

Abstract
Gastric cancer is the second most common cause of cancer-related death in the world. A growing body of evidence indicates that inflammation is closely associated with the initiation, progression, and metastasis of many tumors, including those of gastric cancer. In addition, approximately 60% of the world's population is colonized by Helicobacter pylori, which accounts for more than 50% of gastric cancers. While the role of inflammation in intestinal and colonic cancers is relatively well defined, its role in stomach neoplasia is still unclear because of the limited access of pathogens to the acidic environment and the technical difficulties isolating and characterizing immune cells in the stomach, especially in animal models. In this review, we will provide recent updates addressing how inflammation is involved in gastric malignancies, and what immune characteristics regulate the pathogenesis of stomach cancer. Also, we will discuss potential therapeutics that target the immune system for the efficient treatment of gastric cancer.
Keywords: Stomach neoplasms, Inflammation, Helicobacter pylori, Immune cells
INTRODUCTION
Gastric cancer is the fourth and fifth most frequent cancer in men and women, respectively, and the second most common cause of cancer-related death in the world.1 While the overall incidence of gastric cancer is declining in the United States and Western Europe, it is still relevant in many Asian nations, parts of South America, and Eastern Europe. These regional variations are likely due to differences in a variety of environmental factors and differences in the prevalence of Helicobacter pylori infection.
Anatomically, there are two types of gastric cancer. The first, gastric cardia (proximal) cancer, occurs in the top portion of the stomach near the gastroesophageal junction, and the second, noncardia cancer, may be found in all other areas of the stomach. These two cancer types have different risk factors and incidence patterns. For example, noncardia cancer is commonly associated with H. pylori infection, while gastric cardia cancer seems not to be affected by H. pylori.2 On the other hand, cardia gastric cancer, rare at one time, is now increasing in Western nations for reasons that are still unclear, and it accounts for about 50% of all stomach cancer in men in the United States. Gastric adenocarcinomas are classified into well-differentiated and undifferentiated types, with the undifferentiated type being characterized by a diffuse presentation, i.e., gastritis throughout the stomach but no atrophy, and the differentiated type is classified by intestinal-type tumors and corpus-dominated gastritis with gastric atrophy and intestinal metaplasia.
Recent insight into the tumor microenvironment has begun to uncover the close association between cancer and inflammation, which bears similarities to wounds that fail to heal. Approximately 20% of cancer deaths worldwide are associated with unresolved infection or chronic inflammation, and the prolonged inflammation can lead to gastric cancer, colorectal cancer, inflammatory bowel disease, hepatocellular carcinoma, and chronic pancreatitis.3 Unresolved inflammation generates a microenvironment that facilitates cellular transformation and the propagation of invasive disease, with chronic tissue damage triggering a repair response including growth and survival factors, tissue-remodeling enzymes, and immune regulatory cytokines. These examples of aberrant immunity foster tumors, but appropriate immune responses can suppress or eliminate tumors via a cancer immunosurveillance system.4 However, the contribution of immune cell populations to either the pathogenesis or protection in the stomach environment has been difficult to elucidate. Here, we highlight recent progress in defining the roles of the immune system in cancer pathology with a focus on gastric adenocarcinoma.
HELICOBACTER INFECTION, GASTRITIS, AND STOMACH CANCER
H. pylori is now regarded as the main cause of chronic gastritis and is classified as a gastric carcinogen. Correa5,6 proposed that chronic gastritis progresses to intestinal-type gastric cancer through histological changes that include atrophy, metaplasia, and dysplasia. Indeed, research has demonstrated that the inflammatory response to H. pylori evokes diverse neoplastic changes of the gastric epithelium.7,8 Thus, eradiation of H. pylori infection leads to positive outcomes in regards to atrophy, metaplasia, and genomic instability, accompanied by reduced inflammation in the stomach.9,10 However, antibiotic therapy not only eradicates H. pylori, but also other microorganisms that can drive the inflammatory microenvironment in the stomach, raising the possibility that other pathogens may also be associated with gastric neoplasia. This idea was supported by a study using transgenic FVB/N insulin-gastrin mice that develop spontaneous gastritis and epithelial neoplasia with 80% prevalence after H. pylori infection.11 When the mice were colonized under germ-free conditions, gastric carcinoma development induced by H. pylori infection was substantially delayed compared to the mice maintained with conventional flora.12
While more than 50% of the world's population is infected with H. pylori, only 10% to 15% of individuals infected with H. pylori develop peptic ulcers, and the risk of gastric cancer is estimated to be approximately 1% to 3%.1,13 This observation indicates that the disease progression of H. pylori-infected individuals depends on the presence of bacterial virulence factors and the types of host responses. The bacterial factors that allow H. pylori to colonize in the stomach and induce atrophic gastritis have been studied through the use of isogenic mutants and genomic analyses of the bacteria, and the crucial factors that are required for persistence of the infected bacteria include urease and flagella. H. pylori secretes large amounts of urease to hydrolyze urea to ammonia and carbon dioxide, which neutralizes gastric acid, providing a local environment supporting bacterial survival.14 In addition, colonization of H. pylori on the mucus layer depends on the presence of flagella and the production of enzymes that break down the surfactant layer over the gastric epithelium.14
Two genetic loci, cag and vacA, that link virulence factors to gastritis have been intensively studied. Infection with H. pylori strains that contain the cag locus, a 40 kb region known as the cag pathogenicity island (cag PAI), exhibit a higher risk for gastric cancer (especially intestinal-type adenocarcinoma) than infection with strains that do not contain the cag PAI.15 On the other hand, the cag PAI is unlikely to be responsible for the pathogenesis of diffused-type gastric carcinoma, although it is associated with H. pylori infection.15 The functional protein, CagA, is encoded by the cag PAI, and is an immunodominant protein produced by H. pylori. Infection with H. pylori translocates CagA protein into the host epithelial cells by a type IV secretion system, which is also encoded by cag PAIs. Once injected into the gastric epithelium, CagA is phosphorylated by Src family kinases and then activates SHP2 and Erk, which result in actin-cytoskeletal rearrangements and cell scattering.14,16 Moreover, gene products of cag PAIs can directly elicit inflammation via production of the interleukin (IL)-8 chemokine and through nucleotide-binding oligomerization domain-1 (NOD1). IL-8 induction by H. pylori infection depends on the presence of specific cagA genes and is mediated by MAPK and nuclear factor κB (NF-κB) signaling pathways in a SHP2-independent manner.17,18 NOD1, a pattern recognition receptor for the innate immune response, detects H. pylori infection by sensing a peptidoglycan produced by cag PAIs, and instructs the production of proinflammatory cytokines such as IL-1β, IL-8, and tumor necrosis factor-α (TNF-α).19
One crucial virulence factor secreted for the pathogenesis of stomach dysplasia is the vacuolating cytotoxin (Vac) encoded by the vacA gene.20,21 In a Mongolian gerbil model, infection with an adapted H. pylori strain lacking VacA reduced the incidence of gastric carcinoma.22,23 Interestingly, the loss of CagA was protective in cancer development by attenuating inflammation in this model, while ablation of VacA did not affect inflammation.22 However, the secreted pore-forming VacA toxin can indirectly augment inflammatory responses. For example, human gastric epithelial cells are highly susceptible to VacA-induced cell death, and this programmed necrotic pathway contributes to the pathogenesis of peptic ulceration and gastric cancer by enhancing mucosal inflammation.24,25
In addition to the direct effect of the virulence factors on the gastric epithelium, inflammation induced by H. pylori infection results in altered patterns of host DNA modification, which is closely associated with cancer risk. Temporal analysis showed that levels of DNA methylation in gastric epithelial cells were increased by H. pylori infection, which was paralleled by the induction of inflammation-related genes encoding IL-1β, NOS2, and TNF-α.26 Suppressing inflammation with cyclosporine A treatment did not affect bacterial colonization, but treatment did attenuate the induction of DNA methylation, suggesting that the infection-induced inflammatory response, rather than H. pylori itself, is responsible for the genetic alteration of the gastric epithelium. Despite the notion that H. pylori evokes a strong inflammatory response, the immune system is likely to be insufficient to clear the infection due to the pathogen's immune evasion strategies. The mechanisms for immune evasion include the induction of a strong, polarized immune response, modulation of phagocytosis and neutrophil function, and inhibition of lymphocyte proliferation.14 Thus, prolonged inflammation and direct action of bacterial factors may simultaneously lead to impaired gland function and eventually to carcinogenesis.
HOST IMMUNE SYSTEM AND GASTRIC CANCER
A large body of evidence suggests pivotal roles for host factors in determining the progression of the gastric pathogenesis. The stroma of gastric tumors is frequently filled with a wide range of white blood cells, but whether these inflammatory cells are responsible for the initiation and/or the progression of gastric pathogenesis remains undetermined. Genetic studies of patients with gastric atrophy, hypochlorhydria, and carcinoma revealed an increased incidence of genetic alterations strongly associated with host immune responses. Bone marrow-derived populations that constitute the inflammatory microenvironment of the stomach include granulocytes, macrophages, myeloid-derived suppressor cells (MDSCs), and dendritic cells, as well as adaptive immune cells such as T and B cells (Figs 1 and 2).
Fig. 1.
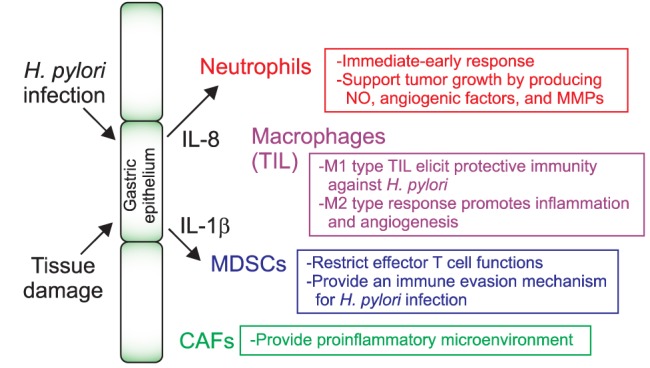
Innate immune cells associated with gastric pathogenesis.
NO, nitric oxide; MMP, matrix metalloproteinase; IL, interleukin; TIL, tissue infiltrating lymphocyte; MDSC, myeloid-derived suppressor cell; CAF, carcinoma-associated fibroblast.
Fig. 2.

Helper T cell subsets and gastric neoplasia.
IL, interleukin; TGF, transforming growth factor; IFN, interferon.
1. Innate immune response
1) Neutrophils
Neutrophils are the most abundant circulating granulocyte population and are recruited to the inflamed tissue immediately or early after infection or tissue damage. Pathogen-induced IL-8 expression from the stroma mobilizes neutrophils into the infected lesion and activates the cells to produce antimicrobial peptides and reactive oxygen species.27 Moreover, the activated neutrophil production of IL-8 is important for the initiation of adaptive immunity by recruiting T cells. Forced expression of human IL-8 in mice is sufficient to accelerate colonic and gastric carcinogenesis induced chemically and by H. felis infection, respectively.28 Of note is that CD11b+ Gr1+ myeloid cell levels increase systemically and locally in IL-8 transgenic mice, and these cells contribute to tumor microenvironment remodeling. These studies suggest that targeting IL-8 may be a useful therapeutic for inflammation-associated carcinogenesis, although further characterization of the myeloid populations is needed.
2) Tumor-associated macrophages
Tumor-associated macrophages (TAMs) are a heterogeneous population, and accumulation of these cells in the neoplastic area indicates a poor prognosis.29 In a gastric cancer model using K19-Wnt1 transgenic mice, macrophages accumulate in the dysplastic mucosa, accompanied by nuclear induction of β-catenin in the epithelium.30 Intriguingly, depletion of macrophages from APC (Δ716) mice mitigates intestinal tumor formation, suggesting that inflammation induced by macrophages contributes to the pathogenesis of the gastric mucosa via Wnt/β-catenin signaling. An example of how macrophages directly affect the pathogenesis of the gastric epithelium is by the production of nitric oxide (NO). Specifically, macrophages activated by H. pylori infection produce NO that causes methylation of genes associated with tumor suppression, such as Runx3, in the epithelial cells. Thus, treatment with an NO-specific inhibitor can reverse H. pylori-induced methylation of the gut epithelium, providing a mechanism by which H. pylori causes epigenetic changes associated with gastric malignancy through inflammation.
Depending on the cytokine expression profiles, there are two distinct types of macrophages.31 M1 macrophages are activated by interferon-γ (IFN-γ) and microbial components such as lipopolysaccharides (LPS), they express high level of class II major histocompatibility complex (MHC) molecules, and they produce proinflammatory cytokines including IFN-γ and TNF-α. Alternatively, M2 macrophages are activated by IL-4, have reduced antigen presentation characteristics and tumoricidal activity, and produce the immunosuppressive cytokine, IL-10. Accordingly, in the area of solid tumors, accumulation of M2 macrophages, which are involved in wound healing and tissue remodeling in normal physiology, is associated with a poor prognosis, whereas tumor infiltration of M1 macrophages could elicit a protective immune responses against the tumor.31,32 However, it is hard to distinguish between M1 and M2 TAMs in gastric tumors because they partially share cytokine profiles that are unlikely to be observed in vitro.
3) IL-1β and MDSCs
Polymorphisms of a gene encoding IL-1β are well known to be high risk factors for intestinal disorders such as Crohn disease.33,34 IL-1β inhibits acid secretion in the stomach, and genetic variations in the IL-1β locus that includes IL-1β and IL-1RN, a soluble IL-1 receptor antagonist, are also associated with gastric atrophy and adenocarcinoma.35 The functional role of IL-1β in gastric inflammation and cancer was reported using a transgenic mouse expressing human IL-1β specifically in the stomach.36 These transgenic mice developed spontaneous inflammation and gastric tumors that correlated with early recruitment and activation of MDSCs, a population having the capacity to inhibit inflammatory T cell responses.37 Interestingly, administration of neutralizing antibody against the IL-1 receptor attenuated the gastric dysplasia, which was accompanied by a marked decrease in MDSC infiltration to the stomach. This study proposed an immune evasion mechanism for gastric tumors with sustained chronic inflammation, and suggested that the proinflammatory cytokine, IL-1β, can elicit immunosuppressive function by recruiting MDSCs. In addition, MDSCs can directly induce tumor progression and metastasis by producing matrix metalloproteinases that facilitate tumor invasion.38 In addition, the conditional deletion of p120 catenin in mice leads to esophageal squamous cancer accompanied by the recruitment of MDSCs to the dysplastic epithelium.39 These results support the finding that mobilization of MDSCs to the gastric epithelium provides a microenvironment to promote inflammation-associated dysplasia.
4) Toll-like receptor
Toll-like receptors (TLRs) sense pathogen-associated molecular patterns to mediate an immediate-early host response to the infectious microorganisms. Specifically, TLR4 recognizes the LPS of gram-negative bacteria and induces expression of proinflammatory cytokines such as IL-1β, TNF-α, and IL-8.40 LPS from H. pylori upregulates TLR4 expression in gastric cancer cells and then induces proliferation of the cells.41 The TLR4 A896G polymorphism is linked to an impaired response to LPS, and genetic analysis of patients with hypochlorhydria and gastric atrophy revealed that this genetic variation is positively correlated with the severity of gastric atrophy and inflammation.40,42 Although it was not associated with gastric acid output in the absence of H. pylori infection, this genetic alteration is a strong risk factor for noncardia gastric carcinoma induced by the host's innate immune response in H. pylori infection. Distinct from TLR4, TLR2 is involved in a wide array of microbial molecules that include peptidoglycans, lipoteichoic acid, lipoproteins, and zymosan.40 Through TLR2 signaling, Listeria monocytogenes stimulates proliferation of tumor cells by increasing production of NO and IL-6, without affecting infiltration of MDSCs or regulatory T cells.43 The effects of H. pylori infection in gastric tumor cells is shared with those of Listeria, such that silencing TLR2 expression abrogates the bacteria-induced tumor cell proliferation.
5) INF-γ receptor 1
Another polymorphism of inflammation-related genes associated with the risk of gastric cancer was found in a gene encoding the INF-γ receptor 1 (Ifngr1). Individuals having polymorphisms in Ifgnr1 are more susceptible to H. pylori infection, and a case-control study revealed a strong correlation between the onset of gastric cancer and a specific genotype (C-56T) in the Ifngr1 promoter region.44 This genetic variation is a relevant host factor that predicts early development of gastric cancer. Using a transgenic mouse model, the potential role of this cytokine in gastric tumorigenesis was evaluated. Under the control of the stomach-specific H+/K+ ATPase promoter, transgenic expression of IFN-γ resulted in proliferation of undifferentiated and metaplastic epithelial cells along with elevated expression of proinflammatory cytokines that include IL-6, IL-1β, and TNF-α.45 The mice exhibited dysplasia at as early as 3 months of age, and some mice developed antral polyps, suggesting IFN-γ as a crucial inflammatory factor that drives preneoplastic progression in the stomach. Nonetheless, IFN-γ involved in the type 1 helper T-cell response may have a protective role in gastric pathogenesis, will be discussed later.
6) Carcinoma-associated fibroblasts
Carcinoma-associated fibroblasts (CAFs) that express α-smooth muscle actin function like a tissue-resident immune cell and contribute to cancer progression. Quante and colleagues46 found that some (about 20%) of the CAFs originate in bone marrow and increased markedly during cancer progression. In response to transforming growth factor β (TGF-β) and stromal cell-derived factor-1α, CAFs are generated and recruited to the dysplastic tumor, suggesting a role for this bone marrow-derived cell in sustaining tumor progression. This suggested role was supported by a report showing that in response to H. pylori infection, bone marrow populations mobilized to the stomach and differentiated into a fibroblast lineage that participated in the gastric neoplasia.47 Chimeric mice transplanted with bone progenitor cells expressing GFP develop gastric dysplasia when infected with H. pylori, in addition to chronic inflammation. More than 20% of the dysplastic lesions were composed of glands that contained epithelial bone marrow-derived cells.
7) IL-6 family
Intracellular signaling mechanisms that specify tissue-specific responses to the IL-6 family of cytokines are emerging to be crucial for inflammation-induced neoplasia. Knocking-in a GP130 mutation, a shared subunit of receptors for IL-6 family cytokines, resulted in gastric adenomas by 3 months of age, and the adenoma displayed many of the characteristics of human intestinal-type gastric cancer.48 Loss of a STAT3 or IL-6 allele in the mutant GP130 mice resulted in a reduced frequency and rate of tumor development due to the inhibition of proliferation-induced glandular hyperplasia.49,50 The mice exhibited a reduction in the recruitment of inflammatory cells such as macrophages and neutrophils, cytokine expression, angiogenesis, and metalloproteinase expression, which was similarly observed in the mice treated with an antimicrobial agent. These results support the notion that IL-6-induced STAT3 activation plays a crucial role in inflammation-induced gastric cancer development. Moreover, another group reported an unexpected correlation between TLR2 and STAT3, in which STAT3 upregulated TLR2 expression in the gastric epithelium.51 Genetic and therapeutic targeting of TLR2 in the gastric cancer mouse model inhibited gastric tumorigenesis, but not inflammation. Furthermore, increased activation of STAT3 and TLR2 were inversely correlated with gastric cancer patient survival.51 Collectively, in addition to the impact on immune function, STAT3-dependent TLR2 activation has an oncogenic function in a gastric epithelial cell-intrinsic manner.
2. Adaptive immune response
1) Effector versus regulatory T cells
Appropriate T cell responses are important for the clearance of chronic bacterial infection, while it is still unclear how H. pylori infection evokes adaptive immunity and how this pathogen evades immune surveillance to survive in specialized organs. For proper infection clearance, naïve T cells differentiate into effector T cell subsets that include type 1 helper T (Th1) cells for intracellular viruses and tumors, type 2 (Th2) cells for helminthic worms, and type 17 (Th17) cells for extracellular bacteria, especially in the intestine.52 On the other hand, regulatory T cells (Treg cells) that express the FoxP3 transcription factor and anti-inflammatory cytokine, IL-10, are important for peripheral tolerance, and balance between effector T cells and Treg cells is crucial for the resolution of chronic inflammation. Recent studies focus on tumor-infiltrating lymphocyte (TIL) populations that are associated with disease outcomes in various human cancers.53,54 However, the prognostic role of TILs in patients with gastric cancer still remains to be determined.
Flow cytometry analysis of human subjects with or without H. pylori infection indicated that the bacteria-induced immune response includes both Th1 and Th2 subsets with high levels of anti-inflammatory Treg cells.55 Intriguingly, individuals with peptic ulcers exhibited reduced IL-10+ Treg responses but increased Th1 and Th2 responses compared to those without ulcers. These results indicate a modulatory mechanism of Treg cells in the H. pylori-induced inflammatory environment, but otherwise, gastritis occurs when this regulatory response is inadequate. Other example that H. pylori infection can induce systemic Treg cells has been shown in a murine asthma model. H. pylori infection efficiently protected mice from airway hypersensitiveness, tissue inflammation, and goblet cell metaplasia, which are hallmarks of asthma.56 The accumulation of highly suppressive Treg cells in the lungs was associated with asthma protection, and this Treg accumulation was accompanied by a reduction in allergen-induced lung infiltration of eosinophils, Th2 cells, and Th17 cells, a response that was abolished with antibiotic eradication of H. pylori or systemic Treg depletion. These results provide experimental evidence for a competitive effect of H. pylori colonization on the development of allergen-induced asthma.
2) TGF-β signaling
TGF-β-deficient mice spontaneously develop gastritis, which is associated with an exaggerated Th1 response, characterized by IFN-γ production and reduced induction of Treg cells.57 Analysis of patient samples infected with H. pylori infection revealed defective TGF-β1-induced Smad3 phosphorylation in the H. pylori-infected whole biopsy specimens and isolated mucosal cells.58 Although activated TGF-β1 was abundant in the mucus, H. pylori infection results in increased expression of Smad7, which inhibits Smad3 functions. Antisense treatment for Smad7 is sufficient for restoring TGF-β1-induced Smad3 phosphorylation in the biopsy specimens, which is followed by reduced expression of the Th1 subset markers IFN-γ and T-bet.58 These data suggest that the downregulation of TGF-β1 signaling by H. pylori infection promotes the ongoing tissue-damaging Th1 response.
3) IFN-γ
Although chronic infection with H. pylori upregulates the proinflammatory Th1 response, the effect of the Th1 cytokine IFN-γ on inflammation-associated gastric carcinogenesis is debatable. In contrast to the expected proinflammatory effect, targeted expression of IFN-γ in a transgenic mouse under the control of the H+/K+-ATPase failed to induce gastritis, and instead, inhibited gastric carcinogenesis driven by IL-1β and/or H. pylori infection.59 Expression of IFN-γ inhibited Th1 and Th17 immune responses through Fas induction and apoptosis of CD4 T cells, and evoked autophagy in gastric epithelial cells by increasing Beclin-1. These results were far different than the results observed in the above-described IFN-γ transgenic mice that spontaneously develop spasmolytic polypeptide expressing metaplasia and dysplasia.45 This discrepancy might be explained by differences in the amount of IFN-γ produced and the cytokine milieu in the mutant mouse lines. Thus, overexpression of IFN-γ in concert with proinflammatory cytokines such as IL-6 can promote gastritis and dysplasia, while moderate expression of IFN-γ can elicit a coordinated function that eliminates the infection and modulates chronic inflammation by inducing an autophagic program.
4) Th17 cells
While Th17 cells are essential for the homeostasis of and protection against intestinal bacterium, implications of this helper T-cell subset in the stomach are unclear. Several lines of experimental evidence recently suggested a potential role for the Th17 cytokine, IL-17, in gastric carcinogenesis. Stimulation of human gastric cancer cell lines, including AGS, with IL-17A induces NF-κB activation and IL-8 production, which is abrogated by inhibition of IL-17 receptors using siRNAs.60 In addition, a genetic polymorphism of the IL-17 gene in gastric carcinogenesis has been reported.61 The study showed that the 197A polymorphism of the IL-17A gene was significantly correlated with both the inflammation score and the risk of developing gastric mucosal atrophy, especially in intestinal-type cancer compared to diffuse-type cancer. Furthermore, the Iida group62 analyzed human gastric tumors by immunohistochemistry, and they observed infiltration of CD4+ IL-17+ cells and upregulation of mRNA of the Th17-associated cytokines IL-17, IL-21, and IL-23 in the tumor tissues compared to normal tissues. It was intriguing that the number of vascular endothelial cells and neutrophils were markedly increased in tumors expressing high level of IL-17 compared those expressing a low IL-17 levels. Overall, these studies suggest a pivotal role for the Th17 subset in the persistence of inflammation and gastric neoplasia.
In the presence of TGF-β, IL-6, and IL-23-naive CD4 T cells differentiate into the Th17 subset, and these factors might be enriched in the tumor microenvironment.63 Gastric myofibrobalsts (GMFs) express high levels of class II MHC and are thought to act as antigen-presenting cells in the gastric mucosa. When CD4 T cells differentiate under the Th17-polarizing conditions, GMFs isolated from patients with gastric cancer or infected with H. pylori substantially enhanced Th17 induction compared to the cells from normal tissues.64 This study provides a mechanism by which Th17 cells are enriched in the inflammatory milieu of the gastric stroma and provide a link between inflammation and carcinogenesis.
In contrast, the Kennedy group65 claimed that Th17 responses are not directly associated with the pathogenesis of gastric tumors. Knock-in mice with the pp130(F/F) allele spontaneously develop gastric inflammation-associated tumors akin to human intestinal-type gastric cancer. This mutation leads to hyperactivation of STAT3 via the stimulation of IL-6 family cytokines.50 In these mice, generation of Th17 cells and the gastric expression of Th17-related factors such as IL-17a, Rorγt, and IL-23 were augmented. However, genetic ablation of IL-17A did not suppress the initiation and growth of gastric tumors in the gp130(F/F) background. Moreover, IL-17A and Rorγt expression was strongly increased in patients with gastritis but not gastric cancer, suggesting that increased expression of Th17-related factors does not correlate with the progression of gastric tumorigenesis. In other reports, however, flow cytometric analysis of peripheral blood from patients with gastric cancer showed that circulating Th17 cells are positively correlated with the stage of gastric cancer,66 implying that Th17 cells contribute to the gastric tumor.
5) B cells and Th2 cells
Although H. pylori-induced chronic atrophic gastritis is characterized by marked infiltration of Th1 and Th17 cells, the majority of the inflamed gastric mucosa also contains focal lymphoid aggregates with germinal centers.67 Autoantibodies against tumor-associated antigens are very attractive biomarkers for the development of serological tests for early cancer detection. Recently, phage display-based serological analyses have identified a representative set of antigens eliciting humoral responses in patients with gastric cancer,68 although it is unknown whether these B-cell responses are sufficient to cause gastric pathology. Typical H. pylori-induced chronic gastritis in children, which is called follicular gastritis, is characterized by B-cell follicles in the gastric mucosa.69 Thus, B-cell activation and cognate Th2 cell's help can be composed in the pathogen-induced inflammatory responses. Kido and his colleagues69 found that H. pylori directly induced production of thymic stromal lymphopoietin (TSLP) from the epithelial cells in human follicular gastritis and that TSLP-mediated dendritic cell activation is involved in the Th2 response to activate B cells in the pathogen-driven gastritis.
DIAGNOSIS AND THERAPEUTIC TARGETING IMMUNITY
A high density of cytotoxic CD8 T cells and memory T cells are usually associated with better outcomes in gastric cancer, indicating the crucial role of adaptive immunity in the tumor immunosurveillance system.70 On the other hand, the prevalence of suppressor cells may prove to be a decisive factor for poor outcomes because MDSC and Treg cells restrain the antitumor activity of cytotoxic T cells. Indeed, analysis of patients with hepatocarcinoma revealed that a high ratio of cytotoxic T cells to Treg cells was linked to increased survival, whereas a low ratio was associated with tumor vascular invasion and inferior survival.71 In this context, the type, density, and intratumoral location of the leukocyte infiltrate has been shown to be a more informative biomarker than the TNM or Duke's classification.72
Programmed death-1 (PD1) is a negative regulator of effector T cell function.73 Signaling through PD1 is triggered by engagement of the ligands PD-L1 or PD-L2, and intriguingly, some tumors upregulate expression of PD-L1, resulting in a dampened cytotoxic T-cell response. Actually, patients with pancreatic adenocarcinoma exhibit high levels of intratumoral PD-L1, which is accompanied by decreased T-cell infiltration and poor survival, suggesting an immune-evasion mechanism.74 One promising therapy is antibody blockade of the interaction between PD1 and PD-L1, which is undergoing clinical trials. Another promising target for cancer therapy involves cytotoxic T cell antigen-4 (CTLA-4). Treatment with monoclonal antibodies against CTLA-4 augments the antitumor activity of effector T cells and results in improved patient survival in advanced melanoma.75 Evaluation of this antibody therapy is ongoing in the adjuvant setting for gastric cancer.
CONCLUDING REMARKS
While bacterial virulence factors are crucial determinants of disease outcomes, the host defense system also plays a role by eliciting an immune response to clear H. pylori infection, which results in a robust gastritis followed by a series of pathological changes that progress to cancer. Antibiotic intervention to eradicate H. pylori early in this pathological process might be protective against more severe disease. However, it is still unclear whether patients with advanced, premalignant lesions can benefit from antibiotic treatment. Alternatively, mediators of chronic inflammation could serve as potential therapeutic targets to suppress the gastric pathogenesis. For example, in the setting of achlorhydria, sustained tissue damage in the stomach due to chronic inflammation leads to cancer in part by affecting recruitment of immune cell populations, thereby altering the tumor microenvironment. Thus, we should translate our knowledge of the tumor microenvironment into clinical trials, such as those that combine drugs targeting inflammation with conventional chemotherapies.
ACKNOWLEDGEMENTS
This study was supported by a faculty research grant of Yonsei University College of Medicine for 2013 (6-2013-0061), a new faculty research seed money grant of Yonsei University College of Medicine for 2013 (2013-32-0031), and the Brain Korea 21 PLUS Project for Medical Science of Yonsei University to K.T.N.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 2.Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 4.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 5.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process. First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 6.Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995;19(Suppl 1):S37–S43. [PubMed] [Google Scholar]
- 7.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 8.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–1179. doi: 10.1016/s0016-5085(98)70422-6. [DOI] [PubMed] [Google Scholar]
- 9.Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005;128:1937–1952. doi: 10.1053/j.gastro.2005.02.066. [DOI] [PubMed] [Google Scholar]
- 10.Nardone G, Staibano S, Rocco A, et al. Effect of Helicobacter pylori infection and its eradication on cell proliferation, DNA status, and oncogene expression in patients with chronic gastritis. Gut. 1999;44:789–799. doi: 10.1136/gut.44.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36–47. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 12.Lofgren JL, Whary MT, Ge Z, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsonnet J, Friedman GD, Vandersteen DP, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 14.Salama NR, Hartung ML, Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–399. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301. doi: 10.1136/gut.40.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA x SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003;278:3664–3670. doi: 10.1074/jbc.M208155200. [DOI] [PubMed] [Google Scholar]
- 17.Crabtree JE, Xiang Z, Lindley IJ, Tompkins DS, Rappuoli R, Covacci A. Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J Clin Pathol. 1995;48:967–969. doi: 10.1136/jcp.48.10.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 20.Yu J, Leung WK, Go MY, et al. Relationship between Helicobacter pylori babA2 status with gastric epithelial cell turnover and premalignant gastric lesions. Gut. 2002;51:480–484. doi: 10.1136/gut.51.4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prinz C, Schoniger M, Rad R, et al. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 2001;61:1903–1909. [PubMed] [Google Scholar]
- 22.Franco AT, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogura K, Maeda S, Nakao M, et al. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med. 2000;192:1601–1610. doi: 10.1084/jem.192.11.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radin JN, Gonzalez-Rivera C, Ivie SE, McClain MS, Cover TL. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect Immun. 2011;79:2535–2543. doi: 10.1128/IAI.01370-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamasaki E, Wada A, Kumatori A, et al. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem. 2006;281:11250–11259. doi: 10.1074/jbc.M509404200. [DOI] [PubMed] [Google Scholar]
- 26.Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 27.Fischer W, Prassl S, Haas R. Virulence mechanisms and persistence strategies of the human gastric pathogen Helicobacter pylori. Curr Top Microbiol Immunol. 2009;337:129–171. doi: 10.1007/978-3-642-01846-6_5. [DOI] [PubMed] [Google Scholar]
- 28.Asfaha S, Dubeykovskiy AN, Tomita H, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology. 2013;144:155–166. doi: 10.1053/j.gastro.2012.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 30.Oguma K, Oshima H, Aoki M, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008;27:1671–1681. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 32.Fehlings M, Drobbe L, Moos V, et al. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect Immun. 2012;80:2724–2734. doi: 10.1128/IAI.00381-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Omar EM, Carrington M, Chow WH, et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412:99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- 34.Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 35.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taketo MM. Role of bone marrow-derived cells in colon cancer: lessons from mouse model studies. J Gastroenterol. 2009;44:93–102. doi: 10.1007/s00535-008-2321-3. [DOI] [PubMed] [Google Scholar]
- 39.Stairs DB, Bayne LJ, Rhoades B, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19:470–483. doi: 10.1016/j.ccr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27:244–252. doi: 10.1038/sj.onc.1210912. [DOI] [PubMed] [Google Scholar]
- 41.Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun. 2010;78:468–476. doi: 10.1128/IAI.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hold GL, Rabkin CS, Chow WH, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132:905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 43.Huang B, Zhao J, Shen S, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007;67:4346–4352. doi: 10.1158/0008-5472.CAN-06-4067. [DOI] [PubMed] [Google Scholar]
- 44.Canedo P, Corso G, Pereira F, et al. The interferon gamma receptor 1 (IFNGR1) -56C/T gene polymorphism is associated with increased risk of early gastric carcinoma. Gut. 2008;57:1504–1508. doi: 10.1136/gut.2007.143578. [DOI] [PubMed] [Google Scholar]
- 45.Syu LJ, El-Zaatari M, Eaton KA, et al. Transgenic expression of interferon-γ in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am J Pathol. 2012;181:2114–2125. doi: 10.1016/j.ajpath.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varon C, Dubus P, Mazurier F, et al. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. 2012;142:281–291. doi: 10.1053/j.gastro.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 48.Judd LM, Alderman BM, Howlett M, et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 49.Howlett M, Judd LM, Jenkins B, et al. Differential regulation of gastric tumor growth by cytokines that signal exclusively through the coreceptor gp130. Gastroenterology. 2005;129:1005–1018. doi: 10.1053/j.gastro.2005.06.068. [DOI] [PubMed] [Google Scholar]
- 50.Jenkins BJ, Grail D, Nheu T, et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med. 2005;11:845–852. doi: 10.1038/nm1282. [DOI] [PubMed] [Google Scholar]
- 51.Tye H, Kennedy CL, Najdovska M, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22:466–478. doi: 10.1016/j.ccr.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br J Cancer. 2011;105:93–103. doi: 10.1038/bjc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson K, Kenefeck R, Pidgeon EL, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57:1375–1385. doi: 10.1136/gut.2007.137539. [DOI] [PubMed] [Google Scholar]
- 56.Arnold IC, Dehzad N, Reuter S, et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest. 2011;121:3088–3093. doi: 10.1172/JCI45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hahm KB, Lee KM, Kim YB, et al. Conditional loss of TGF-beta signalling leads to increased susceptibility to gastrointestinal carcinogenesis in mice. Aliment Pharmacol Ther. 2002;16(Suppl 2):115–127. doi: 10.1046/j.1365-2036.16.s2.3.x. [DOI] [PubMed] [Google Scholar]
- 58.Monteleone G, Del Vecchio Blanco G, Palmieri G, et al. Induction and regulation of Smad7 in the gastric mucosa of patients with Helicobacter pylori infection. Gastroenterology. 2004;126:674–682. doi: 10.1053/j.gastro.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 59.Tu SP, Quante M, Bhagat G, et al. IFN-gamma inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71:4247–4259. doi: 10.1158/0008-5472.CAN-10-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y, Toh ML, Zrioual S, Miossec P. IL-17A versus IL-17F induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in AGS gastric adenocarcinoma cells. Cytokine. 2007;38:157–164. doi: 10.1016/j.cyto.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 61.Shibata T, Tahara T, Hirata I, Arisawa T. Genetic polymorphism of interleukin-17A and -17F genes in gastric carcinogenesis. Hum Immunol. 2009;70:547–551. doi: 10.1016/j.humimm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 62.Iida T, Iwahashi M, Katsuda M, et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep. 2011;25:1271–1277. doi: 10.3892/or.2011.1201. [DOI] [PubMed] [Google Scholar]
- 63.Wilke CM, Bishop K, Fox D, Zou W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011;32:603–611. doi: 10.1016/j.it.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pinchuk IV, Morris KT, Nofchissey RA, et al. Stromal cells induce Th17 during Helicobacter pylori infection and in the gastric tumor microenvironment. PLoS One. 2013;8:e53798. doi: 10.1371/journal.pone.0053798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kennedy CL, Najdovska M, Jones GW, et al. The molecular pathogenesis of STAT3-driven gastric tumourigenesis in mice is independent of IL-17. J Pathol. 2011;225:255–264. doi: 10.1002/path.2933. [DOI] [PubMed] [Google Scholar]
- 66.Liu T, Peng L, Yu P, et al. Increased circulating Th22 and Th17 cells are associated with tumor progression and patient survival in human gastric cancer. J Clin Immunol. 2012;32:1332–1339. doi: 10.1007/s10875-012-9718-8. [DOI] [PubMed] [Google Scholar]
- 67.Shimatani T, Inoue M, Iwamoto K, et al. Gastric acidity in patients with follicular gastritis is significantly reduced, but can be normalized after eradication for Helicobacter pylori. Helicobacter. 2005;10:256–265. doi: 10.1111/j.1523-5378.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 68.Zayakin P, Ancans G, Siliņa K, et al. Tumor-associated autoantibody signature for the early detection of gastric cancer. Int J Cancer. 2013;132:137–147. doi: 10.1002/ijc.27667. [DOI] [PubMed] [Google Scholar]
- 69.Kido M, Tanaka J, Aoki N, et al. Helicobacter pylori promotes the production of thymic stromal lymphopoietin by gastric epithelial cells and induces dendritic cell-mediated inflammatory Th2 responses. Infect Immun. 2010;78:108–114. doi: 10.1128/IAI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sangiolo D. Cytokine induced killer cells as promising immunotherapy for solid tumors. J Cancer. 2011;2:363–368. doi: 10.7150/jca.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao Q, Qiu SJ, Fan J, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–2593. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 72.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 73.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 74.Nomi T, Sho M, Akahori T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 75.Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. 2013;1291:1–13. doi: 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]