

Abstract
Allo-antibodies, particularly when donor specific, are one of the most important factors that cause both early and late graft dysfunction. The authors review the current state of the art concerning this important issue in renal transplantation. Many antibodies have been recognized as mediators of renal injury. In particular donor-specific-Human Leukocyte Antigens antibodies appear to play a major role. New techniques, such as solid phase techniques and Luminex, have revealed these antibodies from patient sera. Other new techniques have uncovered alloantibodies and signs of complement activation in renal biopsy specimens. It has been acknowledged that the old concept of chronic renal injury caused by calcineurine inhibitors toxicity should be replaced in many cases by alloantibodies acting against the graft. In addition, the number of patients on waiting lists with preformed anti-human leukocyte antigens (HLA) antibodies is increasing, primarily from patients with a history of renal transplant failure already been sensitized. We should distinguish early and late acute antibody-mediated rejection from chronic antibody-mediated rejection. The latter often manifets late during the course of the post-transplant period and may be difficult to recognize if specific techniques are not applied. Different therapeutic strategies are used to control antibody-induced damage. These strategies may be applied prior to transplantation or, in the case of acute antibody-mediated rejection, after transplantation. Many new drugs are appearing at the horizon; however, these drugs are far from the clinic because they are in phase I-II of clinical trials. Thus the pipeline for the near future appears almost empty.
Keywords: Donor-specific antibodies, Solid-phase techniques, Complement activation, Renal transplantation, Antibody-mediated rejection, Desensitization, New drugs for B-cells
Core tip: Clear evidence exists that shows that donor-specific-HLA antibodies (DSAs) are the primary players in the acute and chronic deterioration of graft. The emergence of sensitive techniques that detect DSAs, together with advances in the assessment of graft pathology, has enabled an improved understanding of antibody-mediated graft injury. Acute and chronic antibody-mediated rejection conditions have changed the nomenclature during recent Banff conferences and have enabled the dismissal of older terminologies, such as chronic allograft nephropathy. Therapies aimed at B cells and plasma cells and that control complement activation will be extremely important for improving long-term outcomes in kidney transplantations.
INTRODUCTION
Despite improvements in renal transplantation outcomes, kidney allograft loss remains substantial and is associated with increased morbidity, mortality and costs[1,2]. Clearly, the identification of the critical pathologic pathways underlying allograft loss and the development of therapeutic interventions that improve the duration and quality of allograft function are among the most important targets for transplant medicine. One of the most important advances of the past decade has been the realization that the insufficient control of the humoral arm of a recipient’s immune system by current immunosuppressive regimens[3] is the factor primarily responsible for allograft dysfunction and loss[4-6].
ALLOGRAFT ANTIBODY EVOLUTION IN TRANSPLANTATION
The induction of allograft injury alloantibodies induced has now superseded the historical dogma that allograft losses were caused by the toxicity of calcineurin inhibitors (CNIs) and by chronic allograft nephropathy (CAN). Indeed, nephrotoxicity and CAN as causes of late graft failure are being challenged by the findings of the Long-Term Deterioration of Kidney Allograft Function (DeKAF)[6-8] and other studies[9,10].
In addition, recent therapeutic strategies that have permitted the human leukocyte antigens (HLA) to be crossed have created a new population at risk of antibody-mediated rejection (ABMR), which has enabled these patients to be studied over an extended time period.
The emergence of sensitive techniques that detect donor-specific anti-HLA antibodies (DSAs) and other HLA and non-HLA antibodies together with advances in the assessment of graft pathology have expanded the spectrum of ABMR.
The different technologies used by researchers and the significance of alloantibodies found by these technologies led recently to a consensus conference that elaborated upon consensus guidelines for testing and clinical management issues associated with HLA and non-HLA antibodies in transplant recipients[11] .
As a consequence of this increase in knowledge, the term CAN was deleted in the Banff’05 meeting report[12]. In the Banff’07 and Banff’09 conferences[13,14], the concept of ABMR was further evaluated, and ABMR was definitively included in the Banff classifications.
The Banff’11 meeting report[15] and the recent Banff’13 conference (unpublished data) further elaborated upon new concepts in ABMR, which included the significance of C4d-negative and C1q-positive ABMR.
DETECTION OF ANTIBODIES AND THEIR SIGNIFICANCE
Preformed antibodies targeted against HLAs or antibodies formed de novo after transplantation predispose to either acute or chronic ABMR. These antibodies can be detected using several techniques.
A complement-dependent lymphocytotoxicity (CDC) cross-match is typically performed to detect cytotoxic DSAs. The main disadvantages of the CDC assay are that it is subjective and cumbersome and will only detect complement-fixing antibodies[16]. Indeed, ABMR has occurred in patients with a negative cross-match. This observation may indicate that the CDC lacks the sensitivity required to detect some clinically significant antibodies; moreover, acute ABMR can occur in recipients with immunological memory and undetectable levels of circulating HLA antibodies at the time of transplant[17]. Cross-match (XM) and antibody detection techniques have improved with time and show increased sensitivity and specificity[18,19].
Flow cytometry (FC) is another cell-based technique that was introduced more than 20 years ago to improve sensitivity. This test also lacks specificity, and with the introduction of solid-phase assays (SPA), the use of FC has been superseded. The introduction of SPA detection, while providing greater sensitivity than CDC assays, has resulted in a new paradigm with respect to the interpretation of DSAs. Although SPA using the Luminex instrument has permitted the detection of antibodies not detectable by CDC, the clinical significance of these antibodies is not fully understood. In addition, SPA testing raises technical issues that require resolution and careful consideration when interpreting antibody results. SPA, such as flow cytometry using antigen-coated micro particles, enzyme-linked immunosorbent assays (ELISAs) and Luminex, are now used to determine the specificity of anti-HLA antibodies and to better interpret positive CDC-XM results.
ELISA has an advantage over the CDC because is more sensitive and detects antibodies that not fix complement. However, non-specific binding to other immunoglobulins may occur in patients with autoimmune disorders. When detecting antibodies using flow panel-reactive antibody (PRA) beads, micro-particles are coated with purified HLA molecules[19]. The fluorescence is then measured using flow cytometry and the level of fluorescence is indicative of the level of antibody binding.
Luminex technology also uses pools of HLA class Ior II antigen-coated micro-particles. These beads are colored with a combination of two dyes. Serum reactivity is assessed based on the fluorescent signal of each HLA-coated micro particle[19]. The Luminex platform enables the determination of DSAs specificity by using single HLA-coated beads and provides a relative indication of the antibody strength and level in the circulation by returning results to the user in the form of mean fluorescence intensity (MFI)[20]. However, MFI is not standardized across labs, and there is some arbitrariness in determining the MFI thresholds. Molecules with equivalent soluble fluorochrome (MESF) and maximum fluorescence values, obtained using the Luminex machine, enable more standardized measures of antibody strength[21,22].
According to the consensus publication by the National Conference to Assess Antibody-Mediated Rejection in Solid Organ Transplantation[23], a current positive CDC or anti-human immunoglobulin-CDC (CDC-AHG) cytotoxicity cross-match (CXM) predisposes to a high risk of ABMR or early graft loss. A current positive CDC or CDC-AHG CXM is a contraindication for transplantation unless DSAs can be reduced using desensitization protocols. A positive flow CXM or a remote (historic) positive CDC or CDC-AHG CXM poses an intermediate risk for early acute rejection and may require augmented immunosuppression.
The wide use of the Luminex technique with its increased specificity and sensitivity did uncover a new paradigm. Using the Luminex technique DSAs have been found in patients who show a negative classic CDC. Several studies have shown that these results represent a risk factor, but not a formal contraindication for transplantation[24-26].
These facts lead to workshops and Consensus Guidelines to further understand these technologies[11,27].
The recent consensus guidelines highlighted the technological advantages and limitations of Luminex as shown in Table 1.
Table 1.
Technological advantages and limitations of luminex human leukocyte antigens single antigen bead
| Technological advantages | Technological limitations |
| Qualitative: enables precise identification of all antibody specificities in complex sera (DSA) | Some positive results can be caused by antibodies to denatured HLA |
| Comprehensive: distinguishes antibodies to all common alleles for HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DRB3/4/5, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1 | Occasional high background binding requiring repeat testing and absorption protocols |
| Semiquantitative: enables determination of antibody levels (high, intermediate, and low) | Variable HLA protein density on beads. Blocking factors may cause false-negative or misleading low assessment of antibody levels (prozone?); IgM and C1 can block IgG binding |
| Sensitive: enables detection of weak antibody testing | |
| Rapid: enables real-time antibody monitoring for DSA. Pre- transplantation and post-transplantation antibody monitoring (assist diagnosis of ABMR). Virtual XM | Lot-to-lot variation requiring validation. Vendor-specific variation |
| Enables detection of non-HLA-specific antibodies (e.g., MICA) | |
| Detection and differentiation between immunoglobulin class and isotype (e.g., complement fixing and non-complement fixing C4d and C1q | Reagents not standardized |
ABMR: Antibody mediated rejection; DSA: Donor specific HLA antibodies; HLA: Human leukocyte antigens; MICA: Major histocompatibility complex class I-related chain A; SAB: Single-antigen beads; XM: Cross-match.
In addition, the consensus guidelines[11] considered the following modifications to SPA for detecting new antibodies and assessing their functionality.
C4d assay
The C4d assay[28,29] shows superior specificity compared with the CDC. The C4d assay requires complement activation to occur and is influenced by complement regulatory factors. Clinical data obtained using various modifications of the C4d assay have shown that the presence of C4d+ antibodies correlates with graft survival in the kidney and hearts[28,29].
C1q assay
The C1q assay was designed to distinguish complement-fixing from non-complement-fixing antibodies and does not require complement activation other than the binding of C1q to the antibody[30]. It detects antibodies capable of binding complement and initiating the classical pathway.
The results of this technique still remain under debate. Although some authors[31] have reported no correlation with the clinical course in kidney transplant patients, others[32] have reported that both C1q and C4d Luminex assays show increased sensitivity and specificity and that they can be useful for both pre-transplant risk assessment and post-transplant monitoring.
Detection of antibodies targeted to non-HLA antigens
The endothelial cell is the principal target used to detect non-HLA antibodies involved in ABMR. Historically, different assay systems have been used to identify and characterize AECA including CDC[33], flow cytometry[34] and immunofluorescence[35].
The primary limitation is that the endothelial cells used for the detection and characterization of AECA have been derived from third-party donors, and that the cells used show different protein expressions and distinct phenotypes[36]. Surrogates of endothelial cells, such as MICA may be useful. However, MICA is not expressed constitutively on the endothelium; rather, its expression is induced under conditions of cellular stress.
Lymphocyte XM tests fail to detect AECA. The cross-match ONE assay is a Food and Drug Administration (FDA)-approved endothelial ECXM technique that uses endothelial cell precursor cells found in the peripheral blood at a frequency of 1%-2%[37]. An advantage of this test is that it detects DSAs and can be used to test for antibodies targeted to T lymphocytes, B lymphocytes and endothelial cells in the same assay[38].
Proteomics approaches using protein extracts from different sources, including cell lysates and protein microarrays are being used for antibody screening and identification of specificities[39,40].
A variety of non-HLA targets have been identified including MICA, vimentin, angiotensin II type 1 receptor, tubulin, myosin and collagen V. In general, single antigen bead (SAB) testing permits reassessment of the immunologic risk for kidney transplantation. Traditionally, high panel-reactive antibody, re-transplant and deceased donor grafts have been associated with increased risk. However, the risk factors for ABMR are DSAs, reduced HLA matching and evaluation of DSAs using different techniques[41].
PATHOPHYSIOLOGY
An increasing body of evidence suggests that patients with high titers of anti-HLA antibodies (particularly if they are donor-specific) that develop either pre-transplant or post-transplant, show a worse outcome. At any given time, approximately 25% of transplant recipients show antibodies against HLA antigens when evaluated using the newest, highly sensitive and specific techniques for DSAs monitoring[42,43]. Moreover, antibodies against non-HLA have also been implicated in ABMR[44]. Antibodies may mediate endothelial injury via complement-dependent or independent mechanisms by transducing signals that are pro-inflammatory and proliferative[45].
Preformed or de novo DSAs clearly cause acute and chronic ABMR; however the role and scope of non-HLA antibodies in mediating graft injury and loss remains less certain[46].
One hypothesis is that alloantigen sensitization occurs based on non-HLA polymorphic differences between the donor and the recipient [e.g., major-histocompatibility-complex (MHC) class I-related chains A and B (MICA and MICB, respectively)]. Unfortunately progress in this area has been limited by a lack of validated clinical assays for non-HLA alloantibodies, the confounding presence of HLA-DSAs and, in the case of MICA antibodies, a lack of proof of specificity[47].
A second hypothesis is that auto antigen sensitization occurs due to exposure of cryptic epitopes after tissue injury or inflammation (including vimentin, K-α I tubulin, collagen V and agrin).
Although anti-HLA antibodies are responsible for the majority of antibody-mediated injuries, they do not underlie all ABMRs. In addition, as discussed above, the major histocompatibility antigens and a large number of minor antigens have been recognized as possible antibody targets[48-50].
Endothelial cells are targets for immune-mediated assaults via anti-endothelial cell antibodies (AECAs). The de novo development of circulating anti-endothelial cell antibodies, rather than pre-existing antibodies, is associated with post-transplant allograft rejection[51].
Apoptotic endothelial cells (ECs) release a bioactive C-terminal fragment of perlecan called laminin G-like 3 (LG3)[52]. LG3 behaves as a neo-antigen and induces the production of anti-LG-3 antibodies. Recently, these anti LG-3 antibodies have been documented to be novel accelerators of immune-mediated vascular injury and to obliterate remodeling[53].
Vimentin[54], collagen V[55] and K-α 1 tubulin[56] are involved in the ABMR of organ other than kidney as neo-antigens. The apoptosis of ECs and subsequent exposure of neo-antigens may induce an autoimmune response.
An autoantibody specific for angiotensin II receptor type 1 has been associated with the development of hypertensive vasculopathy and acute renal allograft dysfunction[57]. Antibodies directed towards MHC class I polypeptide-related sequences A (MICA) and B (MICB), and not classical HLA molecules, have been implicated in transplant rejection in recipients who were otherwise well-matched for HLA due to the contribution of MICA antigens towards the activation of cellular and humoral immune responses[58].
The HLA complex encodes molecules crucial for the initiation and proliferation of the immune response. It is highly polymorphic and polygenic and its proteins are co-dominantly expressed. The HLA genes that are involved in the immune response belong to classes I and II, which are structurally and functionally different. Recently, DSAs have been reported to activate endothelial cells, thereby increasing their potential to recruit and bind recipient leukocytes and increasing the potential for allograft inflammation[59,60].
Approximately 30% of patients on waiting list show detectable levels of HLA antibodies[61]. After transplantation, 25% of non-sensitized patients develop de novo HLA-DSAs.
In both groups of patients, the presence of these antibodies increases the risk of subsequent ABMR[9]. The development of a histological test to identify antibody-mediated complement activation on transplant biopsies (C4d staining) has provided a method for flagging potentially deleterious interactions between antibodies and the graft endothelium. In addition, molecular techniques, such as gene expression profiling, have enabled the identification of subclinical endothelial cell damage that can be present even in the absence of complement activation or detectable DSA[62]. Recent studies have documented the role of B cells and antibodies in transplantation. A study by Lynch et al63] described a technique that may enable a more global assessment of B-cell reactivity to the allograft. Their results suggest that humoral responses to the allograft may be more common than previously appreciated. Antibodies reactive to donor human leukocyte antigen molecules, minor histocompatibility antigens, endothelial cells, red blood cells or auto antigens may trigger or contribute to rejection at both early and late time points after transplantation[64]. Often, the immune system shows an integrated response that results in allograft rejection involving parallel or simultaneous T cell mediated rejection (TCMR) and ABMR[65]. Antibody-mediated injury to the allograft is initiated by DSAs binding to HLA antigens or to other targets on the allograft endothelium. If DSAs are complement-activating, the classic complement pathway is rapidly activated via IgG binding and C1q activation[66]. This process typically results in the rapid loss of the allograft. Alternatively, DSAs can bind endothelial cell targets and stimulate cell proliferation or induce antibody-dependent cell-mediated cyto-toxicity with interferon γ release[45]. These processes appear to be more important for the development of the chronic antibody-mediated injury that is more dependent on natural killer (NK) cells than the complement[67]. Antibodies may also bind HLA and other targets and incompletely activate the complement system without causing apparent injury. This process is referred to as accommodation[68].
ABMR is a continuous process, and its oscillation is characterized by fluctuations in DSAs, C4d deposition and dynamic and multidirectional glomerulitis and/or capillaritis scores[69]. The time to diagnosis of ABMR is highly dependent on the population studied. Early-onset ABMR (typically occurring within the first months after transplantation) is observed predominantly in patients with preformed DSAs, whereas late acute ABMR occurs primarily in patients who develop de novo DSAs after transplantation. Indeed, the pathologic and clinical manifestations may vary, including hyper-acute humoral rejection, acute humoral rejection, indolent or subclinical humoral rejection, “C4d”-negative humoral rejection and late acute humoral rejection.
ACUTE ABMR
Hyper-acute ABMR
The pathology of hyper-acute rejection overlaps completely with acute ABMR. It arises within minutes or a few hours after transplantation in pre-sensitized patients who have circulating HLA, AB0, or other alloantibodies to the donor endothelial surface antigens[70]. The outcome is always poor.
Acute ABMR
The diagnosis of acute ABMR relies upon the criteria shown: (1) morphologic evidence of acute tissue injury: acute tubular injury, neutrophils and/or mononuclear cells in PTC and/or glomeruli and/or capillary thrombosis, fibrinoid necrosis/intramural or trans-mural inflammation in arteries; (2) immuno-pathologic evidence for antibody action: C4d and/or (rarely) immunoglobulin in PTC; Ig and complement in arterial fibrinoid necrosis; and (3) serologic evidence of circulating antibodies to donor HLA or other anti-donor endothelial antigen. The endothelial injury has been recently reviewed completely by Drachenberg and colleagues[71]. Although acute ABMR generally occurs within the first year after transplantation in pre-sensitized patients[72], it may also develop years after transplantation and is often triggered by a decrease in immunosuppression (iatrogenic, non-compliance or malabsorption)[5,73-75].
Several patients with acute ABMR show a negative cross-match, which may be due to low level DSAs that are undetectable[76] or to de novo DSAs[77].
Recently, an increased risk of acute ABMR has been associated to elevated pre-transplantation soluble B-cell activating factor (BAFF)[78], whose neutralization may be an interesting therapeutic strategy.
Recently, Orandi et al[79] examined the long-term effect of early acute ABMR on kidney allograft and patient survival in 201 adult kidney transplant recipients who developed acute ABMR within the first year after transplantation. Each recipient was matched with 5 control patients. The majority of recipients were sensitized. Allograft survival rates at 1, 5 and 10 years in the group that developed acute ABMR were significantly lower than in the control group.
In another study[60] of a cohort of 355 adult kidney transplant recipients, all with a negative CDC-XM, C1q-fixing DSAs did not predict acute ABMR or allograft loss; however, the presence of class I DSAs (versus class II donor specific antigens) predicted acute ABMR and allograft loss.
Indolent or subclinical acute ABMR
Chronic rejection is often preceded by the occurrence of an acute ABMR due to the fact that modern therapeutic strategies fail to deplete antibody secreting plasma cells from the spleen and bone marrow of patients[80].
In addition, kidney transplant recipients who develop de novo DSAs are now recognized to often show pathologic features of indolent and slowly progressive micro-vascular abnormalities, which are referred referred to occasionally as subclinical acute ABMR[16,77,81]. The appearance of de novo DSAs likely results from inadequate immunosuppression and represents a dynamic process that begins early after transplantation and continues at varying levels thereafter.
C4d negative acute ABMR
Initial evidence for C4d-negative acute ABMR emerged in 2009 based on the work of the teams in Paris[69] and Edmonton[62]. The latter study demonstrated high endothelial-specific gene expression in kidney transplant biopsy samples with DSAs but without C4d. In this study, C4d-negative acute ABMR was characterized by the high intragraft endothelial gene expression of allo-antibodies, by histology typical of chronic or acute ABMR and by poor outcomes. Several hypotheses have been generated to explain the lack of complement deposition despite the evidence of micro-vascular inflammation and persistence of DSAs in the circulation. The low sensitivity of C4d[13,82] could be due to technical issues including the type of fixative used and the different methods used to detect C4d (immunofluorescence versus immunochemistry). Moreover, as documented by the Edmonton study, some DSAs, although showing poor complement-fixing ability, may nonetheless activate endothelial cells[62]. Another possibility is that the various prophylactic strategies used to prevent ABMR may decrease the burden of complement activation within capillaries[80].
Given the concerns over the lack of sensitivity of C4d for kidney transplantations, a working group was established at the 2011 Banff conference to refine the criteria used for diagnosis of ABMR in the kidney[15]. Although the 2013 Banff Conference, held in Fortaleza (Brazil) in August 2013, has ended, to the best of our knowledge, this work remains in progress.
Late acute ABMR
If the majority of early-acute ABMR depends upon preformed DSAs and primarily occurs in sensitized patients, late-acute ABMR often depends upon de novo DSAs.
De novo DSAs appear in 25% of non-sensitized patients[77]. De novo DSAs are often linked to late-acute ABMR and are characterized as occurring in patients who are young, with frequent non-adherence or suboptimal immunosuppression[74]. The observation that many cases of de novo DSAs are associated with prior therapy non-adherence or with a history of a clinical acute cellular rejection episode, suggests that immunosuppression is a potent inhibitor of the activation of mature, naïve B cells[83]. However, the observation that some cases of de novo DSAs formation appear in compliant patients suggests that either T cells capable of helping naïve B cells emerge despite immunosuppression or that some allo-reactive B cells may differentiate into antibody-secreting cells in the absence of T cell assistance. The antibody-producing cells may also originate from an existing population of memory B cells that do not require T-cell mediated activation[84].
CHRONIC ABMR
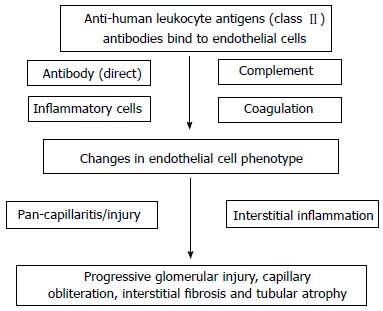
The clinical significance of chronic ABMR has been increasingly documented in recent years with some data suggesting that it may represent the leading cause of late allograft loss[4]. In contrast to acute ABMR, chronic ABMR is a long-term process that develops in sequential steps over a period of months to years[85]. Chronic ABMR has been proposed to arise over a series of stages or states[86]. The first common event is the production of alloantibodies followed by antibody interaction with alloantigens, resulting in the deposition of C4d in peritubular capillaries (PTC) and possibly glomeruli, followed by pathologic changes and graft dysfunction (Figure 1). Diagnostic features of chronic ABMR may include the presence of DSAs, transplant glomerulopathy (TG), peritubular capillary basement multilayering and the presence of C4d[27].
Figure 1.

Stages of chronic antibody-mediated chronic rejection. PTC: Peritubular capillaries.
TG and PTC basement multilayering represent the histological hallmark of chronic ABMR. Transplant glomerulopathy is a morphological pattern of chronic kidney injury that lacks detectable immune-complex deposits and is associated with poor kidney transplant outcomes. It is primarily an endothelial pathology that affects kidney microcirculation endothelium, which is observed as a duplication (double contours) and/or multilamination of capillary basement membranes together with the substantial replacement of endothelial fenestrations with a continuous endothelial lining[87]. DSAs, particularly HLA antigen class II antibodies may cause insidious graft injury and therefore constitute a central causative factor of transplant glomerulopathy (Figure 2). Although the international Banff consensus criteria classify transplant glomerulopathy as chronic ABMR if the pattern is accompanied by detectable DSAs and diffuse or focal linear C4d positivity in peritubular capillaries[4-6], Mauiyyedi et al[88] detected the deposition of C4d in peritubular capillaries in 61% of biopsies from patients showing chronic rejection with transplant glomerulopathy. In addition, a study by Regele et al[89] reported the presence of C4d in peritubular capillaries in 34% of patients with transplant glomerulopathy and this staining presaged the later development of transplant glomerulopathy.
Figure 2.
Proposed pathogenetic mechanisms for transplant glomerulopathy.
Pathologic patterns of chronic ABMR are observed in renal biopsies performed either for clinical indications or for protocol at a much later date after kidney transplantation[5-83]. In addition to reduced immunosuppression and non-adherence, early acute rejection appears to play a relevant role during late chronic ABMR. Indeed, several years ago, Cosio et al[90] documented that in 1-year surveillance biopsies, the degree of inflammation at 1-year post-transplant predicts the loss of graft function and graft failure independently of function and other variables (Figure 3).
Figure 3.
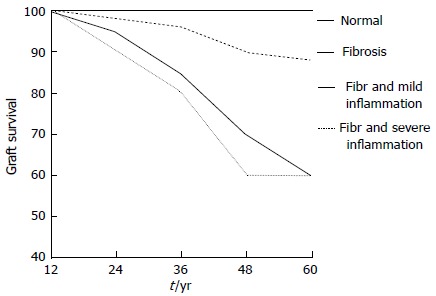
Five year post-transplant graft survival according 1-year post-transplant surveillance biopsy.
Recently El Ters et al[91] reported that early acute rejection, even in the absence of pre-transplant DSAs, increases the risk of alloimmune allograft loss late after transplantation and that the phenotype of this late loss is chronic ABMR. The hypothesis of this study was that the formation of new DSAs, particularly class II DSAs, may be a consequence of early acute rejection[92]. El Ters et al[91] noted that the presence of inflammation in 1-year protocol biopsies correlated with early acute rejection, presensitization, re-transplantation and HLA mismatch. He also observed that chronic ABMR was responsible for 43% of allograft loss.
In surveillance biopsies performed at 3 years after transplantation, Willicombe[93] reported that, despite excellent serum creatinine values, only one-third of biopsies were normal and that lesions appeared to correlate with the risks of immunological injury.
The 5-year follow-up data of the patient cohort from the DeKAF study[94,95] documented the role of antibodies in late graft dysfunction. Indeed, these studies showed a great number of patients with inflammation accompanying fibrosis or scarring, and their graft survival correlated with the presence of DSAs and/or C4d (Figure 4).
Figure 4.
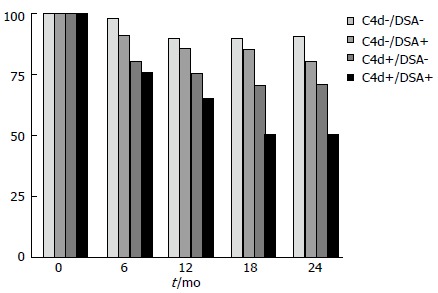
Deterioration of kidney allograft function study. Graft survival at 2-year according presence of donor-specific-HLA antibodies (DSAs) and/or C4d. HLA: Human leukocyte antigens.
The therapeutic approach to these conditions is one of the major challenges to date in the treatment of transplanted patients.
Finally, recent studies[96,97] examining BAFF, a B-cell stimulating molecule, showed that the appearance of soluble BAFF levels early after transplantation correlated with the de novo development of DSAs and, ultimately, with the progression to chronic active ABMR in pediatric and adult first kidney transplant recipients who were highly desensitized prior to transplantation.
Hill et al[98] described a new insight into the pathogenesis of chronic ABMR. DSAs-positive patients showed a striking acceleration of arteriosclerosis. Pathologic examination revealed that the inner intima is hypercellular with actively proliferating myofibroblasts that lay down collagen that often overlies older, condensed collagen of pre-transplantation donor origin.
THERAPY
The primary drugs or systems used are shown in Table 2 and are divided based on their action on the different maturation steps of B cells. The primary therapeutic strategies used are the following: (1) removal of antibodies; (2) inhibition of antibody production; (3) complement inhibitors; (4) intravenous immunoglobulins; and (5) splenectomy.
Table 2.
Anti-antibodies main drugs to date in use and mechanism of action
| Steps | Cells or mechanisms involved | Drugs | Mechanism of action |
| Exposure to antigen | B Cells | Rituximab ivig | Binds CD20 on B cells and mediates cell lysis Multiple B cell apoptosis, decrease in B-cell proliferation |
| Secretion of alloantibodies | Plasma cells; antibodies | Bortezomib | Decrease donor-specific alloantibody production Mechanical removal of alloantibodies |
| Plasma- exchange ivig | Multiple B cell apoptosis, decrease in B-cell proliferation | ||
| Binding of antibodies to the graft | Complement activation | Eculizumab | Blocks cleavage of terminal complement C5 and halts the process of complement-mediated cell destruction |
Removal of antibodies by plasmapheresis or immunoabsorption
Plasmapheresis (PP) and immunoabsorption (IA) techniques have been used to remove alloantibodies. PP is not specific for immunoglobulins (Ig) removal and requires replacement with fresh frozen plasma and albumin. IA shows high affinity for binding Igs and has the advantage of specificity over PP.
However, due to the tendency of DSAs to rebound and return to baseline levels, several repeated treatments are required[99] or an additional inhibitor of antibody production is required.
Inhibition of antibody production
Rituximab (anti-CD20): Rituximab is a chimeric murine/human monoclonal antibody that binds CD20 on pre B and mature B lymphocytes[100,101]. Recently rituximab has been documented to also prevent an anamnestic response in patients with cryptic sensitization to HLA[102].
BAFF blockade: BAFF, also known as B lymphocyte stimulator (Blys), is a member of the tumor necrosis factor cytokine family and is expressed primarily on T cells and dendritic cells for B-cell co-stimulation. BAFF binds to the receptor B-cell maturation antigen (BCMA), to the transmembrane activator (TACI) and to BAFF-receptor (BAFF-R) for B cell survival, proliferation and maturation[103].
BAFF blockade is a possible future therapy for renal transplantation. These drugs are highly promising because they selectively target B cells. Nevertheless no clinical trial is active in the field of transplantation although, these drugs have either been approved or are being examined for other diseases in large studies.
The best BAFF blockade drug is belimumab, which is a fully human recombinant IgG monoclonal antibody targeted against BAFF[104].
Bortezomib: Bortezomib is a proteasome inhibitor that is primarily used to treat acute ABMR or to decrease de novo DSA levels post-transplantation[105,106]. In further pilot studies, the authors used bortezomib in desensitization protocols with encouraging results[107,108].
Complement inhibitors
Eculizumab: Eculizumab is a humanized monoclonal antibody targeted against complement protein C5 that binds the C5 protein with high affinity and inhibits its cleavage to C5a and C5b, thereby preventing the generation of the terminal complement complex C5b-9. Eculizumab is used for the treatment of paroxysmal nocturnal hemoglobinuria and for atypical hemolytic uremic syndrome. Stegall et al[109] documented a decrease in post-transplant acute ABMR in sensitized renal transplant recipients, indicating its usefulness for desensitization protocols. Case reports have documented the effective rescue treatment of severe complement activation and reversal of acute ABMR by eculizumab in AB0-incompatible kidney-pancreas transplants and re-transplanted kidney recipients[110,111].
Intravenous immunoglobulins
Intravenous immunoglobulins show pleiotropic effects: They neutralize circulating anti-HLA antibodies via anti-idiotypic antibodies, inhibit complement activation by binding C3b and C4b and neutralizing C3a and C5a[112]. They also inhibit the expression of CD19 on activated B cells and induce the apoptosis of B cells[113]. Intravenous immunoglobulins (IVIGs) also show inhibitory effects on cellular immune responses with no specific inhibitory effects on the immune system by binding to Fcy receptors on macrophages, neutrophils, platelets, mast cells and NK cells.
IVIGs are used to decrease PRA levels in highly sensitized patients, in desensitization protocols of AB0-incompatible and XM-positive patients and in the treatment of ABMR.
Splenectomy: Splenectomy has been used in desensitization protocols and in the treatment of refractory acute ABMR[114,115]. Splenectomy removes a major source of lymphocytes, but the effect on the immune system is permanent and places the patients at risk for the development of sepsis.
As discussed in the pathophysiology chapter, we should distinguish the following: (1) acute ABMR; and (2) chronic ABMR.
Acute ABMR
Early acute ABMR often occurs in patients with DSAs prior to transplantation with a CDC-XM-positive with the donor. Even after successful desensitization strategies and successful kidney transplantations acute ABMR occurs in up to 40% of recipients. A later occurrence of acute ABMR is typically noted in patients with de novo DSAs and often after the reduction of immunosuppression or non-adherence[116,117].
We should now distinguish between the prevention and the treatment of acute ABMR.
Prevention of acute ABMR: Patients waiting for a transplant may be highly immunized and many show detectable DSAs in their serum. Sensitized patients who are DSAs-negative with negative XM-CDC may be transplanted safely. They will likely require more immunosuppressive therapy and an induction therapy[118-120].
The different desensitization protocols apply primarily to DSA-positive patients who are XM-CDC positive. The majority of the current protocols are modified version of the high-dose IVIG initiated at the Cedars-Sinai Medical Center or of the PP with low-dose IVIG initiated at John Hopkins Hospital[121].
Jordan initially provided[122] high dose IVIGs (2 g/kg) to cross-match-positive recipients, and the patients received a kidney transplant when their CDC T cell XM became negative. Due to the high rate of acute ABMR, Jordan[123] decided to use alemtuzumab induction treatment and added rituximab to the protocol to decrease the acute rejection rate.
More recently, Vo et al[124]at the Cedars-Sinai reported on the 24-mo outcomes of the aforementioned desensitization protocol and showed a 2-years graft survival of 84% in 76 hyper immune XM-positive recipients.
The other approach to desensitization comprises the use of PP and low-dose anti cytomegalovirus IVIG (CMV Ig). This approach was first adopted in 1998 at John Hopkins Hospital in XM-incompatible living donor kidney transplant candidates[125]. Patients received PP and CMV Ig at 100 mg/kg after each PP, combined with tacrolimus and mycophenolate mofetil. In a recent study, Montgomery et al[126] successfully desensitized 211 DSA-positive recipients of living donor kidneys with PP and low-dose IVIG.
A differing approach is the use of peri-transplant immunoabsorption rather than plasmapheresis. In 68 patients with deceased donors, Bartel and colleagues used peri-transplant IA followed by post-transplant IA and obtained excellent transplant outcomes[127].
Overall, over the last 13 years, almost 1000 patients with DSAs underwent kidney transplants and used varying desensitization protocols. The patient and graft survival rates are 95% and 86%, respectively, at the 2-year median follow-up. The primary issue is the high rates of acute rejection and of ABMR in particular (28%)[128]. New drugs are being developed to reduce this high rate of ABMR.
Stegall et al[109] added eculizumab during the pre-post-transplant period in DSAs-positive patients and obtained 7.7% post-transplant acute ABMR compared with 41.2% in the control group. However, at 2-years after transplantation the incidence of chronic ABMR was similar between the two groups. Chronic ABMR remains a major issue when transplanting hyper-immune patients.
A different option is to use the proteasome inhibitor bortezomib. In pilot studies, bortezomib has been used in desensitization protocols with encouraging results[107,108]. It is being used in a current ongoing a prospective iterative trial of proteasome inhibitor-based desensitization[129]. The trial has been approved by the International Review Board (IRB) and is being conducted under the auspices of FDA. Preliminary data suggest that bortezomib-based desensitization regimens comprising only two cycles (8 doses) may consistently reduce immunodominant HLA antibody levels and that multiple treatments with bortezomib (two-cycle regimen) may enable highly sensitized patients to undergo transplantation without IVIGs.
Treatment of acute ABMR: Acute ABMR in kidney recipients responds poorly to corticosteroids and anti-thymocyte agents alone, which are the standard treatment for acute cellular rejection.
International guidelines do not define an evidence-based treatment for acute ABMR. The kidney disease improving global outcomes guidelines (KDIGO) recommend the use of one or more of the following: corticosteroids, PP, IVIG, anti-CD20 antibodies or lymphocyte-depleting antibodies[130].
Two studies have individually reviewed the current approach to the treatment of acute ABMR[46] and the randomized controlled trials treating acute ABMR[131].
Although the literature suggests that plasmapheresis with or without low-dose IVIG or high-dose IVIG alone shows evidence of efficacy against acute ABMR and that they may be considered for the standard of care (SOC), these treatment regimens have not been standardized or optimized.
Approaches vary based on the amount of replacement volume, type of replacement fluids, number of PP sessions and the dose, timing and formulation of IVIGs used.
Other agents, such as rituximab, bortezomib and eculizumab have been used occasionally in conjunction with the above-mentioned therapies.
Of these treatments, rituximab has been used most frequently, and two studies in particular have evaluated rituximab as part of a combination treatment approach[132,133]. The latter study included 54 patients and compared a historical group treated with plasma exchange and IVIGs with a later group receiving a single dose of 500 mg/m2 rituximab in addition. The use of rituximab was associated with a 90% 2-year graft survival compared with 60% in the control group. Nevertheless, the benefit of adding rituximab remains in question when examining all published patient series.
Several case reports and series have been published on the use of bortezomib in the treatment for acute AMBR.
The largest series of 20 patients treated with bortezomib was reported by Flechner[134]. Using this treatment regimen, a graft survival rate of 85% at 10 mo post-transplant was achieved. The mean decrease in the dominant DSA in MFI values was 50%. However, the side effects of the treatment were considerable. One of the most recent studies compared 10 bortezomib-treated patients with a historical group of 9 rituximab-treated patients and achieved a graft survival of 60% with bortezomib compared with only 11% with rituximab at 18 mo[135].
Taken together, these preliminary results on bortezomib in acute ABMR are promising; however carefully performed, controlled studies are required to prove its benefits.
In the setting of kidney transplantation, there is emerging but limited evidence that eculizumab is efficient in treating acute ABMR[136]. Thus far, only a few reports exist in the literature on the use of eculizumab in refractory acute ABMR[110,111]. Stegall et al[109] reported the largest study of eculizumab in renal transplantation in a desensitization strategy. In this study, eculizumab appeared to show no impact on DSAs production after transplantation. In addition, the incidence of chronic ABMR appeared unchanged either by the prevention of early ABMR or by the prolonged complement blockade.
Splenectomy: One last option to salvage a graft with acute therapy-resistant ABMR is rescue splenectomy and its use has been reported by at least three groups[137-139]. The majority of patients underwent this surgery prior to the advent of eculizumab, and in the future, splenectomy may be avoided by using eculizumab instead. Splenectomy is recommended only in resistant cases of acute ABMR where bortezomib or eculizumab have already failed.
In summary, the first step for managing acute ABMR includes steroid pulses and/or antibody removal with PP or IA and IVIGs. The second step in patients with persistent allograft dysfunction includes the use of bortezomib and/or rituximab. The third step in resistant acute ABMR includes eculizumab and rescue splenectomy.
Chronic ABMR: In contrast to acute ABMR, chronic ABMR is a long-term process that develops in sequential steps over months to years[84].
In theory, every option available to treat acute ABMR may also be applied to chronic ABMR. However, there are no controlled trials in the literature regarding the treatment of chronic ABMR. The only treatment option with some reported benefit is a combination of rituximab and IVIGs[140].
With respect to established chronic ABMR, there have only been three case series treated with this combination therapy[141-143]. DSAs decreased only in some patients, and the therapy showed limited effects in cases with massive proteinuria, more severe peritubular capillaritis and previous acute rejection.
Very few patients have received bortezomib as a rescue treatment for chronic ABMR and proteinuria, and they have shown mixed results[144,145].
An interim analysis of a very recent study[146] of eculizumab therapy in chronic ABMR documented an apparent stabilization of renal function.
Taken together, these results indicate that any treatment for chronic ABMR using drugs with potentially high toxicity should only be performed in the context of a randomized controlled trial.
A recent recognized context that should be distinguished from acute or chronic ABMR is the negative impact of de novo DSAs after transplantation on the transplant outcome.
Several authors have reviewed the incidence and impact of de novo donor DSAs, in both adult[77] and pediatric recipients[147].
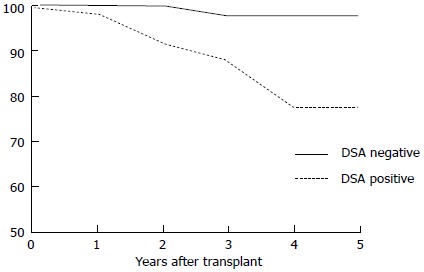
The actual 5-year post-transplantation cumulative incidence of de novo DSAs in a low-risk population is 20% (Figure 5). Once DSAs appear, the probability of graft loss within the 3 years of the appearance of DSAs is 24% (Figure 6). In patients without DSAs, the relative risk of graft loss is 9-fold higher at 1 year after the appearance of DSAs. In a multivariate analysis[77], the primary causes of de novo DSAs were DQ locus mismatches, a younger age at transplantation and transplants from deceased donors. Others claim prior non-adherence or a history of a clinical acute cellular rejection as being causes of de novo DSAs[82].
Figure 5.
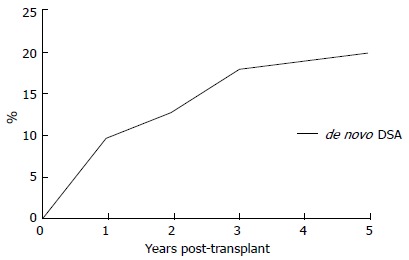
Actual 5-year post-transplantation cumulative incidence of de novo donor-specific-human leukocyte antigens antibodies (DSA). DSA:Donor-specific-human leukocyte antigens antibodies.
Figure 6.
Probability of graft loss within 3 years after de novo donor-specific-human leukocyte antigens antibodies appearance. DSA:Donor-specific-human leukocyte antigens antibodies.
If the appearance of DSAs is associated with the clinical signs of acute ABMR, the treatments used have already been discussed. The primary issue is how to treat when the appearance of DSAs is not associated with acute rejection.
To date, prophylactic treatments, such as rituximab and splenectomy[148] or eculizumab[109], do not appear to induce any effect on the appearance of DSAs.
Monitoring DSAs after transplantation appears to be essential because the appearance of DSAs is associated with a poor prognosis. Because procedures, such as antibody removal by PP or IA and the down regulation of antibody production by B cell- or plasma cell-targeting or complement cascade inhibition show very limited success when employed during the advanced phase of chronic ABMR[142,149,150], the prompt removal of de novo DSAs appears to be essential. However, no SOC exist for this issue. To date, only a multicenter antibody removal trial study in Italy is ongoing; it is using a randomized, prospective PP and low-dose CMV-IVIGs[151].
NEW AND FUTURE THERAPIES
Some of the drugs mentioned above that have been used to prevent or treat acute ABMR remain in pre-marketing clinical trials or have been approved for other diseases.
Drugs already known to control T cells also appear to be active in the long-term control of B cells.
Belatacept, a fusion receptor protein that blocks the co-stimulation pathway CD80/CD86-CD28, was recently approved for the prevention of acute rejection. Belatacept inhibited DSAs in phase 3 trials[152].
Another co-stimulation pathway is the CD40/CD40L pathway. Previous studies with antibodies directed against CD40L failed due to severe episodes of thrombosis. Indeed, CD40L is also expressed on the platelet surface, and its inhibition may induce thrombosis. More recently, the inhibition of the CD40/CD40L pathway by directly targeting CD40 has drawn interest from investigators particularly because CD40 is not expressed on the platelet surface. Humanized anti-CD40 antibodies prevented acute rejection and prolonged renal graft in non-human primates. In addition, these anti-CD40 antibodies appear safe and effective as maintenance immunosuppressive therapies[153-155]. To date, five monoclonal antibodies directed against CD40 have been studied for different diseases including kidney transplantation (Clinical Trial.gov NCT01780844).
Newer drugs that target B cell have been described. The most exciting are likely those that target survival factors and are part of the tumor necrosis factor super family: BAFF, Blys and the proliferation-inducing ligand (APRIL)[103].
Belimumab has already been discussed: it is a fully human antibody that neutralizes BAFF and deprives B cells of this important survival factor. The FDA approved belimumab in March 2011 for systemic lupus erythematosus (SLE). A group from Pennsylvania has enrolled patients in a phase II clinical trial of desensitization in sensitized patients awaiting clinical transplantation (clinicaltrials.gov NCT01025193). In this context, the study was unable to demonstrate the efficacy of belimumab.
Atacicept is a fusion receptor protein that neutralizes both BAFF or Blys and APRIL. In allo-sensitized nonhuman primates, atacicept reduced T-cell and B-cell alloantibodies by 36% and 24%, respectively[156].
A further possibility is complement inhibition by C1 esterase inhibitors, a plasma-derived human C1 esterase inhibitor. Initially used in allotransplantation to protect against ischemia/reperfusion injury[157], it is now under investigation for solid organ transplantations and approved by the FDA for use in other disease states. A trial studying the safety and tolerability of C1 inhibitor therapy in the context of the prevention of acute rejection (clinicaltrial.gov NCT01134510) is now ongoing. However, thus far no patients have been recruited.
CONCLUSION
The relevant graft injury is now well recognized to be caused by alloantibodies. Both acute and chronic graft injury may be caused by alloantibodies, and the most recent Banff classifications have been modified to introduce acute and chronic ABMR. The latter appears to be the most relevant cause of long-term graft injury rather than CNIs nephrotoxicity and “chronic allograft nephropathy”.
In addition to the major histocompatibility antigens, a large number of minor antigens have been recognized as possible antibody targets. The most important and the most widely studied antibodies responsible for graft injury are the HLA-DSAs.
The availability of new techniques for detecting circulating antibodies has enabled better understanding in recent years of the presence and role of antibodies in determining graft injury.
From a clinical point of view, we must distinguish between acute ABMR and chronic ABMR. In addition, we now recognize indolent ABMR and C4d-negative acute ABMR. Indolent ABMR develops sub-clinically. It often manifests in patients with de novo DSAs and causes slowly progressive microvascular abnormalities that lead to chronic ABMR. C4d-negative ABMR is cause for great discussion among scholars. It may be caused by an injury that is non-complement-mediated; however it may also be due to defective techniques. The Banff group is still working to improve understanding of this entity.
Recently, evidence has accumulated on the significant role of HLA-DSAs in the pathogenesis of slowly progressive graft injury and dysfunction. Several studies have shown that circulating DSAs (class I or class II) are found in a substantial fraction of renal allograft recipients and are associated with long-term graft loss.
The primary therapeutic approach comprises antibody removal, B-cells and plasma cells- targeting and inhibition of the complement pathway. The therapeutic approach used is based on the clinical conditions.
In patients waiting for transplantation who show positive XM-CDC, the removal of antibodies with or without B- or plasma cell-inhibition remains the best approach.
Patients with acute ABMR should be treated with a heavy regimen of T/B cell-targeting drugs (pulse corticosteroids and ATG), by removing antibodies, and using specific B- or plasma cell-inhibition or by complement inhibition.
No SOC exists for chronic ABMR, and only randomized controlled trials will indicate the best therapeutic option.
What may we hope for in the future? Unfortunately, the pipeline is almost empty.
Essentially, we may consider two types of drugs that are either already on the market or remain in premarket trials: (1) drugs targeting both T and B cells; Belatacept has already been approved by the FDA for the prevention of acute rejection. In a 3-year follow-up study[152], it proved to be effective on DSAs also in CNIs free protocols. The blockade of CD40-CD154 with humanized anti-CD 40 antibodies has prevented acute rejection[154]. In addition, these antibodies appear safe and effective in maintenance therapy; and (2) drugs targeting B cells or the complement pathway.
BAFF-blocking drugs: Represent new interesting drugs that target B lymphocyte stimulators. Belimumab, a fully human recombinant IgG monoclonal antibody to BAFF, was approved in 2011 for the treatment of SLE; however the above-mentioned phase II trial for desensitization failed. Atacicept was evaluated in diseases including rheumatoid arthritis, SLE, multiple sclerosis and B-cell malignancies. It awaits evaluation in human transplant patients.
While waiting for the approval of eculizumab, the C1 esterase inhibitor is being studied. This drug has been FDA-approved for treating hereditary angioedema; however it appears to be far from approval for use in transplantation.
Footnotes
P- Reviewers: Andrew Scott M, Caigan D, Hilmi IA S- Editor: Qi Y L- Editor: A E- Editor: Wu HL
References
- 1.L’Agence de la biomédecine. Agence de la Biomédecine Annual Report. Available from: http://www. agence-biomedecine.fr/article/111 (2011)
- 2. Available from: http://optn.transplant.hrsa.gov/ar2009/default.htm (2012).
- 3.Morais OO, Costa IM, Gomes CM, Shinzato DH, Ayres GM, Cardoso RM. The use of the Er: YAG 2940nm laser associated with amorolfine lacquer in the treatment of onychomycosis. An Bras Dermatol. 2013;88:847–849. doi: 10.1590/abd1806-4841.20131932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, Kaplan B, Halloran PF. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9:2520–2531. doi: 10.1111/j.1600-6143.2009.02799.x. [DOI] [PubMed] [Google Scholar]
- 5.Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, Hidalgo LG, Famulski K, Matas A, Halloran PF. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12:388–399. doi: 10.1111/j.1600-6143.2011.03840.x. [DOI] [PubMed] [Google Scholar]
- 6.Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R, Cosio FC, Gourishankar S, Grande J, Halloran P, Hunsicker L, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation. 2010;90:68–74. doi: 10.1097/TP.0b013e3181e065de. [DOI] [PubMed] [Google Scholar]
- 7.Gourishankar S, Leduc R, Connett J, Cecka JM, Cosio F, Fieberg A, Gaston R, Halloran P, Hunsicker L, Kasiske B, et al. Pathological and clinical characterization of the ‘troubled transplant’: data from the DeKAF study. Am J Transplant. 2010;10:324–330. doi: 10.1111/j.1600-6143.2009.02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matas AJ, Leduc R, Rush D, Cecka JM, Connett J, Fieberg A, Halloran P, Hunsicker L, Cosio F, Grande J, et al. Histopathologic clusters differentiate subgroups within the nonspecific diagnoses of CAN or CR: preliminary data from the DeKAF study. Am J Transplant. 2010;10:315–323. doi: 10.1111/j.1600-6143.2009.02943.x. [DOI] [PubMed] [Google Scholar]
- 9.Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol. 2012;8:348–357. doi: 10.1038/nrneph.2012.81. [DOI] [PubMed] [Google Scholar]
- 10.Hidalgo LG, Campbell PM, Sis B, Einecke G, Mengel M, Chang J, Sellares J, Reeve J, Halloran PF. De novo donor-specific antibody at the time of kidney transplant biopsy associates with microvascular pathology and late graft failure. Am J Transplant. 2009;9:2532–2541. doi: 10.1111/j.1600-6143.2009.02800.x. [DOI] [PubMed] [Google Scholar]
- 11.Tait BD, Süsal C, Gebel HM, Nickerson PW, Zachary AA, Claas FH, Reed EF, Bray RA, Campbell P, Chapman JR, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non-HLA antibodies in transplantation. Transplantation. 2013;95:19–47. doi: 10.1097/TP.0b013e31827a19cc. [DOI] [PubMed] [Google Scholar]
- 12.Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, Campbell PM, Cascalho M, Collins AB, Demetris AJ, et al. Banff ‘05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’) Am J Transplant. 2007;7:518–526. doi: 10.1111/j.1600-6143.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 13.Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, Halloran PF, Baldwin W, Banfi G, Collins AB, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 14.Sis B, Mengel M, Haas M, Colvin RB, Halloran PF, Racusen LC, Solez K, Baldwin WM, Bracamonte ER, Broecker V, et al. Banff ‘09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant. 2010;10:464–471. doi: 10.1111/j.1600-6143.2009.02987.x. [DOI] [PubMed] [Google Scholar]
- 15.Mengel M, Sis B, Haas M, Colvin RB, Halloran PF, Racusen LC, Solez K, Cendales L, Demetris AJ, Drachenberg CB, et al. Banff 2011 Meeting report: new concepts in antibody-mediated rejection. Am J Transplant. 2012;12:563–570. doi: 10.1111/j.1600-6143.2011.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin S, Claas F. Antibodies and crossmatching for transplantation. In: Dyer PA, Middleton D, eds , editors. Histocompatibility testing: a practical approach. Oxford: IRL Press; 1993. pp. 97–105. [Google Scholar]
- 17.Salvadori M, Bertoni E. Renal transplant allocation criteria, desensitization strategies and immunosuppressive therapy in retransplant renal patients. J Nephrol. 2012;25:890–899. doi: 10.5301/jn.5000207. [DOI] [PubMed] [Google Scholar]
- 18.Eng HS, Bennett G, Bardy P, Coghlan P, Russ GR, Coates PT. Clinical significance of anti-HLA antibodies detected by Luminex: enhancing the interpretation of CDC-BXM and important post-transplantation monitoring tools. Hum Immunol. 2009;70:595–599. doi: 10.1016/j.humimm.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Fuggle SV, Martin S. Tools for human leukocyte antigen antibody detection and their application to transplanting sensitized patients. Transplantation. 2008;86:384–390. doi: 10.1097/TP.0b013e31817c90f5. [DOI] [PubMed] [Google Scholar]
- 20.Vlad G, Ho EK, Vasilescu ER, Colovai AI, Stokes MB, Markowitz GS, D’Agati VD, Cohen DJ, Ratner LE, Suciu-Foca N. Relevance of different antibody detection methods for the prediction of antibody-mediated rejection and deceased-donor kidney allograft survival. Hum Immunol. 2009;70:589–594. doi: 10.1016/j.humimm.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Mizutani K, Terasaki P, Hamdani E, Esquenazi V, Rosen A, Miller J, Ozawa M. The importance of anti-HLA-specific antibody strength in monitoring kidney transplant patients. Am J Transplant. 2007;7:1027–1031. doi: 10.1111/j.1600-6143.2006.01721.x. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz A, Gaigalas AK, Wang L, Marti GE, Vogt RF, Fernandez-Repollet E. Formalization of the MESF unit of fluorescence intensity. Cytometry B Clin Cytom. 2004;57:1–6. doi: 10.1002/cyto.b.10066. [DOI] [PubMed] [Google Scholar]
- 23.Takemoto SK, Zeevi A, Feng S, Colvin RB, Jordan S, Kobashigawa J, Kupiec-Weglinski J, Matas A, Montgomery RA, Nickerson P, et al. National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004;4:1033–1041. doi: 10.1111/j.1600-6143.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 24.Gupta A, Sinnott P. Clinical relevance of pretransplant human leukocyte antigen donor-specific antibodies in renal patients waiting for a transplant: a risk factor. Hum Immunol. 2009;70:618–622. doi: 10.1016/j.humimm.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 25.Gupta A, Iveson V, Varagunam M, Bodger S, Sinnott P, Thuraisingham RC. Pretransplant donor-specific antibodies in cytotoxic negative crossmatch kidney transplants: are they relevant? Transplantation. 2008;85:1200–1204. doi: 10.1097/TP.0b013e31816b1c37. [DOI] [PubMed] [Google Scholar]
- 26.Riethmüller S, Ferrari-Lacraz S, Müller MK, Raptis DA, Hadaya K, Rüsi B, Laube G, Schneiter G, Fehr T, Villard J. Donor-specific antibody levels and three generations of crossmatches to predict antibody-mediated rejection in kidney transplantation. Transplantation. 2010;90:160–167. doi: 10.1097/tp.0b013e3181e36e08. [DOI] [PubMed] [Google Scholar]
- 27.Archdeacon P, Chan M, Neuland C, Velidedeoglu E, Meyer J, Tracy L, Cavaille-Coll M, Bala S, Hernandez A, Albrecht R. Summary of FDA antibody-mediated rejection workshop. Am J Transplant. 2011;11:896–906. doi: 10.1111/j.1600-6143.2011.03525.x. [DOI] [PubMed] [Google Scholar]
- 28.Bartel G, Wahrmann M, Exner M, Regele H, Huttary N, Schillinger M, Körmöczi GF, Hörl WH, Böhmig GA. In vitro detection of C4d-fixing HLA alloantibodies: associations with capillary C4d deposition in kidney allografts. Am J Transplant. 2008;8:41–49. doi: 10.1111/j.1600-6143.2007.01998.x. [DOI] [PubMed] [Google Scholar]
- 29.Smith JD, Hamour IM, Banner NR, Rose ML. C4d fixing, luminex binding antibodies - a new tool for prediction of graft failure after heart transplantation. Am J Transplant. 2007;7:2809–2815. doi: 10.1111/j.1600-6143.2007.01991.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen G, Sequeira F, Tyan DB. Novel C1q assay reveals a clinically relevant subset of human leukocyte antigen antibodies independent of immunoglobulin G strength on single antigen beads. Hum Immunol. 2011;72:849–858. doi: 10.1016/j.humimm.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Otten HG, Verhaar MC, Borst HP, Hené RJ, van Zuilen AD. Pretransplant donor-specific HLA class-I and -II antibodies are associated with an increased risk for kidney graft failure. Am J Transplant. 2012;12:1618–1623. doi: 10.1111/j.1600-6143.2011.03985.x. [DOI] [PubMed] [Google Scholar]
- 32.Lachmann N, Todorova K, Schulze H, Schönemann C. Systematic comparison of four cell- and Luminex-based methods for assessment of complement-activating HLA antibodies. Transplantation. 2013;95:694–700. doi: 10.1097/TP.0b013e31827b3dc3. [DOI] [PubMed] [Google Scholar]
- 33.Glotz D, Lucchiari N, Pegaz-Fiornet B, Suberbielle-Boissel C. Endothelial cells as targets of allograft rejection. Transplantation. 2006;82:S19–S21. doi: 10.1097/01.tp.0000231348.55262.5a. [DOI] [PubMed] [Google Scholar]
- 34.Ronda C, Borba SC, Ferreira SC, Glotz D, Ianhez LE, Rodrigues H, Viggiani CS, Nahas W, David-Neto E, Castro MC, et al. Non-human leukocyte antigen antibodies reactive with endothelial cells could be involved in early loss of renal allografts. Transplant Proc. 2011;43:1345–1348. doi: 10.1016/j.transproceed.2011.03.059. [DOI] [PubMed] [Google Scholar]
- 35.Pontes LF, Carvalho L, Stumbo AC, Porto LC. Detection and localization of non-HLA-ABC antigenic sites relevant to kidney rejection on endothelial cells. J Immunol Methods. 2001;251:73–80. doi: 10.1016/s0022-1759(01)00309-x. [DOI] [PubMed] [Google Scholar]
- 36.Hendrickx J, Doggen K, Weinberg EO, Van Tongelen P, Fransen P, De Keulenaer GW. Molecular diversity of cardiac endothelial cells in vitro and in vivo. Physiol Genomics. 2004;19:198–206. doi: 10.1152/physiolgenomics.00143.2004. [DOI] [PubMed] [Google Scholar]
- 37.Vermehren D, Sumitran-Holgersson S. Isolation of precursor endothelial cells from peripheral blood for donor-specific crossmatching before organ transplantation. Transplantation. 2002;74:1479–1486. doi: 10.1097/00007890-200212150-00001. [DOI] [PubMed] [Google Scholar]
- 38.Alheim M, Johansson SM, Hauzenberger D, Grufman P, Holgersson J. A flow cytometric crossmatch test for simultaneous detection of antibodies against donor lymphocytes and endothelial precursor cells. Tissue Antigens. 2010;75:269–277. doi: 10.1111/j.1399-0039.2009.01439.x. [DOI] [PubMed] [Google Scholar]
- 39.Dinavahi R, George A, Tretin A, Akalin E, Ames S, Bromberg JS, Deboccardo G, Dipaola N, Lerner SM, Mehrotra A, et al. Antibodies reactive to non-HLA antigens in transplant glomerulopathy. J Am Soc Nephrol. 2011;22:1168–1178. doi: 10.1681/ASN.2010111183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sigdel TK, Li L, Tran TQ, Khatri P, Naesens M, Sansanwal P, Dai H, Hsieh SC, Sarwal MM. Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. J Am Soc Nephrol. 2012;23:750–763. doi: 10.1681/ASN.2011060596. [DOI] [PubMed] [Google Scholar]
- 41.Dunn TB, Noreen H, Gillingham K, Maurer D, Ozturk OG, Pruett TL, Bray RA, Gebel HM, Matas AJ. Revisiting traditional risk factors for rejection and graft loss after kidney transplantation. Am J Transplant. 2011;11:2132–2143. doi: 10.1111/j.1600-6143.2011.03640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zito A, Schena A, Grandaliano G, Gesualdo L, Schena FP. Increasing relevance of donor-specific antibodies in antibody-mediated rejection. J Nephrol. 2013;26:237–242. doi: 10.5301/jn.5000240. [DOI] [PubMed] [Google Scholar]
- 43.Terasaki PI, Ozawa M, Castro R. Four-year follow-up of a prospective trial of HLA and MICA antibodies on kidney graft survival. Am J Transplant. 2007;7:408–415. doi: 10.1111/j.1600-6143.2006.01644.x. [DOI] [PubMed] [Google Scholar]
- 44.Rodríguez PC, Arroyave IH, Mejía G, García LF. Detection of alloantibodies against non-HLA antigens in kidney transplantation by flow cytometry. Clin Transplant. 2000;14:472–478. doi: 10.1034/j.1399-0012.2000.140505.x. [DOI] [PubMed] [Google Scholar]
- 45.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 46.Nickerson PW, Rush DN. Antibodies beyond HLA. Am J Transplant. 2013;13:831–832. doi: 10.1111/ajt.12160. [DOI] [PubMed] [Google Scholar]
- 47.Breimer ME, Rydberg L, Jackson AM, Lucas DP, Zachary AA, Melancon JK, Von Visger J, Pelletier R, Saidman SL, Williams WW, et al. Multicenter evaluation of a novel endothelial cell crossmatch test in kidney transplantation. Transplantation. 2009;87:549–556. doi: 10.1097/TP.0b013e3181949d4e. [DOI] [PubMed] [Google Scholar]
- 48.Dragun D. Humoral responses directed against non-human leukocyte antigens in solid-organ transplantation. Transplantation. 2008;86:1019–1025. doi: 10.1097/TP.0b013e3181889748. [DOI] [PubMed] [Google Scholar]
- 49.Regele H. Non-HLA antibodies in kidney allograft rejection: convincing concept in need of further evidence. Kidney Int. 2011;79:583–586. doi: 10.1038/ki.2010.517. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, Reed EF. Non-MHC antigenic targets of the humoral immune response in transplantation. Curr Opin Immunol. 2010;22:682–688. doi: 10.1016/j.coi.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Q, Cheng Z, Cheng D, Chen J, Ji S, Wen J, Zheng C, Liu Z. De novo development of circulating anti-endothelial cell antibodies rather than pre-existing antibodies is associated with post-transplant allograft rejection. Kidney Int. 2011;79:655–662. doi: 10.1038/ki.2010.437. [DOI] [PubMed] [Google Scholar]
- 52.Soulez M, Pilon EA, Dieudé M, Cardinal H, Brassard N, Qi S, Wu SJ, Durocher Y, Madore F, Perreault C, et al. The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ Res. 2012;110:94–104. doi: 10.1161/CIRCRESAHA.111.250431. [DOI] [PubMed] [Google Scholar]
- 53.Cardinal H, Dieudé M, Brassard N, Qi S, Patey N, Soulez M, Beillevaire D, Echeverry F, Daniel C, Durocher Y, et al. Antiperlecan antibodies are novel accelerators of immune-mediated vascular injury. Am J Transplant. 2013;13:861–874. doi: 10.1111/ajt.12168. [DOI] [PubMed] [Google Scholar]
- 54.Mahesh B, Leong HS, McCormack A, Sarathchandra P, Holder A, Rose ML. Autoantibodies to vimentin cause accelerated rejection of cardiac allografts. Am J Pathol. 2007;170:1415–1427. doi: 10.2353/ajpath.2007.060728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braun RK, Molitor-Dart M, Wigfield C, Xiang Z, Fain SB, Jankowska-Gan E, Seroogy CM, Burlingham WJ, Wilkes DS, Brand DD, et al. Transfer of tolerance to collagen type V suppresses T-helper-cell-17 lymphocyte-mediated acute lung transplant rejection. Transplantation. 2009;88:1341–1348. doi: 10.1097/TP.0b013e3181bcde7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ochoa L, Merck B, Geli S, Díaz de Liaño A. Hepatic trauma (90 consecutive cases treated in 9 years. Rev Esp Enferm Dig. 1991;79:393–396. [PubMed] [Google Scholar]
- 57.Reinsmoen NL, Lai CH, Heidecke H, Haas M, Cao K, Ong G, Naim M, Wang Q, Mirocha J, Kahwaji J, et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation. 2010;90:1473–1477. doi: 10.1097/TP.0b013e3181fd97f1. [DOI] [PubMed] [Google Scholar]
- 58.Sánchez-Zapardiel E, Castro-Panete MJ, Castillo-Rama M, Morales P, Lora-Pablos D, Valero-Hervás D, Ruiz-García R, Apaza J, Talayero P, Andrés A, et al. Harmful effect of preformed anti-MICA antibodies on renal allograft evolution in early posttransplantation period. Transplantation. 2013;96:70–78. doi: 10.1097/TP.0b013e3182943506. [DOI] [PubMed] [Google Scholar]
- 59.Naemi FM, Carter V, Kirby JA, Ali S. Anti-donor HLA class I antibodies: pathways to endothelial cell activation and cell-mediated allograft rejection. Transplantation. 2013;96:258–266. doi: 10.1097/TP.0b013e3182985504. [DOI] [PubMed] [Google Scholar]
- 60.Crespo M, Torio A, Mas V, Redondo D, Pérez-Sáez MJ, Mir M, Faura A, Guerra R, Montes-Ares O, Checa MD, et al. Clinical relevance of pretransplant anti-HLA donor-specific antibodies: does C1q-fixation matter? Transpl Immunol. 2013;29:28–33. doi: 10.1016/j.trim.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Clatworthy MR. B cell responses to allograft--more common than we thought? Am J Transplant. 2013;13:1629–1630. doi: 10.1111/ajt.12309. [DOI] [PubMed] [Google Scholar]
- 62.Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant. 2009;9:2312–2323. doi: 10.1111/j.1600-6143.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 63.Lynch RJ, Silva IA, Chen BJ, Punch JD, Cascalho M, Platt JL. Cryptic B cell response to renal transplantation. Am J Transplant. 2013;13:1713–1723. doi: 10.1111/ajt.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wood KJ, Goto R. Mechanisms of rejection: current perspectives. Transplantation. 2012;93:1–10. doi: 10.1097/TP.0b013e31823cab44. [DOI] [PubMed] [Google Scholar]
- 65.Nickerson PW, Rush DN. Rejection: an integrated response. Am J Transplant. 2013;13:2239–2240. doi: 10.1111/ajt.12365. [DOI] [PubMed] [Google Scholar]
- 66.Smith RN, Colvin RB. Chronic alloantibody mediated rejection. Semin Immunol. 2012;24:115–121. doi: 10.1016/j.smim.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirohashi T, Chase CM, Della Pelle P, Sebastian D, Alessandrini A, Madsen JC, Russell PS, Colvin RB. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant. 2012;12:313–321. doi: 10.1111/j.1600-6143.2011.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jordan SC, Toyoda M, Vo AA. Regulation of immunity and inflammation by intravenous immunoglobulin: relevance to solid organ transplantation. Expert Rev Clin Immunol. 2011;7:341–348. doi: 10.1586/eci.11.10. [DOI] [PubMed] [Google Scholar]
- 69.Loupy A, Hill GS, Suberbielle C, Charron D, Anglicheau D, Zuber J, Timsit MO, Duong JP, Bruneval P, Vernerey D, et al. Significance of C4d Banff scores in early protocol biopsies of kidney transplant recipients with preformed donor-specific antibodies (DSA) Am J Transplant. 2011;11:56–65. doi: 10.1111/j.1600-6143.2010.03364.x. [DOI] [PubMed] [Google Scholar]
- 70.Colvin RB, Nickeleit V. Renal transplant pathology. In: Heptinstall’s Pathology of the kidney, 6th Ed, edited by Jennette JC, Olson JL, Scwartz MM, Silva PG, Philadelphia, Lippincott-Raven; 2006. pp. 1347–1490. [Google Scholar]
- 71.Drachenberg CB, Papadimitriou JC. Endothelial injury in renal antibody-mediated allograft rejection: a schematic view based on pathogenesis. Transplantation. 2013;95:1073–1083. doi: 10.1097/TP.0b013e31827e6b45. [DOI] [PubMed] [Google Scholar]
- 72.Caro-Oleas JL, González-Escribano MF, Gentil-Govantes MÁ, Acevedo MJ, González-Roncero FM, Blanco GB, Núñez-Roldán A. Clinical relevance of anti-HLA donor-specific antibodies detected by Luminex assay in the development of rejection after renal transplantation. Transplantation. 2012;94:338–344. doi: 10.1097/TP.0b013e31825ace2c. [DOI] [PubMed] [Google Scholar]
- 73.Hourmant M, Cesbron-Gautier A, Terasaki PI, Mizutani K, Moreau A, Meurette A, Dantal J, Giral M, Blancho G, Cantarovich D, et al. Frequency and clinical implications of development of donor-specific and non-donor-specific HLA antibodies after kidney transplantation. J Am Soc Nephrol. 2005;16:2804–2812. doi: 10.1681/ASN.2004121130. [DOI] [PubMed] [Google Scholar]
- 74.Dörje C, Midtvedt K, Holdaas H, Naper C, Strøm EH, Øyen O, Leivestad T, Aronsen T, Jenssen T, Flaa-Johnsen L, et al. Early versus late acute antibody-mediated rejection in renal transplant recipients. Transplantation. 2013;96:79–84. doi: 10.1097/TP.0b013e31829434d4. [DOI] [PubMed] [Google Scholar]
- 75.Bista B, Jackson S, Issa N, Matas A, Kukla A. High creatinine and advanced chronic rejection (CR) predicts graft loss in non-adherent kiney transplant recipients. Am J Transplant. 2013;13:415. [Google Scholar]
- 76.Lawrence C, Willicombe M, Brookes PA, Santos-Nunez E, Bajaj R, Cook T, Roufosse C, Taube D, Warrens AN. Preformed complement-activating low-level donor-specific antibody predicts early antibody-mediated rejection in renal allografts. Transplantation. 2013;95:341–346. doi: 10.1097/TP.0b013e3182743cfa. [DOI] [PubMed] [Google Scholar]
- 77.Everly MJ, Rebellato LM, Haisch CE, Ozawa M, Parker K, Briley KP, Catrou PG, Bolin P, Kendrick WT, Kendrick SA, et al. Incidence and impact of de novo donor-specific alloantibody in primary renal allografts. Transplantation. 2013;95:410–417. doi: 10.1097/TP.0b013e31827d62e3. [DOI] [PubMed] [Google Scholar]
- 78.Banham G, Prezzi D, Harford S, Taylor CJ, Hamer R, Higgins R, Bradley JA, Clatworthy MR. Elevated pretransplantation soluble BAFF is associated with an increased risk of acute antibody-mediated rejection. Transplantation. 2013;96:413–420. doi: 10.1097/TP.0b013e318298dd65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Orandi B, Chow E, Van Arendonk K, Montgomery J, Gupta N, Montgomery R, Segev D. Early antibody-mediated rejection portends worse long-term renal graft and patient serviva. Am J Transplant. 2013;13:324. [Google Scholar]
- 80.Loupy A, Suberbielle-Boissel C, Zuber J, Anglicheau D, Timsit MO, Martinez F, Thervet E, Bruneval P, Charron D, Hill GS, et al. Combined posttransplant prophylactic IVIg/anti-CD 20/plasmapheresis in kidney recipients with preformed donor-specific antibodies: a pilot study. Transplantation. 2010;89:1403–1410. doi: 10.1097/TP.0b013e3181da1cc3. [DOI] [PubMed] [Google Scholar]
- 81.de Kort H, Willicombe M, Brookes P, Dominy KM, Santos-Nunez E, Galliford JW, Chan K, Taube D, McLean AG, Cook HT, et al. Microcirculation inflammation associates with outcome in renal transplant patients with de novo donor-specific antibodies. Am J Transplant. 2013;13:485–492. doi: 10.1111/j.1600-6143.2012.04325.x. [DOI] [PubMed] [Google Scholar]
- 82.Haririan A, Kiangkitiwan B, Kukuruga D, Cooper M, Hurley H, Drachenberg C, Klassen D. The impact of c4d pattern and donor-specific antibody on graft survival in recipients requiring indication renal allograft biopsy. Am J Transplant. 2009;9:2758–2767. doi: 10.1111/j.1600-6143.2009.02836.x. [DOI] [PubMed] [Google Scholar]
- 83.Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, Nickerson PW. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant. 2012;12:1157–1167. doi: 10.1111/j.1600-6143.2012.04013.x. [DOI] [PubMed] [Google Scholar]
- 84.Stegall MD, Moore N, Taner T, Li H, Dean PG. Down-regulating humoral immune responses: implications for organ transplantation. Transplantation. 2014;97:247–257. doi: 10.1097/TP.0b013e3182a72115. [DOI] [PubMed] [Google Scholar]
- 85.Smith RN, Kawai T, Boskovic S, Nadazdin O, Sachs DH, Cosimi AB, Colvin RB. Four stages and lack of stable accommodation in chronic alloantibody-mediated renal allograft rejection in Cynomolgus monkeys. Am J Transplant. 2008;8:1662–1672. doi: 10.1111/j.1600-6143.2008.02303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. 2005;5:807–817. doi: 10.1038/nri1702. [DOI] [PubMed] [Google Scholar]
- 87.Husain S, Sis B. Advances in the understanding of transplant glomerulopathy. Am J Kidney Dis. 2013;62:352–363. doi: 10.1053/j.ajkd.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 88.Mauiyyedi S, Pelle PD, Saidman S, Collins AB, Pascual M, Tolkoff-Rubin NE, Williams WW, Cosimi AA, Schneeberger EE, Colvin RB. Chronic humoral rejection: identification of antibody-mediated chronic renal allograft rejection by C4d deposits in peritubular capillaries. J Am Soc Nephrol. 2001;12:574–582. doi: 10.1681/ASN.V123574. [DOI] [PubMed] [Google Scholar]
- 89.Regele H, Böhmig GA, Habicht A, Gollowitzer D, Schillinger M, Rockenschaub S, Watschinger B, Kerjaschki D, Exner M. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002;13:2371–2380. doi: 10.1097/01.asn.0000025780.03790.0f. [DOI] [PubMed] [Google Scholar]
- 90.Cosio FG, Grande JP, Wadei H, Larson TS, Griffin MD, Stegall MD. Predicting subsequent decline in kidney allograft function from early surveillance biopsies. Am J Transplant. 2005;5:2464–2472. doi: 10.1111/j.1600-6143.2005.01050.x. [DOI] [PubMed] [Google Scholar]
- 91.El Ters M, Grande JP, Keddis MT, Rodrigo E, Chopra B, Dean PG, Stegall MD, Cosio FG. Kidney allograft survival after acute rejection, the value of follow-up biopsies. Am J Transplant. 2013;13:2334–2341. doi: 10.1111/ajt.12370. [DOI] [PubMed] [Google Scholar]
- 92.Casey E, Gandhi M, Dean P, Stegall M, Cosio F Class I HLA mismatches increase risk of acute rejection, graft loss and formation of de novo lass II antibody. Am J Transplant. 2013;19:42. [Google Scholar]
- 93.Willicombe M, Roufosse C, Brookes P, Galliford J, McLean A, Cook T, Taube D. Potential importance of 3 year surveillance biopsies in renal transplantation. Am J Transplant. 2013;19:41. [Google Scholar]
- 94.Gaston RS. Our evolving understanding of late kidney allograft failure. Curr Opin Organ Transplant. 2011;16:594–599. doi: 10.1097/MOT.0b013e32834c23a7. [DOI] [PubMed] [Google Scholar]
- 95.Rush D, Leduc R, Connett J, Cosio F, Gaston R, Mannon R, Hunsicker L, Gourishankar S, Halloran P, Cecka M, et al. DeKAF pathology clusters: follow up at 5 years. Am J Transplant. 2013;19:417. [Google Scholar]
- 96.Comoli P, Quartuccio G, Cioni M, Parodi A, Fontana I, Basso S, Magnasco A, Tagliamacco A, Nocera A, Cardillo M, et al. Soluble BAFF levels correlate with de novo DSA development and progression to chronic active antibody-mediated rejection in pediatric recipients of first kidney transplants. Am J Transplant. 2013;19:113. [Google Scholar]
- 97.Thibault-Espitia A, Foucher Y, Danger R, Migone T, Pallier A, Castagnet S, G-Gueguen C, Devys A, C-Gautier A, Giral M, et al. BAFF and BAFF-R levels are associated with risk of long-term kidney graft dysfunction and development of donor-specific antibodies. Am J Transplant. 2012;12:2754–2762. doi: 10.1111/j.1600-6143.2012.04194.x. [DOI] [PubMed] [Google Scholar]
- 98.Hill GS, Nochy D, Bruneval P, Duong van Huyen JP, Glotz D, Suberbielle C, Zuber J, Anglicheau D, Empana JP, Legendre C, et al. Donor-specific antibodies accelerate arteriosclerosis after kidney transplantation. J Am Soc Nephrol. 2011;22:975–983. doi: 10.1681/ASN.2010070777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hakim RM, Milford E, Himmelfarb J, Wingard R, Lazarus JM, Watt RM. Extracorporeal removal of anti-HLA antibodies in transplant candidates. Am J Kidney Dis. 1990;16:423–431. doi: 10.1016/s0272-6386(12)80054-0. [DOI] [PubMed] [Google Scholar]
- 100.Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant. 2006;6:859–866. doi: 10.1111/j.1600-6143.2006.01288.x. [DOI] [PubMed] [Google Scholar]
- 101.van den Hoogen MW, Hilbrands LB. Use of monoclonal antibodies in renal transplantation. Immunotherapy. 2011;3:871–880. doi: 10.2217/imt.11.72. [DOI] [PubMed] [Google Scholar]
- 102.Zachary AA, Lucas DP, Montgomery RA, Leffell MS. Rituximab prevents an anamnestic response in patients with cryptic sensitization to HLA. Transplantation. 2013;95:701–704. doi: 10.1097/TP.0b013e31827be3c1. [DOI] [PubMed] [Google Scholar]
- 103.Page EK, Dar WA, Knechtle SJ. Biologics in organ transplantation. Transpl Int. 2012;25:707–719. doi: 10.1111/j.1432-2277.2012.01456.x. [DOI] [PubMed] [Google Scholar]
- 104.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, León MG, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 105.Everly MJ, Everly JJ, Susskind B, Brailey P, Arend LJ, Alloway RR, Roy-Chaudhury P, Govil A, Mogilishetty G, Rike AH, et al. Bortezomib provides effective therapy for antibody- and cell-mediated acute rejection. Transplantation. 2008;86:1754–1761. doi: 10.1097/TP.0b013e318190af83. [DOI] [PubMed] [Google Scholar]
- 106.Everly MJ, Terasaki PI, Trivedi HL. Durability of antibody removal following proteasome inhibitor-based therapy. Transplantation. 2012;93:572–577. doi: 10.1097/TP.0b013e31824612df. [DOI] [PubMed] [Google Scholar]
- 107.Everly MJ, Everly JJ, Terasaki PI. Role of proteasome inhibition in sensitized transplant candidates. Chin Med J (Engl) 2011;124:771–774. [PubMed] [Google Scholar]
- 108.Kute VB, Vanikar AV, Trivedi HL, Shah PR, Goplani KR, Patel HV, Gumber MR, Patel RD, Kanodia KV, Suthar KS, et al. Desensitization protocol for highly sensitized renal transplant patients: a single-center experience. Saudi J Kidney Dis Transpl. 2011;22:662–669. [PubMed] [Google Scholar]
- 109.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, Cosio FG, Gandhi MJ, Kremers W, Gloor JM. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 110.Biglarnia AR, Nilsson B, Nilsson T, von Zur-Mühlen B, Wagner M, Berne C, Wanders A, Magnusson A, Tufveson G. Prompt reversal of a severe complement activation by eculizumab in a patient undergoing intentional ABO-incompatible pancreas and kidney transplantation. Transpl Int. 2011;24:e61–e66. doi: 10.1111/j.1432-2277.2011.01290.x. [DOI] [PubMed] [Google Scholar]
- 111.Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, King KE, Kraus E, Lees LM, Melancon JK, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9:231–235. doi: 10.1111/j.1600-6143.2008.02451.x. [DOI] [PubMed] [Google Scholar]
- 112.Watanabe J, Scornik JC. IVIG and HLA antibodies. Evidence for inhibition of complement activation but not for anti-idiotypic activity. Am J Transplant. 2005;5:2786–2790. doi: 10.1111/j.1600-6143.2005.01056.x. [DOI] [PubMed] [Google Scholar]
- 113.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 114.Locke JE, Zachary AA, Mohammed BS, Warren DS, Montgomery RA. Rescue splenectomy for severe acute antibody-mediated rejection. Clin Transpl. 2006:518–520. [PubMed] [Google Scholar]
- 115.Locke JE, Zachary AA, Haas M, Melancon JK, Warren DS, Simpkins CE, Segev DL, Montgomery RA. The utility of splenectomy as rescue treatment for severe acute antibody mediated rejection. Am J Transplant. 2007;7:842–846. doi: 10.1111/j.1600-6143.2006.01709.x. [DOI] [PubMed] [Google Scholar]
- 116.Lefaucheur C, Suberbielle-Boissel C, Hill GS, Nochy D, Andrade J, Antoine C, Gautreau C, Charron D, Glotz D. Clinical relevance of preformed HLA donor-specific antibodies in kidney transplantation. Am J Transplant. 2008;8:324–331. doi: 10.1111/j.1600-6143.2007.02072.x. [DOI] [PubMed] [Google Scholar]
- 117.Kayler LK, Kiss L, Sharma V, Mohanka R, Zeevi A, Girnita A, Shapiro R, Randhawa PS. Acute renal allograft rejection: diagnostic significance of focal peritubular capillary C4d. Transplantation. 2008;85:813–820. doi: 10.1097/TP.0b013e3181669194. [DOI] [PubMed] [Google Scholar]
- 118.Noël C, Abramowicz D, Durand D, Mourad G, Lang P, Kessler M, Charpentier B, Touchard G, Berthoux F, Merville P, et al. Daclizumab versus antithymocyte globulin in high-immunological-risk renal transplant recipients. J Am Soc Nephrol. 2009;20:1385–1392. doi: 10.1681/ASN.2008101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gurk-Turner C, Airee R, Philosophe B, Kukuruga D, Drachenberg C, Haririan A. Thymoglobulin dose optimization for induction therapy in high risk kidney transplant recipients. Transplantation. 2008;85:1425–1430. doi: 10.1097/TP.0b013e31816dd596. [DOI] [PubMed] [Google Scholar]
- 120.Cai J, Terasaki PI. Current trend of induction and maintenance treatment in positive panel-reactive antibody patients: a report on OPTN/UNOS kidney transplant registry data. Chin Med J (Engl) 2011;124:649–654. [PubMed] [Google Scholar]
- 121.Montgomery RA, Zachary AA, Racusen LC, Leffell MS, King KE, Burdick J, Maley WR, Ratner LE. Plasmapheresis and intravenous immune globulin provides effective rescue therapy for refractory humoral rejection and allows kidneys to be successfully transplanted into cross-match-positive recipients. Transplantation. 2000;70:887–895. doi: 10.1097/00007890-200009270-00006. [DOI] [PubMed] [Google Scholar]
- 122.Jordan SC, Vo A, Bunnapradist S, Toyoda M, Peng A, Puliyanda D, Kamil E, Tyan D. Intravenous immune globulin treatment inhibits crossmatch positivity and allows for successful transplantation of incompatible organs in living-donor and cadaver recipients. Transplantation. 2003;76:631–636. doi: 10.1097/01.TP.0000080685.31697.FC. [DOI] [PubMed] [Google Scholar]
- 123.Vo AA, Lukovsky M, Toyoda M, Wang J, Reinsmoen NL, Lai CH, Peng A, Villicana R, Jordan SC. Rituximab and intravenous immune globulin for desensitization during renal transplantation. N Engl J Med. 2008;359:242–251. doi: 10.1056/NEJMoa0707894. [DOI] [PubMed] [Google Scholar]
- 124.Vo AA, Peng A, Toyoda M, Kahwaji J, Cao K, Lai CH, Reinsmoen NL, Villicana R, Jordan SC. Use of intravenous immune globulin and rituximab for desensitization of highly HLA-sensitized patients awaiting kidney transplantation. Transplantation. 2010;89:1095–1102. doi: 10.1097/TP.0b013e3181d21e7f. [DOI] [PubMed] [Google Scholar]
- 125.Montgomery RA, Zachary AA. Transplanting patients with a positive donor-specific crossmatch: a single center’s perspective. Pediatr Transplant. 2004;8:535–542. doi: 10.1111/j.1399-3046.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 126.Montgomery RA, Lonze BE, King KE, Kraus ES, Kucirka LM, Locke JE, Warren DS, Simpkins CE, Dagher NN, Singer AL, et al. Desensitization in HLA-incompatible kidney recipients and survival. N Engl J Med. 2011;365:318–326. doi: 10.1056/NEJMoa1012376. [DOI] [PubMed] [Google Scholar]
- 127.Bartel G, Wahrmann M, Regele H, Kikić Z, Fischer G, Druml W, Mühlbacher F, Böhmig GA. Peritransplant immunoadsorption for positive crossmatch deceased donor kidney transplantation. Am J Transplant. 2010;10:2033–2042. doi: 10.1111/j.1600-6143.2010.03226.x. [DOI] [PubMed] [Google Scholar]
- 128.Marfo K, Lu A, Ling M, Akalin E. Desensitization protocols and their outcome. Clin J Am Soc Nephrol. 2011;6:922–936. doi: 10.2215/CJN.08140910. [DOI] [PubMed] [Google Scholar]
- 129.Woodle ES, Alloway RR, Girnita A. Proteasome inhibitor treatment of antibody-mediated allograft rejection. Curr Opin Organ Transplant. 2011;16:434–438. doi: 10.1097/MOT.0b013e328348c0e5. [DOI] [PubMed] [Google Scholar]
- 130.Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9 Suppl 3:S1–155. doi: 10.1111/j.1600-6143.2009.02834.x. [DOI] [PubMed] [Google Scholar]
- 131.Roberts DM, Jiang SH, Chadban SJ. The treatment of acute antibody-mediated rejection in kidney transplant recipients-a systematic review. Transplantation. 2012;94:775–783. doi: 10.1097/TP.0b013e31825d1587. [DOI] [PubMed] [Google Scholar]
- 132.Lefaucheur C, Nochy D, Andrade J, Verine J, Gautreau C, Charron D, Hill GS, Glotz D, Suberbielle-Boissel C. Comparison of combination Plasmapheresis/IVIg/anti-CD20 versus high-dose IVIg in the treatment of antibody-mediated rejection. Am J Transplant. 2009;9:1099–1107. doi: 10.1111/j.1600-6143.2009.02591.x. [DOI] [PubMed] [Google Scholar]
- 133.Kaposztas Z, Podder H, Mauiyyedi S, Illoh O, Kerman R, Reyes M, Pollard V, Kahan BD. Impact of rituximab therapy for treatment of acute humoral rejection. Clin Transplant. 2009;23:63–73. doi: 10.1111/j.1399-0012.2008.00902.x. [DOI] [PubMed] [Google Scholar]
- 134.Flechner SM, Fatica R, Askar M, Stephany BR, Poggio E, Koo A, Banning S, Chiesa-Vottero A, Srinivas T. The role of proteasome inhibition with bortezomib in the treatment of antibody-mediated rejection after kidney-only or kidney-combined organ transplantation. Transplantation. 2010;90:1486–1492. doi: 10.1097/TP.0b013e3181fdd9b0. [DOI] [PubMed] [Google Scholar]
- 135.Waiser J, Budde K, Schütz M, Liefeldt L, Rudolph B, Schönemann C, Neumayer HH, Lachmann N. Comparison between bortezomib and rituximab in the treatment of antibody-mediated renal allograft rejection. Nephrol Dial Transplant. 2012;27:1246–1251. doi: 10.1093/ndt/gfr465. [DOI] [PubMed] [Google Scholar]
- 136.Legendre C, Sberro-Soussan R, Zuber J, Rabant M, Loupy A, Timsit MO, Anglicheau D. Eculizumab in renal transplantation. Transplant Rev (Orlando) 2013;27:90–92. doi: 10.1016/j.trre.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 137.Tzvetanov I, Spaggiari M, Jeon H, Roca RG, Bhati C, Oberholzer J, Benedetti E. The role of splenectomy in the setting of refractory humoral rejection after kidney transplantation. Transplant Proc. 2012;44:1254–1258. doi: 10.1016/j.transproceed.2012.01.109. [DOI] [PubMed] [Google Scholar]
- 138.Kaplan B, Gangemi A, Thielke J, Oberholzer J, Sankary H, Benedetti E. Successful rescue of refractory, severe antibody mediated rejection with splenectomy. Transplantation. 2007;83:99–100. doi: 10.1097/01.tp.0000243739.31440.2b. [DOI] [PubMed] [Google Scholar]
- 139.Roberti I, Geffner S, Vyas S. Successful rescue of refractory acute antibody-mediated renal allograft rejection with splenectomy--a case report. Pediatr Transplant. 2012;16:E49–E52. doi: 10.1111/j.1399-3046.2011.01518.x. [DOI] [PubMed] [Google Scholar]
- 140.Fehr T, Gaspert A. Antibody-mediated kidney allograft rejection: therapeutic options and their experimental rationale. Transpl Int. 2012;25:623–632. doi: 10.1111/j.1432-2277.2012.01453.x. [DOI] [PubMed] [Google Scholar]
- 141.Billing H, Rieger S, Ovens J, Süsal C, Melk A, Waldherr R, Opelz G, Tönshoff B. Successful treatment of chronic antibody-mediated rejection with IVIG and rituximab in pediatric renal transplant recipients. Transplantation. 2008;86:1214–1221. doi: 10.1097/TP.0b013e3181880b35. [DOI] [PubMed] [Google Scholar]
- 142.Fehr T, Rüsi B, Fischer A, Hopfer H, Wüthrich RP, Gaspert A. Rituximab and intravenous immunoglobulin treatment of chronic antibody-mediated kidney allograft rejection. Transplantation. 2009;87:1837–1841. doi: 10.1097/TP.0b013e3181a6bac5. [DOI] [PubMed] [Google Scholar]
- 143.Ahn G, Chung B, Park C, Choi B, Kim Y, Yang C. Effectiveness of Rituximab and intravenous immunoglobulin for the treatment of chronic antibody mediated rejection in kidney transplant recipients. Am J Transplant. 2013;19:328. [Google Scholar]
- 144.Schwaiger E, Regele H, Wahrmann M, Werzowa J, Haidbauer B, Schmidt A, Böhmig GA. Bortezomib for the treatment of chronic antibody-mediated kidney allograft rejection: a case report. Clin Transpl. 2010:391–396. [PubMed] [Google Scholar]
- 145.Lonze BE, Dagher NN, Simpkins CE, Singer AL, Segev DL, Zachary AA, Montgomery RA. The fate of anti-HLA antibody among renal transplantation recipients treated with bortezomib. Clin Transpl. 2009:377–384. [PubMed] [Google Scholar]
- 146.Kulkarni S, Kirkles-Smith , Tomlin R, Formica R, Moeckel G, Pober J. Eculizumab therapy for chronic antibody-mediated injury in kidney transplantation: an interim assessment. Am J Transplant. 2013;19:86. doi: 10.1111/ajt.14001. [DOI] [PubMed] [Google Scholar]
- 147.Ginevri F, Nocera A, Comoli P, Innocente A, Cioni M, Parodi A, Fontana I, Magnasco A, Nocco A, Tagliamacco A, et al. Posttransplant de novo donor-specific hla antibodies identify pediatric kidney recipients at risk for late antibody-mediated rejection. Am J Transplant. 2012;12:3355–3362. doi: 10.1111/j.1600-6143.2012.04251.x. [DOI] [PubMed] [Google Scholar]
- 148.Ashimine S, Watarai Y, Yamamoto T, Hiramitsu T, Tsujita M, Nanmoku K, Goto N, Takeda A, Katayama A, Uchida K, et al. Neither pre-transplant rituximab nor splenectomy affects de novo HLA antibody production after renal transplantation. Kidney Int. 2014;85:425–430. doi: 10.1038/ki.2013.291. [DOI] [PubMed] [Google Scholar]
- 149.Vincenti F, Cohen SD, Appel G. Novel B cell therapeutic targets in transplantation and immune-mediated glomerular diseases. Clin J Am Soc Nephrol. 2010;5:142–151. doi: 10.2215/CJN.04580709. [DOI] [PubMed] [Google Scholar]
- 150.Jordan SC, Reinsmoen N, Peng A, Lai CH, Cao K, Villicana R, Toyoda M, Kahwaji J, Vo AA. Advances in diagnosing and managing antibody-mediated rejection. Pediatr Nephrol. 2010;25:2035–2045; quiz 2035-2045. doi: 10.1007/s00467-009-1386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seveso M, Rigotti P, Schena PF, Stefoni S, Sandrini S, Salvadori M, Stratta P, Citterio F, Segoloni GP, Ruffoni E, et al. Preliminary results from a prospective multi-center study reveal the early appearance of de novo anti-HLA antibodies following renal transplantation. Transpl Int. 2011;24:38. [Google Scholar]
- 152.Vincenti F, Larsen CP, Alberu J, Bresnahan B, Garcia VD, Kothari J, Lang P, Urrea EM, Massari P, Mondragon-Ramirez G, et al. Three-year outcomes from BENEFIT, a randomized, active-controlled, parallel-group study in adult kidney transplant recipients. Am J Transplant. 2012;12:210–217. doi: 10.1111/j.1600-6143.2011.03785.x. [DOI] [PubMed] [Google Scholar]
- 153.Badell IR, Thompson PW, Turner AP, Russell MC, Avila JG, Cano JA, Robertson JM, Leopardi FV, Strobert EA, Iwakoshi NN, et al. Nondepleting anti-CD40-based therapy prolongs allograft survival in nonhuman primates. Am J Transplant. 2012;12:126–135. doi: 10.1111/j.1600-6143.2011.03736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Goldwater R, Keirns J, Blahunka P, First R, Sawamoto T, Zhang W, Kowalski D, Kaibara A, Holman J. A phase 1, randomized ascending single-dose study of antagonist anti-human CD40 ASKP1240 in healthy subjects. Am J Transplant. 2013;13:1040–1046. doi: 10.1111/ajt.12082. [DOI] [PubMed] [Google Scholar]
- 155.Watanabe M, Yamashita K, Suzuki T, Kamachi H, Kuraya D, Koshizuka Y, Ogura M, Yoshida T, Aoyagi T, Fukumori D, et al. ASKP1240, a fully human anti-CD40 monoclonal antibody, prolongs pancreatic islet allograft survival in nonhuman primates. Am J Transplant. 2013;13:1976–1988. doi: 10.1111/ajt.12330. [DOI] [PubMed] [Google Scholar]
- 156.Boskovic S, Smith RN, Kawai T, Nadazdin , O; Sachs, D; Cosimi, A B ; Colvin, R. Inhibitory effects of atacicept (TACI-Ig) on circulating antibodies, B cells and plasma cells in allosensitized cynomolgus monkeys. Transplantation. 2008;86:163. [Google Scholar]
- 157.Kirschfink M. C1-inhibitor and transplantation. Immunobiology. 2002;205:534–541. doi: 10.1078/0171-2985-00152. [DOI] [PubMed] [Google Scholar]