not been shown.
In addition to binding and repressing the iuc and feo promoters, ArcA was found to bind to the fur promoter of S. flexneri and repress fur expression under anoxic conditions. ArcA regulation of fur was also observed in E. coli. There is an ArcA binding site upstream of fur, and the fur transcript was derepressed in an arcA deletion strain of E. coli [98].
Somewhat surprisingly, the sit genes were induced aerobically in S. flexneri [78] and Salmonella [109], even though they encode a ferrous iron transporter. Induction of the S. flexneri sit genes required ArcA, but ArcA binding to the promoter was not observed, suggesting that the effect was indirect [78]. Induction of the S. flexneri sit operon was also observed when the bacteria are growing intracellularly [110,111], and as noted above, the Sit transporter is required for optimal intracellular growth. These observations suggest that the sit operon is expressed under conditions where oxygen is present, but iron is in the ferrous form, an environment that is likely found in the cytoplasm of the eukaryotic cells that Shigella inhabits. Thus, the up-regulation of sit gene expression noted upon S. flexneri entry into epithelial cells indicates an adaptation to a microoxic environment in which iron is limiting, but the available iron is ferrous, rather than ferric [78,111]. The expression of the sit genes in the intracellular environment may also reflect their role in manganese transport, since sit expression is regulated by the manganese response regulator MntR in both Salmonella [109] and Shigella [74]. Salmonella and Shigella would be able to use the imported manganese to increase their ability to survive under oxidative stress, particularly in macrophages [112].
The effects of oxygen on iron transport systems appear to operate primarily at the level of transcription, but oxygen may also influence the activity of iron transport proteins. In Klebsiella pneumoniae, the FeoC protein contains a [4Fe-4S] cluster, which degraded to [3Fe-4S] upon exposure to oxygen [113]. The presence of an oxygen-sensitive iron-sulfur cluster might allow FeoC to serve as an oxygen sensor for regulation of Feo activity. This model would not apply to FeoC in all species, since the V. cholerae FeoC is required for Feo iron transport but has no cysteines for binding an iron-sulfur cluster.
7. Conclusions
Iron is an essential element for most bacteria, and bacteria have developed systems for transporting both ferrous and ferric forms of iron. The regulation of iron transport systems has been linked both to iron regulators, Fur and RhyB, and to aerobic and anaerobic regulators, including ArcA and Fnr. By using both iron and oxygen responsive regulators, bacteria can optimize their transport of iron while minimizing the potential damaging effects of Fenton radicals.
Table 1.
| Gene | Role in Iron Transport |
|---|---|
| tonB, exbB, exbD | Provides energy for siderophore or heme transport across the outer membrane |
| entABCDEF | Enterobactin production |
| ahpC | Enhances enterobactin production |
| fepABCDE | Enterobactin transport |
| iroB | Glucosylates enterobactin |
| iroCDEN | Salmochelin transport |
| iucABCD | Aerobactin production |
| iutA | Aerobactin outer membrane receptor |
| fhuCDB | Aerobactin periplasmic ABC permease |
| ybtE, HMWP1, HMWP1, ybtU | Yersiniabactin production |
| feoABC | Ferrous iron transport |
| sitABCD | Ferrous iron and manganese transport |
| yfe | Ferrous iron and manganese transport |
Highlights.
Bacteria have multiple systems for transporting iron.
Bacterial iron transport genes are regulated in response to iron and oxygen.
ArcA and Fnr regulate transcription of iron transport genes in response to oxygen.
The peroxidase, AhpC, is required for optimal enterobactin production.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guerinot ML. Annu Rev Microbiol. 1994;48:743–772. doi: 10.1146/annurev.mi.48.100194.003523. [DOI] [PubMed] [Google Scholar]
- 2.Sansonetti PJ. Jpn J Med Sci Biol. 1998;51(Suppl):S69–80. doi: 10.7883/yoken1952.51.supplement1_s69. [DOI] [PubMed] [Google Scholar]
- 3.Philpott DJ, Edgeworth JD, Sansonetti PJ. Philos Trans R Soc Lond B Biol Sci. 2000;355:575–586. doi: 10.1098/rstb.2000.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibarra JA, Steele-Mortimer O. Cell Microbiol. 2009;11:1579–1586. doi: 10.1111/j.1462-5822.2009.01368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rollins SE, Rollins SM, Ryan ET. Am J Clin Pathol. 2003;119(Suppl):S78–85. doi: 10.1309/DQM9-3R8Q-NQWB-FYU8. [DOI] [PubMed] [Google Scholar]
- 6.Clements A, Young JC, Constantinou N, Frankel G. Gut Microbes. 2012;3:71–87. doi: 10.4161/gmic.19182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neilands JB. Struct Bond. Vol. 11. Springer; Berlin Heidelberg: 1972. pp. 145–170. [Google Scholar]
- 8.Wooldridge KG, Williams PH. FEMS Microbiol Rev. 1993;12:325–348. doi: 10.1111/j.1574-6976.1993.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 9.Hall DO, Cammack R, Rao KK. In: Iron Bochemistry Med. Jacobs A, Worwood M, editors. Academic Press; New York: 1974. [Google Scholar]
- 10.Crichton RR, Charloteaux-Wauters M. Eur J Biochem FEBS. 1987;164:485–506. doi: 10.1111/j.1432-1033.1987.tb11155.x. [DOI] [PubMed] [Google Scholar]
- 11.Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 12.Mietzner TA, Morse SA. Annu Rev Nutr. 1994;14:471–493. doi: 10.1146/annurev.nu.14.070194.002351. [DOI] [PubMed] [Google Scholar]
- 13.Wyckoff EE, Boulette ML, Payne SM. Biometals. 2009;22:43–51. doi: 10.1007/s10534-008-9188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touati D. Arch Biochem Biophys. 2000;373:1–6. doi: 10.1006/abbi.1999.1518. [DOI] [PubMed] [Google Scholar]
- 15.Kehrer JP. Toxicology. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- 16.Neilands JB. Siderophores: structure and function of microbial iron transport compounds. J Biol Chem. 1995;270:26723–26726. doi: 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- 17.Andrews SC, Robinson AK, Rodríguez-Quiñones F. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 18.Braun V, Braun M, Killmann H. In: Iron Transp Bact. Crosa JH, Mey AR, Payne SM, editors. ASM Press; Washington, DC: 2004. pp. 158–177. [Google Scholar]
- 19.Krewulak KD, Peacock S, Vogel HJ. In: Iron Transp Bact. Crosa JH, Mey AR, Payne SM, editors. ASM Press; Washington, DC: 2004. pp. 113–129. [Google Scholar]
- 20.Langman L, Young IG, Frost GE, Rosenberg H, Gibson F. J Bacteriol. 1972;112:1142–1149. doi: 10.1128/jb.112.3.1142-1149.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontecave M, Covès J, Pierre JL. Biometals Med. 1994;7:3–8. doi: 10.1007/BF00205187. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien IG, Gibson F. Biochim Biophys Acta. 1970;215:393–402. doi: 10.1016/0304-4165(70)90038-3. [DOI] [PubMed] [Google Scholar]
- 23.Pollack JR, Neilands JB. Biochem Biophys Res Commun. 1970;38:989–992. doi: 10.1016/0006-291x(70)90819-3. [DOI] [PubMed] [Google Scholar]
- 24.Crosa JH, Walsh CT. Microbiol Mol Biol Rev MMBR. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh CT, Marshall GC. In: Iron Transp Bact. Crosa JH, Mey AR, Payne SM, editors. ASM Press; Washington, DC: 2004. pp. 18–37. [Google Scholar]
- 26.Dosselaere F, Vanderleyden J. Crit Rev Microbiol. 2001;27:75–131. doi: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- 27.Kwon O, Hudspeth ME, Meganathan R. J Bacteriol. 1996;178:3252–3259. doi: 10.1128/jb.178.11.3252-3259.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahm C, Müller R, Schulte G, Schmidt K, Leistner E. Biochim Biophys Acta. 1998;1425:377–386. doi: 10.1016/s0304-4165(98)00089-0. [DOI] [PubMed] [Google Scholar]
- 29.Ma L, Payne SM. J Bacteriol. 2012;194:6748–6757. doi: 10.1128/JB.01574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellis HR, Poole LB. Biochemistry (Mosc) 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 31.Jönsson TJ, Ellis HR, Poole LB. Biochemistry (Mosc) 2007;46:5709–5721. doi: 10.1021/bi7001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waters LS, Sandoval M, Storz G. J Bacteriol. 2011;193:5887–5897. doi: 10.1128/JB.05872-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerstle K, Klätschke K, Hahn U, Piganeau N. RNA Biol. 2012;9:458–468. doi: 10.4161/rna.19065. [DOI] [PubMed] [Google Scholar]
- 34.Müller SI, Valdebenito M, Hantke K. Biometals. 2009:691–695. doi: 10.1007/s10534-009-9217-4. [DOI] [PubMed] [Google Scholar]
- 35.Payne SM, Wyckoff EE, Murphy ER, Oglesby AG, Boulette ML, Davies NML. Biometals. 2006:173–180. doi: 10.1007/s10534-005-4577-x. [DOI] [PubMed] [Google Scholar]
- 36.Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 37.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 38.Fischbach MA, Lin H, Zhou L, Yu Y, Abergel RJ, Liu DR, Raymond KN, Wanner BL, Strong RK, Walsh CT, Aderem A, Smith KD. Proc Natl Acad Sci U S A. 2006;103:16502–16507. doi: 10.1073/pnas.0604636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achard MES, Chen KW, Sweet MJ, Watts RE, Schroder K, Schembri MA, McEwan AG. Biochem J. 2013;454:543–549. doi: 10.1042/BJ20121771. [DOI] [PubMed] [Google Scholar]
- 40.Konopka K, Neilands JB. Biochemistry (Mosc) 1984;23:2122–2127. doi: 10.1021/bi00305a003. [DOI] [PubMed] [Google Scholar]
- 41.Moore DG, Earhart CF. Infect Immun. 1981;31:631–635. doi: 10.1128/iai.31.2.631-635.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neilands JB. SMicrobiol Sci. 1984;1:9–14. [PubMed] [Google Scholar]
- 43.de Lorenzo V, Bindereif A, Paw BH, Neilands JB. J Bacteriol. 1986;165:570–578. doi: 10.1128/jb.165.2.570-578.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Lorenzo V, Neilands JB. J Bacteriol. 1986;167:350–355. doi: 10.1128/jb.167.1.350-355.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haag H, Hantke K, Drechsel H, Stojiljkovic I, Jung G, Zähner H. J Gen Microbiol. 1993;139:2159–2165. doi: 10.1099/00221287-139-9-2159. [DOI] [PubMed] [Google Scholar]
- 46.Chambers CE, Sokol PA. J Clin Microbiol. 1994;32:32–39. doi: 10.1128/jcm.32.1.32-39.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perry RD, Balbo PB, Jones HA, Fetherston JD, DeMoll E. Microbiol Read Engl. 1999;145(Pt 5):1181–1190. doi: 10.1099/13500872-145-5-1181. [DOI] [PubMed] [Google Scholar]
- 48.Schubert S, Rakin A, Karch H, Carniel E, Heesemann J. Infect Immun. 1998;66:480–485. doi: 10.1128/iai.66.2.480-485.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koczura R, Kaznowski A. J Med Microbiol. 2003;52:637–642. doi: 10.1099/jmm.0.05219-0. [DOI] [PubMed] [Google Scholar]
- 50.Clermont O, Bonacorsi S, Bingen E. FEMS Microbiol Lett. 2001;196:153–157. doi: 10.1111/j.1574-6968.2001.tb10557.x. [DOI] [PubMed] [Google Scholar]
- 51.Schubert S, Picard B, Gouriou S, Heesemann J, Denamur E. Infect Immun. 2002;70:5335–5337. doi: 10.1128/IAI.70.9.5335-5337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bach S, de Almeida A, Carniel E. FEMS Microbiol Lett. 2000;183:289–294. doi: 10.1111/j.1574-6968.2000.tb08973.x. [DOI] [PubMed] [Google Scholar]
- 53.Geoffroy VA, Fetherston JD, Perry RD. Infect Immun. 2000;68:4452–4461. doi: 10.1128/iai.68.8.4452-4461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gehring AM, DeMoll E, Fetherston JD, Mori I, Mayhew GF, Blattner FR, Walsh CT, Perry RD. Chem Biol. 1998;5:573–586. doi: 10.1016/s1074-5521(98)90115-6. [DOI] [PubMed] [Google Scholar]
- 55.Gehring AM, Mori I, Perry RD, Walsh CT. Biochemistry (Mosc) 1998;37:17104. doi: 10.1021/bi9850524. [DOI] [PubMed] [Google Scholar]
- 56.Miller DA, Luo L, Hillson N, Keating TA, Walsh CT. Chem Biol. 2002;9:333–344. doi: 10.1016/s1074-5521(02)00115-1. [DOI] [PubMed] [Google Scholar]
- 57.Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP. Nat Chem Biol. 2012;8:731–736. doi: 10.1038/nchembio.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skaar EP. PLoS Pathog. 2010;6:e1000949. doi: 10.1371/journal.ppat.1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Létoffé S, Delepelaire P, Wandersman C. J Bacteriol. 2004;186:4067–4074. doi: 10.1128/JB.186.13.4067-4074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mills M, Payne SM. J Bacteriol. 1995;177:3004–3009. doi: 10.1128/jb.177.11.3004-3009.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wandersman C, Stojiljkovic I. Curr Opin Microbiol. 2000;3:215–220. doi: 10.1016/s1369-5274(00)00078-3. [DOI] [PubMed] [Google Scholar]
- 62.Stojiljkovic I, Cobeljic M, Hantke K. FEMS Microbiol Lett. 1993;108:111–115. doi: 10.1111/j.1574-6968.1993.tb06082.x. [DOI] [PubMed] [Google Scholar]
- 63.Kammler M, Schön C, Hantke K. J Bacteriol. 1993;175:6212–6219. doi: 10.1128/jb.175.19.6212-6219.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cartron ML, Maddocks S, Gillingham P, Craven CJ, Andrews SC. Biometals. 2006;19:143–157. doi: 10.1007/s10534-006-0003-2. [DOI] [PubMed] [Google Scholar]
- 65.Marlovits TC, Haase W, Herrmann C, Aller SG, Unger VM. Proc Natl Acad Sci U S A. 2002;99:16243–16248. doi: 10.1073/pnas.242338299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim H, Lee H, Shin D. Biochem Biophys Res Commun. 2012;423:733–738. doi: 10.1016/j.bbrc.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 67.Weaver EA, Wyckoff EE, Mey AR, Morrison R, Payne SM. J Bacteriol. 2013 doi: 10.1128/JB.00738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim H, Lee H, Shin D. J Bacteriol. 2013;195:3364–3370. doi: 10.1128/JB.00343-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perry RD, Mier I, Jr, Fetherston JD. Biometals. 2007;20:699–703. doi: 10.1007/s10534-006-9051-x. [DOI] [PubMed] [Google Scholar]
- 70.Janakiraman A, Slauch JM. Mol Microbiol. 2000;35:1146–1155. doi: 10.1046/j.1365-2958.2000.01783.x. [DOI] [PubMed] [Google Scholar]
- 71.Runyen-Janecky LJ, Reeves SA, Gonzales EG, Payne SM. Infect Immun. 2003;71:1919–1928. doi: 10.1128/IAI.71.4.1919-1928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fetherston JD, Mier I, Jr, Truszczynska H, Perry RD. Infect Immun. 2012;80:3880–3891. doi: 10.1128/IAI.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kehres DG, Janakiraman A, Slauch JM, Maguire ME. J Bacteriol. 2002;184:3159–3166. doi: 10.1128/JB.184.12.3159-3166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Runyen-Janecky L, Dazenski E, Hawkins S, Warner L. Infect Immun. 2006;74:4666–4672. doi: 10.1128/IAI.00562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsolis RM, Bäumler AJ, Heffron F, Stojiljkovic I. Infect Immun. 1996;64:4549–4556. doi: 10.1128/iai.64.11.4549-4556.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyer E, Bergevin I, Malo D, Gros P, Cellier MFM. A Infect Immun. 2002;70:6032–6042. doi: 10.1128/IAI.70.11.6032-6042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fisher CR, Davies NMLL, Wyckoff EE, Feng Z, Oaks EV, Payne SM. Infect Immun. 2009;77:1992–1999. doi: 10.1128/IAI.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boulette ML, Payne SM. J Bacteriol. 2007;189:6957–6967. doi: 10.1128/JB.00621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fridovich I. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 80.Hantke K. Mol Gen Genet MGG. 1981;182:288–292. doi: 10.1007/BF00269672. [DOI] [PubMed] [Google Scholar]
- 81.Hantke K. Mol Gen Genet MGG. 1984;197:337–341. doi: 10.1007/BF00330982. [DOI] [PubMed] [Google Scholar]
- 82.Bagg A, Neilands JB. Biochemistry (Mosc) 1987;26:5471–5477. doi: 10.1021/bi00391a039. [DOI] [PubMed] [Google Scholar]
- 83.Escolar L, Pérez-Martín J, de Lorenzo V. J Bacteriol. 1999;181:6223–6229. doi: 10.1128/jb.181.20.6223-6229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Escolar L, de Lorenzo V, Pérez-Martín J. Mol Microbiol. 1997;26:799–808. doi: 10.1046/j.1365-2958.1997.6211987.x. [DOI] [PubMed] [Google Scholar]
- 85.Escolar L, Pérez-Martín J, de Lorenzo V. J Bacteriol. 1998;180:2579–2582. doi: 10.1128/jb.180.9.2579-2582.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hantke K. Curr Opin Microbiol. 2001;4:172–177. doi: 10.1016/s1369-5274(00)00184-3. [DOI] [PubMed] [Google Scholar]
- 87.Massé E, Gottesman S. Proc Natl Acad Sci U S A. 2002;99:4620–4625. doi: 10.1073/pnas.032066599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Massé E, Vanderpool CK, Gottesman S. J Bacteriol. 2005;187:6962–6971. doi: 10.1128/JB.187.20.6962-6971.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Lorenzo V, Herrero M, Giovannini F, Neilands JB. Eur J Biochem FEBS. 1988;173:537–546. doi: 10.1111/j.1432-1033.1988.tb14032.x. [DOI] [PubMed] [Google Scholar]
- 90.Zheng M, Doan B, Schneider TD, Storz G. J Bacteriol. 1999;181:4639–4643. doi: 10.1128/jb.181.15.4639-4643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Green J, Paget MS. Nat Rev Microbiol. 2004;2:954–966. doi: 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- 92.Iuchi S, Weiner L. J Biochem (Tokyo) 1996;120:1055–1063. doi: 10.1093/oxfordjournals.jbchem.a021519. [DOI] [PubMed] [Google Scholar]
- 93.Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ. J Bacteriol. 2003;185:204–209. doi: 10.1128/JB.185.1.204-209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gunsalus RP, Park SJ. Res Microbiol. 1994;145:437–450. doi: 10.1016/0923-2508(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 95.Park SJ, McCabe J, Turna J, Gunsalus RP. J Bacteriol. 1994;176:5086–5092. doi: 10.1128/jb.176.16.5086-5092.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park SJ, Tseng CP, Gunsalus RP. Mol Microbiol. 1995;15:473–482. doi: 10.1111/j.1365-2958.1995.tb02261.x. [DOI] [PubMed] [Google Scholar]
- 97.Chao G, Shen J, Tseng CP, Park SJ, Gunsalus RP. J Bacteriol. 1997;179:4299–4304. doi: 10.1128/jb.179.13.4299-4304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu X, De Wulf P. J Biol Chem. 2004;279:12588–12597. doi: 10.1074/jbc.M313454200. [DOI] [PubMed] [Google Scholar]
- 99.Lynch AS, Lin EC. J Bacteriol. 1996;178:6238–6249. doi: 10.1128/jb.178.21.6238-6249.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Georgellis D, Kwon O, Lin EC. Science. 2001;292:2314–2316. doi: 10.1126/science.1059361. [DOI] [PubMed] [Google Scholar]
- 101.Bekker M, Alexeeva S, Laan W, Sawers G, Teixeira de Mattos J, Hellingwerf K. J Bacteriol. 2010;192:746–754. doi: 10.1128/JB.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alvarez AF, Rodriguez C, Georgellis D. J Bacteriol. 2013;195:3054–3061. doi: 10.1128/JB.00406-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Unden G, Bongaerts J. Biochim Biophys Acta. 1997;1320:217–234. doi: 10.1016/s0005-2728(97)00034-0. [DOI] [PubMed] [Google Scholar]
- 104.Melville SB, Gunsalus RP. Proc Natl Acad Sci U S A. 1996;93:1226–1231. doi: 10.1073/pnas.93.3.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. J Biol Chem. 1996;271:2762–2768. doi: 10.1074/jbc.271.5.2762. [DOI] [PubMed] [Google Scholar]
- 106.Green J, Bennett B, Jordan P, Ralph ET, Thomson AJ, Guest JR. Biochem J. 1996;316(Pt 3):887–892. doi: 10.1042/bj3160887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cotter PA, Melville SB, Albrecht JA, Gunsalus RP. Mol Microbiol. 1997;25:605–615. doi: 10.1046/j.1365-2958.1997.5031860.x. [DOI] [PubMed] [Google Scholar]
- 108.Govantes F, Albrecht JA, Gunsalus RP. Mol Microbiol. 2000;37:1456–1469. doi: 10.1046/j.1365-2958.2000.02100.x. [DOI] [PubMed] [Google Scholar]
- 109.Ikeda JS, Janakiraman A, Kehres DG, Maguire ME, Slauch JM. J Bacteriol. 2005;187:912–922. doi: 10.1128/JB.187.3.912-922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Runyen-Janecky LJ, Payne SM. Infect Immun. 2002;70:4379–4388. doi: 10.1128/IAI.70.8.4379-4388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lucchini S, Liu H, Jin Q, Hinton JCD, Yu J. Infect Immun. 2005;73:88–102. doi: 10.1128/IAI.73.1.88-102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kehres DG, Maguire ME. FEMS Microbiol Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 113.Hsueh KL, Yu LK, Chen YH, Cheng YH, Hsieh YC, Ke S, Hung KW, Chen CJ, Huang TH. J Bacteriol. 2013;195:4726–4734. doi: 10.1128/JB.00687-13. [DOI] [PMC free article] [PubMed] [Google Scholar]