

Abstract
The efficient production of biofuels from cellulosic feedstocks will require the efficient fermentation of the sugars in hydrolyzed plant material. Unfortunately, plant hydrolysates also contain many compounds that inhibit microbial growth and fermentation. We used DNA-barcoded mutant libraries to identify genes that are important for hydrolysate tolerance in both Zymomonas mobilis (44 genes) and Saccharomyces cerevisiae (99 genes). Overexpression of a Z. mobilis tolerance gene of unknown function (ZMO1875) improved its specific ethanol productivity 2.4-fold in the presence of miscanthus hydrolysate. However, a mixture of 37 hydrolysate-derived inhibitors was not sufficient to explain the fitness profile of plant hydrolysate. To deconstruct the fitness profile of hydrolysate, we profiled the 37 inhibitors against a library of Z. mobilis mutants and we modeled fitness in hydrolysate as a mixture of fitness in its components. By examining outliers in this model, we identified methylglyoxal as a previously unknown component of hydrolysate. Our work provides a general strategy to dissect how microbes respond to a complex chemical stress and should enable further engineering of hydrolysate tolerance.
Keywords: biofuels, chemogenomics, plant hydrolysate, systems biology, tolerance
Introduction
Concerns over energy security, global warming, and rising petroleum prices have led to a renewed interest in the development of technologies for cost-effective production of ethanol or other biofuels from renewable resources (Hill et al, 2006; US Department of Energy, 2011). Lignocellulosic biomass, such as wood and grasses, can provide a sufficient quantity of feedstock material that can be converted into biofuels, and a variety of energy crops are currently being considered for use in the United States, such as Miscanthus giganteus (miscanthus) and Panicum virgatum (switchgrass) (Somerville et al, 2010; Youngs and Somerville, 2012). The grand challenge is to produce large quantities of a commodity chemical given economic constraints. In the case of cellulosic ethanol, technoeconomic analysis has been used to determine which steps of the production process, if optimized, can have the greatest impact on the minimum selling price (Klein-Marcuschamer et al, 2010, 2011; Kumar and Murthy, 2011; Tao et al, 2011; Vicari et al, 2012). Two critical steps are the conversion of biomass into fermentable hexose and pentose sugars (mainly glucose and xylose) and their subsequent fermentation into ethanol.
Conversion of biomass into sugars is typically a two-step process involving pretreatment and enzymatic hydrolysis. Although many variations exist, we have focused on the dilute-acid hydrolysis method, which is considered as a viable option for commercial-scale cellulosic ethanol production (Wyman et al, 2005). The high temperatures and pressures typically used for dilute-acid pretreatment result in the co-production of inhibitory compounds (derived from sugar and lignin degradation) in addition to fermentable sugars. This resulting mixture, or plant hydrolysate, contains at least 60 inhibitory compounds (Clark and Mackie, 1984; Palmqvist and Hahn-Hägerdal, 2000a, 2000b). These inhibitors have a significant impact on growth and fermentation of bacteria and yeast, thus preventing efficient biofuel production. There are three common approaches to deal with fermentation inhibitors: prevent their formation by reducing the severity of pretreatment, remove inhibitors by detoxification, or improve the tolerance of the host organism (Palmqvist and Hahn-Hägerdal, 2000a; Nilvebrant et al, 2001; Martín et al, 2007a; Parawira and Tekere, 2011; Stoutenburg et al, 2011). All three methods can be successful and are expected to have a significant economic impact (Klein-Marcuschamer et al, 2010; Tao et al, 2011), but here we will focus on improving tolerance.
There are two complementary approaches to develop more tolerant strains: laboratory evolution in the presence of plant hydrolysates and rational strain engineering (Larsson et al, 2001; Heer and Sauer, 2008; Chen, 2010; Yang et al, 2010a, 2010b; Liu, 2011; Agrawal et al, 2012; Fujitomi et al, 2012). Regardless of the approach, the resulting strains are often not fully tolerant, and further improvements in ethanol yield and productivity are needed (Keller et al, 1998; Martín et al, 2007b; Heer and Sauer, 2008). Many of these studies have focused on single inhibitory compounds present in hydrolysate or simple model mixtures as a proxy for plant hydrolysates (Palmqvist et al, 1999; Liu, 2011). Given the complexity of inhibitors in actual hydrolysate, it is challenging to predict and engineer the necessary genetic changes for tolerance based on these previous studies.
Given this challenge, we present here an experimental and computational approach to dissect the effects of complex chemical mixtures, such as plant hydrolysates, on the growth and fermentation of the bacterium Zymomonas mobilis and the yeast Saccharomyces cerevisiae. Both microbes are being considered for commercial-scale cellulosic ethanol production. Z. mobilis is currently being used as part of a DuPont industrial-scale cellulosic ethanol process ( http://www.ddce.com/), and S. cerevisiae has a long history of industrial-scale ethanol production from corn in the United States and from sugarcane in Brazil (Wheals et al, 1999).
First, we used a functional genomics approach, based on chemogenomic profiling of mutant libraries in Z. mobilis and S. cerevisiae, to identify genes that are important for growth in plant hydrolysates. Chemogenomic profiling with DNA barcodes was first pioneered in S. cerevisiae and we have recently adapted the technology for use in bacteria (Giaever et al, 2002, 2004; Oh et al, 2010; Deutschbauer et al, 2011). These previous studies, and other technologies for profiling large mutant pools, have led to key insights into the function of unknown genes and the mechanism of action of inhibitory compounds (Sassetti et al, 2001; Giaever et al, 2004; Langridge et al, 2009; Smith et al, 2009; Van Opijnen et al, 2009; Hillenmeyer et al, 2010; Deutschbauer et al, 2011). The putative hydrolysate tolerance genes that we identified are attractive targets for strain improvement programs, and we demonstrate here, by systematic overexpression of Z. mobilis tolerance genes, that we can rationally engineer improved fermentation in miscanthus hydrolysate. Most of our fitness experiments were carried out under aerobic growth conditions because this was most compatible with our high-throughput protocols. However, we did perform some experiments under anaerobic growth conditions to better match the environment of an industrial bioreactor, and identified seven additional Z. mobilis tolerance genes (see Discussion).
Second, we deconstructed the complex biological response to plant hydrolysates by obtaining individual chemogenomic profiles for each of 37 hydrolysate-derived inhibitors and in synthetic mixtures of known components. We modeled fitness in hydrolysate as a mixture of fitness in individual components and identified outliers in our model that led to the discovery of a previously unknown chemical component, methylglyoxal, which is present in our miscanthus plant hydrolysate and contributes to overall toxicity. In sum, our combined experimental and computational approach provides a general strategy for understanding how microbes respond to a complex chemical stress, for identifying critical unknown chemical components in plant hydrolysates, and for rationally engineering strains with improved hydrolysate tolerance and fermentation properties.
Results
Plant hydrolysate composition and effects on Z. mobilis growth and fermentation
To explore the natural diversity of hydrolysate composition and to determine the effect of feedstock-derived inhibitors on the growth and fermentation of Z. mobilis, we developed a microwave oven-based protocol to hydrolyze M. giganteus (miscanthus) and P. virgatum (switchgrass) plant material at high temperature and pressure. The procedure was designed to mimic, at the laboratory scale, a dilute-acid hydrolysis pretreatment method that can be used for the industrial-scale production of cellulosic biofuels (Tao et al, 2011). We prepared six hydrolysate samples from miscanthus or switchgrass grown at different field sites in Illinois and two additional hydrolysate samples from a mixture of miscanthus, from multiple field sites. All eight hydrolysate samples were analyzed using a combination of GC/MS and LC-RID/DAD; the composition of these mixtures, including 4 sugars (glucose, xylose, arabinose, and cellobiose) and 37 potential inhibitors, is shown in Supplementary Table 1. As expected, the harsh hydrolysis conditions (low pH and high temperature) resulted in the production of sugar dehydration products, such as furfural and 5-hydroxymethylfurfural (5-HMF) and their degradation products, formic acid and levulinic acid, respectively. In addition, we detected a wide variety of phenolic compounds derived from lignin degradation (Palmqvist and Hahn-Hägerdal, 2000b; Klinke et al, 2004). Based on clustering of the inhibitor and sugar concentrations, we identified three groups of samples with distinct chemical compositions: miscanthus, switchgrass, and the batch miscanthus samples (Supplementary Figure 1). Despite the different field locations, miscanthus hydrolysates were more similar to each other than switchgrass, and vice versa. By contrast, the two batch miscanthus samples (batch 1 andbatch 2) formed a third independent cluster, and this is likely explained by the higher temperature used for their processing (200°C versus 180°C) that led to higher concentrations of inhibitors (Supplementary Table 1).
To understand the inhibitory effect of hydrolysate, we first determined the concentration of hydrolysate that, when added to rich media, significantly inhibited the growth of Z. mobilis. Consistent with their similar, but not identical, chemical compositions, all eight hydrolysate samples inhibited growth of Z. mobilis at concentrations ranging from about 8 to 20% (v/v) (Supplementary Figure 2). The most potent hydrolysate samples were the batch miscanthus that was prepared at a higher temperature, consistent with their higher inhibitor concentrations (Supplementary Table 1). We also determined the effects of plant hydrolysate on the fermentation profile of Z. mobilis by carrying out small-scale aerobic batch fermentations in the absence (Figure 1A) or presence of 8% (v/v) batch 2 hydrolysate (Figure 1B). In the presence of hydrolysate, specific ethanol productivity was reduced about three-fold (0.10 versus 0.27 g/l/h/OD600). Taken together, our hydrolysate preparations provide a complex mixture of inhibitors that serve as a model for industrial-scale dilute-acid hydrolysates.
Figure 1.

Miscanthus hydrolysate inhibits Z. mobilis growth and ethanol production. Batch fermentation profiles for wild-type Z. mobilis strain carrying an empty control plasmid (WT+pJS71) either in (A) rich media (RM) or in (B) rich media supplemented with 8% (v/v) batch 2 miscanthus hydrolysate (HZ). Data shown are the average of four replicates and error bars indicate standard deviation.
Generating a genome-wide Z. mobilis barcoded transposon library
To understand the genetic basis of hydrolysate tolerance, we first mapped 14 008 random DNA-barcoded transposon insertions in Z. mobilis ZM4 using protocols recently developed in our laboratory for Shewanella oneidensis MR-1 (Deutschbauer et al, 2011). From our collection of 14 008 mutants, we derived two Z. mobilis mutant pools of 3716 barcoded strains each that together represent 1620 of the 1892 (86%) annotated protein-coding genes (Materials and methods; Supplementary Table 2). We designed two mutant pools because it provided optimal genome-wide coverage given a limited set of barcodes (about 4200), and it allowed us to include some strains in both pools and measure them twice, which provided an internal control for experimental consistency. These mutant pools were used to perform competitive growth assays, or pooled fitness experiments, where the relative abundance of strains in our two pools was quantified (Giaever et al, 2002; Pierce et al, 2006; Oh et al, 2010). Before all experiments, we recovered the mutant pools from frozen stocks and used these cells as the starting inoculum for setting up fitness experiments in experimental conditions. By comparing strain abundance after growth in the experimental condition (END) to strain abundance in the starting inoculum (START), we calculated a log2 ratio, or strain fitness value for each mutant strain in that condition. We define ‘gene fitness’ as the average strain fitness value for insertions within that gene (see Materials and methods for details). Negative gene fitness values indicate that a gene is important for growth in the condition of interest, that is, transposon mutants of that gene should grow poorly in that condition. In contrast, positive gene fitness values indicate that the gene is detrimental to growth in the condition of interest, that is, transposon mutants of that gene have improved growth relative to the typical strain. After final data filtering, we obtained gene fitness data for 1578 of 1892 (83%) protein-coding genes. Because many genes had multiple transposon insertions (1336/1578 had >1 insertion), and because the same strain was sometimes present in both pools, we were able to make an average of 3.5 fitness measurements per gene.
We were surprised to find transposon insertions within the central 5–80% of most genes regardless of whether they were expected to be essential. In fact, we mapped insertions in 82% of predicted essential genes (Supplementary Table 3), which is not significantly less than the rate of 85% for other genes (P=0.10, Fisher’s exact test). To our knowledge, this is the first example of a large-scale transposon mutagenesis study in bacteria with this unusual distribution of insertion sites. To study this further, we first examined six mutants by PCR using primers that flanked each transposon site. Three of these mutants were in predicted essential genes (leuS::TN5, ftsZ::TN5, and rpoB::TN5), and three mutants were in non-essential genes (ZMO0759::TN5, ZMO1490::TN5, and ZMO1723::TN5). In the predicted essential gene mutants, we amplified two bands that correspond to the wild-type and mutant copies of leuS, ftsZ, and rpoB, respectively (Supplementary Figure 4A and B). By contrast, transposon insertions in non-essential genes only had a single band by PCR analysis, corresponding to the mutant copy. Mutants with two bands by PCR were classified as ‘mixed’ and were examined further by using a transposon stability assay and comparative genome hybridization (Materials and methods; Supplementary Figure 4C–L). Based on these experiments, we concluded that Z. mobilis is polyploid (i.e., has multiple copies of its main 2 Mbp chromosome), and that insertions in essential genes are heterozygous and unstable in the absence of kanamycin selection, whereas insertions in non-essential genes are homozygous and stable (Supplementary Note 1; Supplementary Figure 5). Alternatively, polyploidy might be a rare event that is selected for only when an essential gene is mutated; however, because the rate of insertion in essential genes is similar to non-essential genes, we believe that polyploidy is the normal state for Z. mobilis. In this study, we focused our single-mutant follow-up experiments on stable, homozygous KanR mutants. Although unusual, other bacteria, such as Deinococcus radiodurans, Thermus thermophilus, Synechococcus spp., Sinorhizobium meliloti bacteroids, and Epulopiscium sp. type B are known to be polyploid and to have multiple copies of their chromosome (Masters et al, 1991; Mergaert et al, 2006; Ohtani et al, 2010; Griese et al, 2011; Angert, 2012).
We validated our pooled fitness assay by growing the mutant pools in minimal media where we had a strong prediction of which genes should have fitness defects. Most of the 53 annotated amino-acid synthesis genes had strong fitness defects in minimal media and were rescued by the addition of casamino acids (CAA) (Supplementary Figure 6A–C; Supplementary Table 4). We were unable to rescue mutants of genes required for tryptophan biosynthesis (aroABCDE and trpDE), whichcan be explained because CAA does not contain tryptophan; it is lost during the preparation of CAA, which involves acid hydrolysis of casein. As a second test of auxotroph rescue, we grew the Z. mobilis transposon pools in the presence of methionine, and this specifically rescued the fitness defect ofsix genes (metCEFWXZ) all predicted to be required for methionine biosynthesis (Supplementary Figure 6C). We then carried out single-mutant follow-up studies on a metC::TN5 mutant to confirm that fitness defects in the pooled assay can be recapitulated at the single strain level (Supplementary Figure 6D). As expected, growth of metC::TN5 on minimal media could be rescued by the addition of methionine.
In addition to our biological tests, we calculated two metrics (strain and operon correlation), as previously described (Deutschbauer et al, 2011) that were used to measure overall experiment quality and to flag potential experimental errors (Supplementary Figure 6E–G). In the typical experiment, the correlation of the 1057 identical strains contained in both pools was 0.89 and the correlation of fitness between adjacent genes in the same operon was 0.50. Experiments with poor quality metrics (operon correlation <0.4 or strain correlation <0.75) were repeated, and kept if reproducible across multiple biological replicates. In sum, our validation confirms that our Z. mobilis pooled fitness assay provides accurate and biologically meaningful results that could be used to study hydrolysate tolerance.
Genome-wide fitness profiling of Z. mobilis mutants in plant hydrolysates and 37 chemical components
Using high-throughput culturing methods we recently developed for Shewanella oneidensis MR-1 (Deutschbauer et al, 2011), we performed 189 whole-genome mutant fitness experiments with our two Z. mobilis pools. These 189 experiments represent 58 unique experimental conditions including miscanthus and switchgrass hydrolysates, 2 types of synthetic hydrolysate mixtures (SYN-37 and SYN-10), 37 individual compounds that are present in miscanthus hydrolysate, and 11 other stress conditions (Supplementary Table 5). Most fitness experiments were performed in rich media without inhibitors and in rich media supplemented with hydrolysate or specific compounds of interest. We tested each potential growth inhibitor at several different concentrations to identify the suitable concentration for mutant fitness experiments. In general, we selected the concentration that caused about a two-fold increase in doubling time and gave a fitness pattern that was reproducibly different from the baseline rich media condition (see Materials and methods). An overview of our fitness data set is shown as a clustered heat map of average gene fitness values (Figure 2). Gene fitness values were averaged across replicate conditions, such as rich media (24 replicates) or identical concentrations of the same inhibitor. We also averaged the gene fitness values for 37 hydrolysate experiments (Supplementary Figure 7) that included both miscanthus and switchgrass samples (from single field sites or batch material). The complete, non-averaged data set can be found in Supplementary Dataset 1 and online ( http://genomics.lbl.gov/supplemental/zm4hydrolysate/).
Figure 2.

Genome-wide fitness profiling of Z. mobilis in 58 experimental conditions, including plant hydrolysate and 37 individual components of hydrolysate. Average gene fitness data are represented as a two-dimensional heat map for 1586 genes (X axis) and 58 experimental conditions (Y axis). For each transposon mutant in our pool, strain fitness is calculated as log2 ratio of (END/START). Gene fitness values are the average of per-strain fitness across all insertions within that gene and are displayed according to the color bar at the top right of the heat map. In addition, gene fitness values have also been averaged across replicate experimental conditions. Chemicals with similar structures cluster together on the Y axis (labeled 1–10) and differ by a single functional group, as colored by the key at the top left. For example, compounds 2,5-dihydroxybenzoic acid and 3-hydroxybenzoic acid (cluster 1) differ by a single hydroxyl group (see Supplementary Figure 8 for more examples). The fitness data were clustered in both dimensions by hierarchical agglomerative clustering with complete linkage. Euclidean distance was used as the distance metric for genes and Pearson’s correlation was used as the similarity metric for experimental conditions. Hydrolysate components are indicated by red text.
Two-dimensional hierarchical clustering of the averaged gene fitness data revealed two broad categories. One large group represents genes that when mutated have little or no fitness defect in the 58 conditions we tested (left half, Figure 2). Additional experimental conditions might uncover phenotypes for these mutants. The second large group had fitness defects in nearly all 58 conditions, including rich media (right half, Figure 2). With a fitness value of −1 or less, we identified 402 genes that were important for growth in rich media and 1184 that are not. Of these 402 genes, 185 (46%) are expected to be essential (see Materials and methods), while just 5% of the other 1184 genes are expected essentials (P<10−15, Fisher’s exact test). Because this large cluster of genes had negative fitness values in our baseline condition (rich media), and many are likely to be essential, they were not pursued further in this study. In addition, we found that mutants in expected essential genes were more likely to have a low abundance in our starting pool (Supplementary Figure 3B), making it more difficult to perform accurate fitness measurements on these strains.
Clustering the fitness data by condition revealed a few large groups of similar conditions, such as organic acids (Y -axis, near bottom, Figure 2). In many cases, these broad groups included chemicals of different classes, and it was not clear why they formed a cluster. However, within these broad groups, we found that chemicals with highly similar structures clustered closely (Clusters 1–10 on Y axis, Figure 2). In 10 cases, these closely related chemicals differed by a single functional group, such as a single hydroxyl, ester, methyl, or C=C group (Supplementary Figure 8). For example, cluster 1 on the Y axis contains two chemicals (2,5-dihydroxybenzoic acid and 3-hydroxybenzoic acid) that differ by a single hydroxyl group. In sum, chemicals with closely related structures had very similar fitness profiles. Our results are consistent with previous chemogenomic studies and likely reflect the underlying similarity in mechanism of action for structurally related inhibitory compounds (Hillenmeyer et al, 2010).
Identification of 44 putative hydrolysate tolerance genes in Z. mobilis
To identify genes that are important for growth in hydrolysate, we searched for mutants that showed a significant difference in fitness between rich media supplemented with hydrolysate and plain rich media. Based on this criterion (fitnesshydrolysate <−1 and fitnesshydrolysate<fitnessrich −1), we identified 44 putative hydrolysate tolerance genes that were further grouped into 7 functional categories (Figure 3A; Table I; Supplementary Dataset 2). In contrast, using a second criterion (fitnesshydrolysate>0.5 and fitnesshydrolysate>fitnessrich+0.75), we found only one gene, ZMO1496, that was detrimental for growth in hydrolysate, which encodes phosphoenolpyruvate (PEP) carboxylase (fitness in hydrolysate=0.72 versus rich media=−0.48). Because signal intensity on microarrays can saturate, we believe that our pooled fitness assays have a reduced sensitivity for mutants with positive fitness, which may explain why we only identified one mutant in the positive fitness category. In this study, we only pursued the negative fitness mutants.
Figure 3.

Identification of 44 Z. mobilis genes that are important for growth and 1 gene that is detrimental for growth in plant hydrolysate. (A) Scatter plot of gene fitness values in rich media (average of 24 experiments) versus gene fitness in hydrolysate (average of 37 experiments) for 1586 Z. mobilis genes. A dashed grey line indicates X=Y and dashed black lines indicate the cutoffs used to select tolerance genes. Putative tolerance genes have a more negative fitness value in hydrolysate than in rich media and are indicated by colored symbols. They are further classified based on their predicted function, as indicated in the legend. A single gene (ZMO1496), indicated by a black circle, was found to be detrimental for growth in plant hydrolysate. (B) Subset of average gene fitness data from Figure 2, showing only the fitness data for the 44 tolerance genes. The 37 hydrolysate components are indicated by red text. Arrows indicate the baseline condition without any added inhibitors (rich media), or rich media supplemented with DMSO (DMSO) or plant hydrolysate (hydrolysate). Gene fitness values are colored according to the color bar at the top right of the heat map. Each tolerance gene on the X axis is labeled by its systematic gene name (ZMOxxxx) and the clustering is colored based on predicted functional classes, as in (A). Tolerance genes were clustered by Euclidean distance, and conditions were clustered as in Figure 2.
Table 1. Table of 44 Z. mobilis hydrolysate tolerance genes identified in this study.
| Gene | Class | Annotation |
|---|---|---|
| ZMO0003 | auxo | Adenylylsulfate kinase |
| ZMO0007 | auxo | Phosphoadenosine phosphosulfate reductase |
| ZMO0008 | auxo | Sulfite reductase subunit beta |
| ZMO0009 | auxo | Sulfite reductase subunit alpha |
| ZMO0100 | reg | Transcriptional regulator, HxlR family |
| ZMO0101 | mem | NAD-dependent epimerase/dehydratase |
| ZMO0192 | mem | Peptidase M48 Ste24p |
| ZMO0200 | auxo | Anthranilate phosphoribosyltransferase |
| ZMO0201 | auxo | Glutamine amidotransferase of anthranilate synthase |
| ZMO0418 | other | Hypothetical protein |
| ZMO0429 | other | Iron-sulfur cluster assembly accessory protein |
| ZMO0468 | auxo | Anthranilate synthase component I |
| ZMO0472 | other | rpsU-divergently transcribed protein |
| ZMO0483 | auxo | Homoserine dehydrogenase |
| ZMO0675 | other | Transcriptional regulator, LysR family |
| ZMO0759 | glt | Hydroxyacylglutathione hydrolase |
| ZMO0760 | glt | Glyoxalase/bleomycin resistance protein/dioxygenase |
| ZMO0763 | other | Tetratricopeptide domain protein |
| ZMO0774 | reg | Transcriptional regulator, LysR family |
| ZMO0846 | mem | Sodium/hydrogen exchanger |
| ZMO0975 | mem | Conserved hypothetical membrane spanning protein |
| ZMO1067 | other | Fe-S metabolism-associated SufE |
| ZMO1136 | cytc | Cytochrome c peroxidase |
| ZMO1221 | other | Hypothetical protein |
| ZMO1252 | cytc | Cytochrome c biogenesis factor-like protein |
| ZMO1253 | cytc | Cytochrome c biogenesis protein |
| ZMO1255 | cytc | Cytochrome c assembly protein |
| ZMO1389 | cytc | Cytochrome c biogenesis protein transmembrane region |
| ZMO1391 | mem | Diacylglycerol kinase catalytic region |
| ZMO1404 | reg | RNA polymerase, sigma-24 subunit, ECF subfamily |
| ZMO1429 | eff | RND efflux system, outer membrane lipoprotein, NodT family |
| ZMO1430 | eff | Efflux transporter, RND family, MFP subunit |
| ZMO1431 | eff | Hypothetical protein |
| ZMO1432 | eff | Fusaric acid resistance protein conserved region |
| ZMO1490 | mem | Hypothetical protein |
| ZMO1520 | other | Hypothetical protein |
| ZMO1541 | other | Ferrous iron transport protein B |
| ZMO1590 | mem | ABC transporter related protein |
| ZMO1659 | other | ATP-dependent metalloprotease FtsH |
| ZMO1715 | mem | Biopolymer transport protein ExbD/TolR |
| ZMO1722 | glt | S-(hydroxymethyl)glutathione dehydrogenase/class III alcohol dehydrogenase |
| ZMO1723 | glt | Hypothetical protein |
| ZMO1874 | glt | BolA family protein |
| ZMO1875 | glt | Hypothetical protein |
Systematic gene names, their functional class, and annotation are listed. Functional classes are auxo (auxotroph), cytc (cytochrome c), eff (efflux), glt (glutathione related), mem (membrane), other (other), and reg (regulator). Annotations were obtained from RefSeq ( http://www.ncbi.nlm.nih.gov/RefSeq/).
The 44 putative tolerance genes included 8 auxotrophs, 5 genes involved in cytochrome c biogenesis or cytochrome c-containing proteins, 4 efflux pump-related genes, 6glutathione-related genes, 8 genes related to membranes or the cell wall, 3 regulators, and 10 other genes (Table I). The variety of predicted gene functions suggests that the cellular response to growth in hydrolysate is complex and that hydrolysate tolerance is a multigenic trait. This is consistent with the fact that plant hydrolysates are complex mixtures containing a variety of chemical classes (e.g., weak organic acids, furans, aldehydes, and phenolics) that likely affect cell physiology by different mechanisms of action.
To confirm the pooled fitness results, 25 transposon mutants were selected from our set of 44 tolerance genes for detailed follow-up studies (Supplementary Table 6). This group of strains represents examples from each of the seven functional classes we identified. Each mutant was re-streaked to a single colony isolate and their transposon insertion sites were verified by PCR and DNA sequencing. We then used our transposon stability assay to check whether the insertion was stable or mixed. Nine of the twenty-five mutants had mixed transposon insertions and were not studied further. Four of these nine mixed mutants were in a single operon encoding an efflux pump (ZMO1429-ZMO1432). Of the remaining 16 stable mutants, we confirmed negative fitness defects for 13 of them, when grown in batch 1 or batch 2 miscanthus hydrolysate (e.g., see Supplementary Figure 9). In addition, we complemented three mutants (ZMO0100::TN5, ZMO1722::TN5, and ZMO0759::TN5) by expression of the corresponding wild-type gene on a plasmid, demonstrating that the observed phenotype is due to a single gene defect (Supplementary Figure 10). In sum, these data demonstrate that our pooled fitness assay can be used to identify bona fide hydrolysate tolerance genes that are critical for growth in plant hydrolysate.
To further understand the specific function of each putative tolerance gene, we examined the fitness pattern for these 44 genes in each of the 37 components (colored red) known to be present in miscanthus and switchgrass hydrolysates (Figure 3B). Broadly, most of the tolerance genes had fitness defects in many of the individual hydrolysate components, which makes it difficult to infer a specific detoxification function for any particular tolerance gene. Some of the tolerance genes might respond to or detoxify a class of compounds, such as aldehydes, so this could explain the lack of a one-to-one relationship between gene fitness and hydrolysate component. While more detailed follow-up studies will be required to determine the specific biochemical functions of these tolerance genes, we find that hydrolysate tolerance genes with related functions cluster together on our fitness data heatmap (X axis, Figure 3B). For example, one cluster contains four auxotrophs that are part of the sulfate assimilation pathway (cysCHIJ encoded by ZMO0003, ZMO0007, ZMO0008, and ZMO0009), and a second cluster contains cytochrome c peroxidase (CCP) (ZMO1136) and two genes that are involved in cytochrome c biogenesis (ZMO1389 and ZMO1252). Many of these fitness clusters contain genes that are predicted to form operons, which are also consistent with their shared function (ZMO0100-ZMO0101, ZMO0007-ZMO0009, ZMO1874-ZMO1875, ZMO0200-ZMO0201, ZMO1429-ZMO1432).
Five hydrolysate tolerance genes (ZMO0760, ZMO0100, ZMO0101, ZMO1722, and ZMO1723) did not exhibit a fitness defect in any of the 37 inhibitors we tested. These hydrolysate-specific mutants might be affected by some unknown component of hydrolysate or only by a combination of inhibitors. To test the latter possibility, we made two synthetic hydrolysate mixtures based on the composition of miscanthus batch 1; one contained the 10 most abundant inhibitors (SYN-10; furfural, acetic acid, formic acid, levulinic acid, succinic acid, 5-HMF, 2-furoic acid, vanillin, vanillic acid, and syringaldehyde), and the second contained the full set of 37 inhibitors and four sugars: glucose, xylose, arabinose, and cellobiose (SYN-37). The composition of SYN-10 and SYN-37 was verified using GC/MS and LC-RID and closely matched the values for batch 1 hydrolysate (Supplementary Table 1).
Fitness profiling of synthetic hydrolysate mixtures in Z. mobilis and S. cerevisiae
We first examined the effect of SYN-10 and SYN-37 on the growth of Z. mobilis and found that both mixtures were less potent than the batch 1 and batch 2 hydrolysates and inhibited growth in a similar manner (Supplementary Figure 11A, P<10−5, analysis of variance (ANOVA)). This suggests that our synthetic mixtures are missing critical inhibitory components. To further understand this difference, we performed pooled fitness assays in the presence of SYN-10 or SYN-37 (Supplementary Figure 12A; Figure 4A). The fitness profiles of SYN-10 and SYN-37 were very similar (R2=0.807, Supplementary Figure 12B), consistent with their similar growth effects on Z. mobilis. However, a plot of average fitness in SYN-37 versus average fitness in hydrolysate shows that nine genes are outliers (fitnesshydrolysate <−1 and fitnessSYN-37>−1/3, enclosed by dashed black lines in Figure 4A). Of the 9 genes, 5 are important for growth in hydrolysate but not in any of the 37 components (ZMO0760, ZMO1722, ZMO1723, ZMO0100, and ZMO0101, all fitnesscomponents>−1, leftmost cluster in Figure 3B). In addition, a heatmap of the 44 tolerance genes shows that SYN-10 and SYN-37 are more alike each other than hydrolysate (Figure 4B). In sum, our synthetic mixtures do not fully recapitulate the growth and fitness effects of real hydrolysate and the presence of outliers indicates that there are unidentified hydrolysate components that contribute to its overall toxicity.
Figure 4.

Synthetic hydrolysate mixtures do not fully explain the fitness profile of real hydrolysate. Two synthetic hydrolysate mixtures containing either 37 components (SYN-37) or the 10 most abundant components (SYN-10) were made based on the composition of miscanthus batch 1 (Supplementary Table 1). Data for SYN-10 are shown in Supplementary Figure 12. (A) Scatterplot of Z. mobilis gene fitness data in SYN-37 (average of 4 experiments) versus in hydrolysate (average of 37 experiments). The 44 Z. mobilis tolerance genes are color coded by category. Nine outlier genes (defined by two dashed black lines) have more negative gene fitness values in hydrolysate than in SYN-37, and are listed in a black box and color coded by category. (B) Heatmap of gene fitness data for the 44 Z. mobilis tolerance genes. Genes were clustered by Euclidean distance with complete linkage using all non-averaged fitness data. Fitness values are colored according to the color bar. The baseline conditions are rich media (ZRMG) and rich media supplemented with DMSO (DMSO). (C) Scatterplot of S. cerevisiae gene fitness data in SYN-37 (average of 6 experiments) versus in batch 1 miscanthus hydrolysate (average of 6 experiments). In all, 99 putative tolerance genes are color coded according to their function as indicated on the graph legend. Twenty of these genes are outliers (defined by two dashed black lines) and have more negative fitness values in hydrolysate than in SYN-37. Outlier genes are listed in a black box and color coded according to the legend. (D) Heatmap of gene fitness data for the 99 S. cerevisiae tolerance genes. Genes were clustered as in (B). Fitness values are colored according to the color bar. Two baseline conditions are shown: YPD is the rich media used for S. cerevisiae growth (n=3) and ZRMG is the rich media used for Z. mobilis growth that was also used to prepare the SYN-10 and SYN-37 synthetic hydrolysate mixtures (n=2, see Materials and methods).
To determine whether synthetic mixtures can recapitulate the fitness profile of plant hydrolysates in other organisms, we chose Saccharomyces cerevisiae, for which a genome-wide collection of DNA-barcoded deletion strains is available (Giaever et al, 2002). First, we profiled the S. cerevisiae homozygous deletion library (as a pool) in the presence of batch 1 hydrolysate. To identify the putative tolerance genes in S. cerevisiae, we searched for mutants with a significant fitness defect in rich media (YPD) supplemented with hydrolysate, but not in the rich media baseline condition. Using the same selection criterion as for Z. mobilis (fitnesshydrolysate <−1 and fitnesshydrolysate<fitnessrich −1), we identified 99 yeast hydrolysate tolerance genes. As in Z. mobilis, the S. cerevisiae tolerance genes represent a variety of functional categories and pathways, including 12 regulatory genes, 12 amino-acid biosynthesis genes, 4 pentose phosphate pathway genes, 15 membrane/secretion-related genes, and 5 oxidant-induced cell-cycle arrest (OCA) genes (Table II; Supplementary Figure 13; Supplementary Dataset 3). The overlap withthe Z. mobilis tolerance genes was just two genes involved in amino-acid biosynthesis (see Discussion).
Table 2. Table of 99 S. cerevisiae hydrolysate tolerance genes identified in this study.
| Class | ORF | Gene |
|---|---|---|
| AA biosynthesis | YDR127W | ARO1 |
| AA biosynthesis | YGL148W | ARO2 |
| AA biosynthesis | YPR060C | ARO7 |
| AA biosynthesis | YAL012W | CYS3 |
| AA biosynthesis | YJL101C | GSH1 |
| AA biosynthesis | YDR158W | HOM2 |
| AA biosynthesis | YER052C | HOM3 |
| AA biosynthesis | YJR139C | HOM6 |
| AA biosynthesis | YER086W | ILV1 |
| AA biosynthesis | YCR053W | THR4 |
| AA biosynthesis | YKL211C | TRP3 |
| AA biosynthesis | YGL026C | TRP5 |
| Membrane | YBR036C | CSG2 |
| Membrane | YLR436C | ECM30 |
| Membrane | YMR202W | ERG2 |
| Membrane | YGL012W | ERG4 |
| Membrane | YMR015C | ERG5 |
| Membrane | YPL057C | SUR1 |
| Membrane | YBR058C-A | TSC3 |
| OCA | YNL099C | OCA1 |
| OCA | YNL056W | OCA2 |
| OCA | YCR095C | OCA4 |
| OCA | YHL029C | OCA5 |
| OCA | YDR067C | OCA6 |
| Other | YLR338W | OPI9 |
| Other | YDR136C | VPS61 |
| Other | YLR261C | VPS63 |
| Other | YNL148C | ALF1 |
| Other | YML035C | AMD1 |
| Other | YGL105W | ARC1 |
| Other | YHR013C | ARD1 |
| Other | YBR068C | BAP2 |
| Other | YPL161C | BEM4 |
| Other | YFL023W | BUD27 |
| Other | YFR036W | CDC26 |
| Other | YPL241C | CIN2 |
| Other | YGR092W | DBF2 |
| Other | YFL001W | DEG1 |
| Other | YMR126C | DLT1 |
| Other | YDL172C | Dubious |
| Other | YJL120W | Dubious |
| Other | YDR048C | Dubious |
| Other | YML122C | Dubious |
| Other | YDR008C | Dubious |
| Other | YDL050C | Dubious |
| Other | YDR125C | ECM18 |
| Other | YGL168W | HUR1 |
| Other | YJL082W | IML2 |
| Other | YOL081W | IRA2 |
| Other | YOR322C | LDB19 |
| Other | YDL051W | LHP1 |
| Other | YDR378C | LSM6 |
| Other | YNL147W | LSM7 |
| Other | YBR255W | MTC4 |
| Other | YDL040C | NAT1 |
| Other | YOR209C | NPT1 |
| Other | YDR360W | OPI7 |
| Other | YDL173W | PAR32 |
| Other | YHR071W | PCL5 |
| Other | YGR240C | PFK1 |
| Other | YGR233C | PHO81 |
| Other | YPR153W | Putative protein |
| Other | YDR529C | QCR7 |
| Other | YMR283C | RIT1 |
| Other | YJR063W | RPA12 |
| Other | YOL138C | RTC1 |
| Other | YGL100W | SEH1 |
| Other | YOR035C | SHE4 |
| Other | YNL032W | SIW14 |
| Other | YCR033W | SNT1 |
| Other | YMR304W | UBP15 |
| Other | YKR020W | VPS51 |
| Other | YNL246W | VPS75 |
| Other | YML097C | VPS9 |
| Other | YNL107W | YAF9 |
| PPP | YJL121C | RPE1 |
| PPP | YLR354C | TAL1 |
| PPP | YPR074C | TKL1 |
| PPP | YNL241C | ZWF1 |
| Regulatory | YPL202C | AFT2 |
| Regulatory | YDR173C | ARG82 |
| Regulatory | YIR023W | DAL81 |
| Regulatory | YOL051W | GAL11 |
| Regulatory | YGR163W | GTR2 |
| Regulatory | YOL012C | HTZ1 |
| Regulatory | YDR162C | NBP2 |
| Regulatory | YDR195W | REF2 |
| Regulatory | YDR028C | REG1 |
| Regulatory | YDL020C | RPN4 |
| Regulatory | YHR178W | STB5 |
| Regulatory | YML007W | YAP1 |
| Secretion | YGL054C | ERV14 |
| Secretion | YML121W | GTR1 |
| Secretion | YOR070C | GYP1 |
| Secretion | YDR137W | RGP1 |
| Secretion | YLR039C | RIC1 |
| Secretion | YMR183C | SSO2 |
| Secretion | YOL018C | TLG2 |
| Secretion | YLR262C | YPT6 |
Functional class (Class), systematic gene name (ORF), and gene name (Gene) are listed. The six broad functional classes are AA biosynthesis (amino-acid biosynthesis), membrane, secretion, OCA (Oxidant-induced Cell-cycle Arrest), PPP (pentose phosphate pathway), and regulatory. These classes are based on GO annotations obtained from the Saccharomyces Genome Database ( http://www.yeastgenome.org). See Supplementary Dataset 2 for detailed annotations.
We then examined the effects of SYN-10 and SYN-37 on S. cerevisiae growth. In contrast to Z. mobilis, we found the primary effect of either synthetic or genuine hydrolysates was an increase in the length of lag phase rather than a reduction in growth rate. SYN-10 and SYN-37 increased the length of lag phase less than batch 1 or batch 2 hydrolysate did (Supplementary Figure 11B; P<10−15, ANOVA). To examine this difference in detail, we profiled the S. cerevisiae mutant pool in SYN-10 and SYN-37 (Figure 4C; Supplementary Figure 12C). The fitness profiles of SYN-10 and SYN-37 are highly similar (Figure 4D; Supplementary Figure 12D, R2=0.860) consistent with their similar effects on lag phase length. Similarly to Z. mobilis, a plot of average fitness in SYN-37 versus average fitness in batch 1 hydrolysate uncovered 20 outliers among our set of 99 tolerance genes (fitnesshydrolysate <−1 and fitnessSYN-37>−1/3) that have significant fitness defects in real hydrolysate but not in SYN-37 (listed on plot in Figure 4C).
Taken together, our results demonstrate that in both bacteria and yeast, synthetic hydrolysate mixtures of up to 37 compounds do not fully recapitulate the fitness effects of real hydrolysate, strongly suggesting that we are missing critical inhibitors from our synthetic mixtures. Consistent with our fitness data, there are many unidentified peaks in our hydrolysate GC/MS chromatograms. Prioritizing which peaks to study further using analytical chemistry is difficult without additional knowledge regarding the potential contribution of each peak to overall hydrolysate toxicity. To address this problem, we used chemogenomic profiling of our 37 known hydrolysate components and a computational model to search for key missing inhibitors.
Modeling Z. mobilis hydrolysate fitness and identification of methylglyoxal as a previously unknown hydrolysate component
Using the Z. mobilis fitness data for the 37 compounds, we first modeled average gene fitness in hydrolysate as a linear combination of its fitness in each component. Our model included fitness in the baseline condition (rich media) and in the 16 (of 37 tested) components that significantly improved the fit. The full linear model, including the list of components, can be found in Supplementary Table 7. Our model (Model-16) correlates well with fitness in hydrolysate (R2=0.880, Figure 5A). This is as good a fit as we obtained with experimental fitness data from SYN-37 (R2=0.810, Figure 4A). To test for possible synergistic effects of inhibitors, we also tested a model that included non-linear interactions. We identified three significant pairs (see Materials and methods): formic acid × levulinic acid (P<10−13), furfural × 4-hydroxyphenylacetic acid (P<10−15), and furfural × vanillin (P<10−5). Adding these terms to our linear model makes relatively little difference overall (adjusted R2 rises from 0.880 to 0.893). Unlike previous growth and fermentation studies that have documented synergistic inhibitor combinations (Zaldivar and Ingram, 1999; Zaldivar et al, 1999; Oliva et al, 2004), our modeling suggests that fitness effects of hydrolysate are primarily additive (see Discussion).
Figure 5.

Linear model of hydrolysate fitness based on fitness profiles of chemical components and identification of a new component, methylglyoxal. (A) Scatterplot of actual and predicted gene fitness in Z. mobilis. The model is a linear combination of fitness in rich media and in 16 inhibitors (Model-16). Arrows indicate three outlier genes, ZMO0759, ZMO0760, and ZMO0846. Four additional outlier genes, ZMO1429-ZMO1432, are enclosed by an ellipse. (B) Scatterplot of actual and predicted gene fitness in hydrolysate using a 17-component model that includes methylglyoxal fitness data (Model-17). (C) Scatterplot of actual and predicted gene fitness in hydrolysate using a 24-component model that includes methylglyoxal (MG) and 7 additional significant conditions (Model-24). (D) Plot of average gene fitness (n=4) for a10-component synthetic hydrolysate mixture (SYN-10) versus average gene fitness in hydrolysate (n=37). Arrows indicate two outlier genes, ZMO0759 and ZMO0760, which encode the GloAB detoxification system. Mutants in these genes are sensitive to hydrolysate but not to SYN-10. (E) In SYN-10 with methylglyoxal added (n=2), ZMO0759 and ZMO0760 are now important for fitness, which suggests that methylglyoxal stress contributes to hydrolysate toxicity. In all panels, the 44 Z. mobilis tolerance genes are color coded according to their function as indicated on the graph legend.
Our linear model does not fully explain the fitness profile of real hydrolysates. There are still several outliers where the fitness defect of the mutant predicted by our model is not as severe as observed in the real hydrolysate (Figure 5A). Two of these outliers (ZMO0760-ZMO0759) form an operon that encodes a putative GloAB glutathione-dependent enzyme system, which is required for detoxification of methylglyoxal (or other 2-oxoaldehydes) in a wide variety of organisms (Ozyamak et al, 2010). Typically, methylglyoxal is formed during unbalanced metabolism (Freedberg et al, 1971), which suggests that growth on hydrolysate leads to a methylglyoxal stress that is detoxified by the GloAB system. However, the Z. mobilis genome appears to lack a methyglyoxal synthase gene; thus, it is not clear why Z. mobilis needs a GloAB enzyme system or whether significant amounts of methyglyoxal can be formed intracellularly during unbalanced metabolism. The other outliers in our model encode a predicted efflux pump of unknown function (ZMO1429-ZMO1432) and a sodium/hydrogen exchanger (ZMO0846) (Figure 5A). ZMO0846 is a member of the KefB superfamily and may play a role in pH regulation. The presence of these outliers together with gloAB strongly suggests that we are missing critical hydrolysate components.
To investigate this, we obtained fitness profiles in methylglyoxal and a number of additional compounds and stress conditions that might result from hydrolysate exposure or from unbalanced metabolism (Supplementary Table 5). For example, furfural is known to induce the formation of reactive oxygen species (ROS) (Allen et al, 2010), so we tested two types of oxidative stress (hydrogen peroxide and sodium hypochlorite). Glycolaldehyde was recently reported to be a new component of hydrolysate, so we added this to our list of conditions (Jayakody et al, 2011). In total, we performed Z. mobilis pooled fitness assays in 11 additional conditions, including salt stress (KCl, NaCl), oxidative stress (hydrogen peroxide, sodium hypochlorite), glycerol, acetaldehyde, methylglyoxal, glycolaldehyde, and organic acids (acetic, formic, levulinic) at pH 6 to match the pH of our hydrolysate. The fitness data for these additional conditions are included in Figure 2 and 3B and Supplementary Dataset 1. Adding methylglyoxal to the regression significantly improved the fit (adjusted R2=0.903, P<10−15, ANOVA) and explained the two outliers in the GloAB system (Model-17 in Figure 5B and Supplementary Table 7). Adding all of the extra components, but removing insignificant ones, further improved the adjusted R2 to 0.919 (Model-24 in Figure 5C and Supplementary Table 7). In Model-24, the biggest improved prediction was for ZMO0846, which tends to be sensitive to organic acids at pH 6 but not in our unbuffered organic acid experiments (Figure 3B), consistent with its predicted role in pH regulation.
Our modeling results suggested that methylglyoxal might be present in our plant hydrolysates. We reanalyzed our batch 2 hydrolysate sample using a modified derivatization protocol and detected 41.4 μg/ml (0.56 mM) methylglyoxal (see Materials and methods). To our knowledge, methylglyoxal has not been previously reported in dilute-acid plant hydrolysates; however, the formation of methylglyoxal from glucose degradation has been detected in a model buffer system, and after supercritical water treatment of cellulose derived from red cedar (Thornalley et al, 1999; Nakata et al, 2006). To examine the impact of methylglyoxal in the context of hydrolysate, we measured the genome-wide fitness profile of SYN-10 alone or with 0.56 mM methylglyoxal added and compared it with the fitness profile of genuine hydrolysate. Although SYN-10 with methylglyoxal had a poorer fit to fitness in hydrolysate than SYN-10 did (R2=0.600 versus 0.669), the addition of methylglyoxal recapitulated the fitness defects of the gloAB operon (ZMO0759 and ZMO0760, compare arrows in Figure 5D and E). This suggests that ZMO0759 and ZMO0760 are important for growth in real hydrolysate because they are directly involved in methylglyoxal detoxification. Finally, we tested the effect of 0.56 mM methylglyoxal on the growth of wild-type Z. mobilis and found that addition of this hydrolysate-relevant concentration resulted in a small, but significant growth defect (Supplementary Figure 18, unpaired t-test, P<10−5). In sum, we used our modeling, fitness, and growth experiments to identify methylglyoxal as a previously unknown component of hydrolysate and to demonstrate that it contributes to overall hydrolysate toxicity in Z. mobilis.
Rational engineering of Z. mobilis for improved fermentation performance
We hypothesized that overexpression of putative tolerance genes in Z. mobilis might improve its growth and ethanol production in hydrolysate. To test this idea, we systematically overexpressed 21 tolerance genes (Supplementary Table 6), which were selected because they represented examples of the various functional classes we identified. These genes were overexpressed using an arabinose inducible Pbad promoter and a broad host plasmid system that we developed for this purpose (Supplementary Figure 14). Each tolerance gene was tagged with an N-terminal FLAG tag (DDDDYDK) that allowed us to examine relative protein levels by western blot. We first screened our overexpression strains for the correct molecular weight proteins and lack of significant protein degradation (Supplementary Figure 15). Based on these data, and on our prior verification of their negative fitness phenotypes, we selected 10 transposon mutant strains for complementation studies (Supplementary Table 6). For these experiments, we asked whether each Pbad expression construct was sufficient to complement the fitness defect of the corresponding transposon mutant (e.g., see Supplementary Figure 10). Based on the complementation data, we then selected four strains for detailed fermentation studies to measure ethanol productivity in the presence of miscanthus batch hydrolysate (WT+Pbad-ZMO1722, WT+Pbad-ZMO1875, WT+Pbad-ZMO0760, and WT+Pbad-ZMO0100). Small-scale aerobic batch fermentations were performed to determine specific ethanol productivity (g/l/h/OD600). We found that overexpression of ZMO1875 improved hydrolysate tolerance and increased specific ethanol productivity 2.4-fold (0.38 versus 0.16 g/l/h/OD600), whereas overexpression of ZMO1722, ZMO0760, and ZMO0100 had no significant effect on growth or ethanol production (Figure 6; Supplementary Figure 16). Glucose is fully consumed in both the wild-type and overexpression strains, yet the wild-type strain makes both less biomass and less ethanol. This suggests that the improvements in ethanol productivity in the ZMO1875 overexpression strain are due to a metabolic shift resulting in the production of less byproducts (Amin et al, 1983; Yang et al, 2009b).
Figure 6.

Overexpression of ZMO1875 improves ethanol productivity in the presence of miscanthus hydrolysate. Batch fermentation profile of the Z. mobilis wild-type+Pbad-ZMO1875 overexpression strain grown in rich media supplemented with 8% (v/v) batch 2 miscanthus hydrolysate (HZ). A control fermentation (WT+pJS71, colored symbols over dotted grey lines) is also shown for comparison (data taken from Figure 1B). Data shown are the average of four replicates and error bars indicate standard deviation.
Discussion
Deconstructing a complex chemical stress using chemogenomic profiling
In this study, we present a combined experimental and computational approach to address two challenges: (1) to understand how a complex chemical stress affects the growth and fermentation of Z. mobilis and S. cerevisiae and (2) to rationally engineer Z. mobilis for increased ethanol production in plant hydrolysate. Using chemogenomic profiling, we identified hydrolysate tolerance genes in Z. mobilis and S. cerevisiae and we used this information to rationally improve the fermentation performance of Z. mobilis in miscanthus hydrolysate. By modeling the Z. mobilis hydrolysate fitness data and then examining outliers in the regression, we identified methylglyoxal as an unknown component of miscanthus hydrolysate that contributes to its toxicity. Although we have focused on miscanthus and switchgrass hydrolysates prepared using dilute acid at high temperature, our experimental approach is generally applicable to any plant hydrolysate regardless of method of pretreatment and hydrolysis.
To our knowledge, this study is the first use of chemogenomic profiling to deconstruct the biological response to a highly complex chemical mixture. Previous large-scale studies in yeast have used fitness profiling to understand the mechanism of action of single compounds (Jansen et al, 2009; Cokol et al, 2011). Similarly, most previous studies of hydrolysate inhibitors in bacteria and yeast have focused on single compounds or simple binary mixtures (Palmqvist et al, 1999; Zaldivar and Ingram, 1999; Zaldivar et al, 1999, 2000; Klinke et al, 2003; Oliva et al, 2004). Only a few studies have looked at more complex inhibitor mixtures or at mixtures of hydrolysate fractions (Clark and Mackie, 1984; Koppram et al, 2012). Here, we examined synthetic mixtures of up to 37 inhibitors (SYN-37), greatly extending previous work, and determined that for Z. mobilis SYN-37 is a reasonable proxy (R2=0.810) for a dilute-acid miscanthus hydrolysate. Although not tested, addition of methylglyoxal to the SYN-37 mixture might further improve this correlation. Our Model-17 results (adjusted R2=0.903) suggest that it may be possible to recapitulate the biological effects of hydrolysate with a mixture of only 17 compounds. Thus, our work provides a good starting point for developing new synthetic hydrolysate mixtures that mimic the real material and for enabling the rational engineering of hydrolysate tolerance.
Because our high-throughput fitness protocols were developed for aerobic conditions, most of the experiments in this study were performed in the presence of oxygen. However, we recognize that most industrial biofuel fermentations will likely be microaerobic or anaerobic; therefore, we performed anaerobic hydrolysate experiments in Z. mobilis to determine whether the tolerance genes we identified in this study can be used to engineer tolerance under these growth conditions. Using the same criterion for identifying aerobic tolerance genes (fitnesshydrolysate <−1 and fitnesshydrolysate<fitnessrich −1), we identified 11 genes that are important for growth in anaerobic hydrolysate (Supplementary Figure 17). Four of these genes were found in our aerobic studies (ZMO0100-ZMO0101, ZMO0759, and ZMO1490). In addition to these four genes, we identified seven new anaerobic tolerance genes (ZMO1015, ZMO1016, ZMO1017, ZMO1018, ZMO1355, ZMO1548, and ZMO1556), which provide a basis for future engineering of anaerobic hydrolysate tolerance. These results emphasize the need to match laboratory hydrolysate tolerance studies with the specific growth and environmental conditions of an industrial-scale cellulosic biofuel process. However, our approach for dissecting a complex chemical stress is general, and by collecting fitness data for the 37 components under anaerobic conditions, it should be possible to model anaerobic hydrolysate stress.
Mechanisms of hydrolysate tolerance in Z. mobilis and S. cerevisiae
In both Z. mobilis and S. cerevisiae, we identified severalbroad categories of gene functions required for hydrolysate tolerance. We also identified many genes of unknown function, or unrelated to any previously known tolerance mechanism, demonstrating that our experimental approach is a rich source of new knowledge for understanding the biological response to a complex chemical stress. Of the 44 tolerance genes we identified in Z. mobilis, only 1 (ZMO1432) has previously been reported in the patent WO 2012/082711 A1 (Caimi and Hitz, 2012). In this patent, they identified a point mutation in ZMO1432 after evolving Z. mobilis for improved fermentation performance in hydrolysate. ZMO1432 is part of a four gene operon (ZMO1429-ZMO1432) that encodes a predicted efflux pump. In our study, we identified transposon insertions in all four of these genes as sensitive to hydrolysate, which suggests that efflux of inhibitory compounds is an important mechanism for hydrolysate tolerance. Upregulation of efflux pump genes has recently been reported in a transcriptome study of E. coli growth in corn stover hydrolysate (Schwalbach et al, 2012). In S. cerevisiae, only 9 of 99 tolerance genes that we identified were previously reported in single inhibitor or hydrolysate tolerance studies (BAP2, ERG2, GTR2, LSM6, RPN4, TAL1, TKL1, YAP1, and ZWF1) (Jeppsson et al, 2003; Gorsich et al, 2006; Kawahata et al, 2006; Ma and Liu, 2010; Sundström et al, 2010; Liu, 2011; Pereira et al, 2011; Sanda et al, 2011; Gao and Xia, 2012; Hueso et al, 2012). Broadly, there is little overlap in the genes we identified with previous tolerance studies in Z. mobilis, E. coli, and S. cerevisiae (Petersson et al, 2006; Almeida et al, 2007; Miller et al, 2009b, 2010; Yang et al, 2010a, 2010b; Parawira and Tekere, 2011; Drobna et al, 2012), which likely reflects the underlying genetic complexity of tolerance, the different experimental protocols used for tolerance gene identification, and the different plant feedstocks and methods used for hydrolysate preparation. However, despite these differences, we did identify genes in two pathways (oxidative stress response and amino-acid biosynthesis) that overlap with previous studies, which suggest a fundamental role for these pathways in hydrolysate tolerance in bacteria and yeast (Miller et al, 2009a; Allen et al, 2010; Warner et al, 2010). In addition, our work in Z. mobilis has uncovered genes involved in sulfate assimilation and iron-sulfur (Fe-S) cluster assembly and repair that represent new potential gene targets for strain engineering.
Oxidative stress response
In both Z. mobilis and S. cerevisiae, we identified tolerance genes involved in oxidative stress response, which suggests that growth in hydrolysate induces the intracellular formation of ROS, which includes hydrogen peroxide, superoxide anion, and hydroxyl radicals. Although not tested, these ROS are not likely to be present in our plant hydrolysates due to their chemical instability. Furfural, which is an abundant component of plant hydrolysates, can induce the formation of ROS in S. cerevisiae, and provides a direct link between oxidative stress and hydrolysate toxicity (Allen et al, 2010). In our Z. mobilis gene fitness data, we identified CCP (ZMO1136), and a number of genes involved in cytochrome c biogenesis (ZMO1252, ZMO1253, ZMO1255, and ZMO1389) that are important for growth in hydrolysate. CCP converts hydrogen peroxide to water (Mishra and Imlay, 2012), and likely has a direct role in hydrolysate tolerance by reducing the levels of this ROS. Consistent with previous studies of Z. mobilis CCP (Charoensuk et al, 2011), we find that mutants in ZMO1136 are sensitive to hydrogen peroxide (average fitness in rich media=0.003, average fitness in hydrogen peroxide=−3.82, Supplementary Dataset 1). Previous work in E. coli identified three genes involved in peroxide detoxification (ahpC, tpx, and bcp) that were important for growth in corn stover hydrolysate (Warner et al, 2010). In our S. cerevisiae hydrolysate fitness data, we identified YAP1, which is a known regulator of the oxidative stress response, including the response to peroxides (Veal et al, 2003; Drobna et al, 2012), and previously found to be important for growth in 5-HMF, an abundant component of hydrolysate (Ma and Liu, 2010). We also identified two transcriptional targets (GSH1 and CYS3) of the YAP1 regulator (Nisamedtinov et al, 2011), further implicating the YAP1 pathway. In addition, we identified five OCA genes (OCA1, OCA2, OCA4, OCA5, and OCA6) that are important for growth in hydrolysate, which are involved in the repair of lipids after oxidative damage (Alic et al, 2001). Together, our data suggest that in both bacteria and yeast, growth in hydrolysate leads to the formation of intracellular peroxides and that peroxide detoxification is an important mechanism of hydrolysate tolerance.
Amino-acid biosynthesis
We also implicated amino-acid biosynthesis as a mechanism of hydrolysate tolerance in both Z. mobilis and S. cerevisiae. We found that both homoserine dehydrogenase (ZMO0483 or HOM6), which is required for methionine biosynthesis, and glutamine amidotransferase (ZMO0201 or TRP3), which is required for tryptophan biosynthesis, are important for growth in hydrolysate. In addition, we identified four tolerance genes involved in sulfate assimilation, which also suggests a role for cysteine biosynthesis in hydrolysate tolerance; although this pathway may play other roles in hydrolysate tolerance (see next section). Our work is consistent with previous studies in E. coli which found that addition of cysteine and methionine to the growth media helped alleviate furfural, acetic acid, and hydrolysate toxicity (Roe et al, 2002; Miller et al, 2009a; Nieves et al, 2011; Sandoval et al, 2011). In E. coli, reduction of furfural depletes NADPH, which limits sulfate assimilation by the NADPH-dependent enzyme sulfite reductase encoded by cysIJ. Acetate appears to block methionine biosynthesis downstream of homocysteine, which leads to accumulation of this toxic intermediate (Roe et al, 2002). Our studies also suggest that addition of methionine and cysteine might improve hydrolysate tolerance; however, our fitness experiments were conducted in rich media (ZRMG or YPD), which should have high levels of these amino acids, in contrast to previous studies that were conducted in minimal media (Nieves et al, 2011). It is not clear why we identified amino-acid biosynthesis genes in our rich media growth conditions, but the overlap with previous studies strongly suggests a fundamental role for amino-acid biosynthesis in hydrolysate tolerance in both bacteria and yeast.
Sulfate assimilation
Our fitness data in Z. mobilis also suggest that growth in hydrolysate induces ROS that lead to an increased demand for cysteine biosynthesis and for sulfide. We identified four Z. mobilis auxotrophs in the sulfate assimilation pathway (ZMO0003, ZMO0007, ZMO0008, and ZMO0009) encoding CysC, CysH, CysI, and CysJ, respectively, which are needed for de novo cysteine biosynthesis and are important for growth in hydrolysate. In Salmonella typhimurium, the CysB regulon is induced by oxidants, such as hydrogen peroxide or menadione, and cysCIJ mutants have reduced levels of glutathione and induce an oxidative stress response (Turnbull and Surette, 2010). Similarly, growth of Z. mobilis in hydrolysate may lead to reduced levels of glutathione, which is formed from glutamate and cysteine; thus, this might explain the increased demand for cysteine. In addition, the sulfate assimilation pathway also functions to provide sulfur for assembly of Fe-S clusters; thus, it is likely that this pathway has multiple roles in hydrolysate tolerance.
Fe-S clusters
Fe-S clusters are essential enzyme prosthetic groups that can be damaged by ROS (Djaman, 2004). A number of pathways exist in E. coli for the repair of Fe-S clusters that have been damaged by oxidative stress (Yang, 2002; Djaman, 2004; Bitoun et al, 2008). We identified four tolerance genes in Z. mobilis (ZMO0429, ZMO1067, ZMO1874, and ZMO1875) that provide a causal link between growth in hydrolysate, oxidative stress, and Fe-S clusters. In Z. mobilis, ZMO0429 and ZMO1067 encode predicted Fe-S assembly proteins, and ZMO1874 encodes a predicted BolA family member, which has also been linked to Fe-S cluster assembly (Li and Outten, 2012); however, ZMO1875 has no predicted function. The fitness profiles of ZMO1067::TN5, ZMO1874::TN5, and ZMO1875::TN5 are similar and cluster together (Figure 3B), suggesting a shared function in Fe-S cluster repair and hydrolysate tolerance.
BolA family members have recently been shown to interact with GrxD family monothiol glutaredoxins in both E. coli and S. cerevisiae (Huynen et al, 2005; Koch and Nybroe, 2006; Rouhier et al, 2010; Shakamuri et al, 2012). These complexes directly bind Fe-S clusters, and are thought to function in both Fe-S cluster assembly and regulation of new Fe-S cluster synthesis (Kumanovics et al, 2008; Cameron et al, 2011; Shakamuri et al, 2012; Willems et al, 2012). Consistent with this role, ZMO1874 is in the same operon with a predicted monothiol glutaredoxin (ZMO1873) and an Fe-S containing enzyme quinolinate synthetase (ZMO1871), which is involved in NAD biosynthesis. The ZMO1874-ZMO1873 complex may function to assemble Fe-S clusters in the quinolinate synthetase enzyme, or regulate its activity.
ZMO1875 encodes a gene of unknown function (DUF1476), which, in this study, was used to improve ethanol productivity in the presence of hydrolysate. One recent study identified a DUF1476 protein that functions as an inhibitory subunit of the FoF1 ATP synthase complex (Morales-Rios et al, 2010). It is not clear how inhibiting ATP synthase would improve hydrolysate tolerance, and ATP synthase is not consistently detrimental to fitness in hydrolysate. Instead, we propose that ZMO1875 functions together with BolA-GrxD like complexes in Z. mobilis (ZMO1874-ZMO1873) to assemble Fe-S clusters in the enzyme quinolinate synthetase or to regulate the de novo biosynthesis of new Fe-S clusters, to replace those that have been damaged by ROS. This is the first example, to our knowledge, of targeting Fe-S cluster pathways for engineering improved hydrolysate tolerance.
Synergistic effects of inhibitors
Previous studies have explored synergistic interactions between hydrolysate inhibitors, based on effects on growth and fermentation (Palmqvist et al, 1999; Zaldivar and Ingram, 1999; Zaldivar et al, 1999, 2000). Some combinations of compounds inhibit growth or ethanol production more than one would expect from the activity of the compounds individually. The mechanisms behind this synergistic inhibition of growth are not well understood. More broadly, synergistic inhibition on growth appears to be common but not predominant—a recent study found that 38 of 200 pairs of antifungal drugs synergistically inhibited the growth of S. cerevisiae (Cokol et al, 2011).
Here, we focused on a related but different question: do inhibitors have synergistic effects on a mutant's growth relative to wild type? We found that gene fitness on hydrolysate can be modeled as a linear combination of gene fitness on the individual compounds. Furthermore, although some genes were important for fitness in plant hydrolysate without being important for fitness on any of the known components, these genes were not important for fitness in synthetic hydrolysate mixtures, which suggests that they are important for resisting unidentified components. Similarly, we did not identify any genes with much lower fitness in the defined mixtures SYN-10 or SYN-37 than in any of their components (the biggest reductions in fitness, relative to the minimum on the components, were –1.1 and –0.6, respectively). Finally, we considered adding interaction terms of the form X × Y to our regression. These would be necessary if combining inhibitors converts some mutants’ mild phenotypes into severe ones. Although we found interaction terms that were statistically significant, adding them did not lead to a notable improvement in the fit of the model.
Implications for rational engineering of hydrolysate tolerance
The success of our linear model for predicting hydrolysate fitness suggests that we can separately engineer tolerance to individual components, and then combine these improvements into a single strain. The successful evolution of a S. cerevisiae strain for improved fermentation in spruce hydrolysate by adaptation to a cocktail of 12 inhibitors is consistent with our hypothesis that the biological response to hydrolysate can be understood as the combination of responses to the individual compounds (Koppram et al, 2012). A combinatorial engineering approach has already proved successful for improving tolerance to a binary mixture of inhibitors (Sommer et al, 2010). In their study, they identified three genes, recA, orfX, and udpE that conferred resistance to either 2-furoic acid or syringaldehyde. When these three genes were co-expressed, the combination conferred tolerance to the mixture of both compounds. Based on these results and our data, we believe that a strain engineered for improved tolerance to the main chemical classes found in hydrolysate, such as weak organic acids, phenolics, aldehydes, and furans, might be sufficient to confer tolerance to the full, complex chemical mixture present in real plant hydrolysates. For example, two well-known tolerance mechanisms, furfural reduction and laccase detoxification of phenolics (Parawira and Tekere, 2011) could be combined into a single strain for further improvements in hydrolysate tolerance. In addition, based on our work, this strain could be further engineered by modifying pathways for peroxide detoxification and Fe-S cluster repair. It is possible that complex epistatic relationships exist between tolerance gene pathways, making combinatorial engineering more difficult (Sandoval et al, 2012); however, to a first approximation our linear fitness model and previous work (Wood et al, 2012) argues against this possibility.
Conclusions
In this study, we have shown that chemogenomic profiling and modeling can be used to deconstruct a complex chemical stress and that this information can be used to rationally engineer a strain for improved fermentation in plant hydrolysate. More broadly, our approach can be used to model other complex stresses, such as the array of environmental stresses (including product toxicity) encountered by microbes inside an industrial bioreactor (Gibson et al, 2007, 2008). Understanding how the production host deals with this complex environmental stress has significant implications for improving the yield and productivity of any industrial-scale fermentation process.
Materials and methods
Strains, media, plasmids, and primers
Bacterial strains, primers, and plasmids used in this study are listed in Supplementary Table 6. Zymomonas mobilis strain ZM4 obtained from ATCC (ATCC 31821) was the parent strain for our studies. E. coli strains TOP10 (Invitrogen), NEB 5-alpha (New England Biolabs), and WM3064 (W. Metcalf, University of Illinois at Urbana-Champaign) were used as needed. Z. mobilis was cultured in ZRMG, Zymomonas Rich Medium Glucose (25 g/l glucose, 10 g/l yeast extract, and 2 g/l KH2PO4) or ZMMG, Zymomonas Minimal Medium Glucose (Goodman et al, 1982), and grown aerobically at 30°C. Anaerobic growth was performed in sealed Hungate tubes after degassing media with nitrogen or on ZRMG agar plates in an anaerobic chamber (Coy Lab Products). For Z. mobilis, plates or liquid media were supplemented with 100 μg/ml kanamycin, chloramphenicol, or spectinomycin as necessary. E. coli strains were grown in Luria-Bertani (LB) broth at 37°C, supplemented with 30 μg/ml kanamycin, 20 μg/ml chloramphenicol, or 50 μg/ml spectinomycin as needed. The S. cerevisiae homozygous barcoded deletion mutant collection (Giaever et al, 2002) was a gift of Ron Davis (Stanford Genome Technology Center). Yeast strains were grown aerobically at 30°C in Difco Yeast Extract Peptone Dextrose (YPD) media.
Preparation and analysis of plant hydrolysates
A dilute-acid pretreatment method was used to release fermentable sugars from plant biomass. M. giganteus (miscanthus) and P. virgatum (switchgrass) were grown in Illinois at four different field sites: Brownstown (BTN), Fairfield (FRF), Havana (HAV), and Orr (ORR). ‘Batch’ hydrolysates were prepared from a mixed miscanthus sample that was derived from several field sites (Batch February 2009, EBI South 2006 Season, obtained from UIUC). After harvesting, samples were air dried, processed with a SM 2000 cutting mill and a 2-mm sieve (Retsch, Haan, Germany), and then finally ground to pass a 120-μm sieve screen using a SR 200 rotor beater mill (Retsch). Pretreatment and hydrolysis were performed in an Ethos Z microwave (Milestone Inc., Shelton, CT) equipped with six closed reaction vessels (each 100 ml in volume). In brief, each vessel contained 5 g ground miscanthus and 45 g of 1% (w/w) sulfuric acid. The temperature was increased in 4 min to 180°C (in 4.5 min to 200°C for batch 1 and batch 2) and held for 2 min. The mixture was cooled in an ice bath to 95°C,the vessels were opened and the contents rapidly transferred into a beaker and the mixture was further cooled to room temperature. The supernatant was collected after centrifugation (5000 × g) and adjusted to pH 6.0 with KOH. Samples were filter sterilized, 10 ml aliquots were flash frozen in a dry-ice ethanol bath, and stored at −80°C, protected from light. Since the stability of compounds present in hydrolysate is unknown, hydrolysate aliquots used in pool and growth experiments were thawed and used only once.
The supernatant was analyzed for soluble carbohydrates, organic acids and furans using an Agilent 1200 series liquid chromatography system (Agilent Technologies, Santa Clara, CA) equipped with a refractive index detector and a diode-array detector. Samples were injected onto an Aminex HPX-87H column (Bio-Rad, Hercules, CA) and compounds were eluted at 50°C and a flow rate of 0.6 ml/min by a mobile phase consisting of 0.005 M sulfuric acid. For detection and quantification of methylglyoxal, 1 ml of the hydrolysate was mixed with 0.5 ml of a solution of 1% orthophenylenediamine in 0.5 M sodium phosphate (pH 6.5). The mixture was incubated at room temperature in the dark for 16 h and then 5 μl was injected onto a Zorbax SB-C18 Rapid Resolution column (Agilent Technologies) and eluted at 40°C by a gradient of solvent A (0.1% (v/v) formic acid) and solvent B (acetonitrile containing 0.1% (v/v) formic acid). The gradient program was: 5–50% B in 10 min, 50–90% B in 4 min, 90–5% B in 1 min, then 3 min isocratic elution. Detection was at 312 nm. A standard of methylglyoxal (40% solution, Sigma-Aldrich, St. Louis, MO) was derivatized in the same way and used to confirm the retention time and UV spectra. GC/MS was used for analysis of phenolic compounds: to 1 ml of neutralized hydrolysate, 30 μl of 72% sulfuric acid and 20 μl of internal standard (iso-propylphenol, 1 mg/ml in 0.1% sodium hydroxide) were added. This mixture was vigorously mixed three times each with 0.5 ml of ethyl acetate. After phase separation, the upper ethyl acetate layer was removed and collected. All ethyl acetate phases were combined and dried over sodium sulfate. An aliquot of the combined dried ethyl acetate phase (100 μl) was incubated with 50 μl of N,O-bis(trimethylsilyl)trifluoroacetamide containing 1% trimethylchlorosilane (Sigma-Aldrich) at 70°C for 30 min. In all, 1 μl was injected in splitless mode onto a VF5-MS capillary column (Varian, Palo Alto, CA). An Agilent 7890A gas chromatograph coupled to an Agilent 5975C single quadrupole mass spectrometer with the following settings was used: injector temperature 280°C, carrier gas: helium at 1 ml/min, temperature program: 3 min isocratic 75°C, 5°C/min to 150°C, 0.5°C/min to 160°C, 2°C/min to 190°C, 5°C/min to 240°C, 70°C/min to 325°C, 3 min isocratic, ions were detected by electron impact ionization (70 eV) in full scan mode m/z 35–500. Compounds were identified by matching their mass spectra with NIST database entries and by comparing their retention times with commercially available standards (Sigma-Aldrich). Peak areas were quantified using selected extracted ions for the compounds in internal standard calibration mode (m/z 193 for iso-propylphenol). We quantified 37 inhibitors and 4 sugars for each plant hydrolysate sample (Supplementary Table 1). Only batch 2 hydrolysate was analyzed for methylglyoxal.
Preparation and analysis of synthetic hydrolysate mixtures (SYN-37 and SYN-10)
Two synthetic hydrolysate mixtures were prepared containing the 10 most abundant compounds (SYN-10) or the 37 most abundant compounds (SYN-37) present in batch 1 miscanthus hydrolysate. SYN-37 also contained four sugars (xylose, glucose, arabinose, and cellobiose). All chemicals were purchased from Sigma-Aldrich. Each mixture was prepared by mixing 1 M DMSO (or Milli-Q water) stock solutions directly into 2 × ZRMG media, and then the final solution adjusted to 1 × ZRMG with water and the pH was adjusted to 6.0 with KOH. Liquid chemicals such as furfural were added directly to the mixture. SYN-37 and SYN-10 were analyzed by LC-RID and GC/MS to determine their compositions (Supplementary Table 1). We found that the concentrations of vanillin, syringaldehyde, vanillic acid, and furoic acid in the SYN-10 mixture were higher than expected. For SYN-37, we found that benzoic acid and cellobiose were higher than the values in batch 1 hydrolysate. Despite these differences in composition, the fitness profiles of SYN-37 and SYN-10 are highly correlated (in Z. mobilis, R2=0.81; in S. cerevisiae, R2=0.86), and we chose to continue our studies with these mixtures. In addition, this high correlation implies that the presence of three non-metabolizable sugars in SYN-37 (xylose, arabinose, and cellobiose) versus SYN-10 (glucose only) had little effect on their genome-wide fitness profiles. Once made, the synthetic mixtures were flash frozen into 10 and 50 ml aliquots and stored at −80°C. These stock solutions were considered as 100% by volume and used accordingly for both Z. mobilis and S. cerevisiae fitness and growth experiments. The final concentration of DMSO in SYN-37 was 0.16%. SYN-10 did not contain any DMSO. The fitness profile of ZRMG+2% DMSO was measured as a control for any possible effects of DMSO on fitness, but little effect was seen (e.g., Figure 4B).
Generation of a Z. mobilis barcoded transposon library and chemogenomic profiling
Our laboratory has previously described detailed methods for TagModule construction, the generation of a genome-wide barcoded transposon library, pooled fitness assays, data normalization and analysis in Shewanella oneidensis MR-1 (Oh et al, 2010; Deutschbauer et al, 2011). The same methods were used to create a barcoded transposon library in Z. mobilis, and perform fitness assays, with a few modifications. Each mutant in our Z. mobilis pool contains a barcoded transposon, which is a KanR Tn5 containing a TagModule (each TagModule contains two 20 bp DNA barcodes, called UPTAG and DNTAG). Briefly, to build the transposon mutant collection in Z. mobilis, we used a mini-Tn5 delivery system based on the suicide plasmid pRL27 (Larsen et al, 2002). Transposons were delivered into Z. mobilis by plate mating and conjugation using E. coli WM3064. Kanamycin-resistant colonies were picked and mutants were stored at −80°C in 384-well plates. Gene disruptions were mapped using a two-step arbitrary PCR method previously described (Deutschbauer et al, 2011), using the primers (Round 1: pRL27_IE_rev1+ARB8, ARB11, Round 2: U2_comp+ARB2). A custom Perl program was designed to track each transposon insertion and TagModule identity. Perl scripts are available upon request. Z. mobilis gene annotations for the main chromosome and five plasmids (pZZM401-pZZM405) were obtained from RefSeq ( http://www.ncbi.nlm.nih.gov/RefSeq/) and a recent annotation paper (Yang et al, 2009a). Two Z. mobilis pools (up-pool and dn-pool; Supplementary Table 2) were designed using a custom Perl script that contained 7432 strains total and 6302 unique strains. These strains were cherry-picked from the 384-well plates using a Biomek FxP Liquid Handling Robot (Beckman Instruments) into 96-well ‘pool plates’ and grown to saturation in ZRMG+100 μg/ml kanamycin. In all, 25 μl of each mutant was transferred and pooled together. The mixed pool was pelleted by centrifugation, and resuspended in fresh ZRMG media+10% glycerol. In all, 100 μl pool aliquots were made and frozen at −80 °C.
For chemogenomic profiling, the up-pool and dn-pool were recovered by thawing 100 μl aliquots and inoculating into 10 ml of ZRMG. Cells were grown for 5 h, shaking at 30°C, until they reached an OD600 of 0.5. These cultures called ‘START’ were used to initiate pool experiments, which were done either in 10 ml or in 24-well plate format, starting at 0.02 OD600. Several concentrations of each condition were prescreened until a concentration that showed about 50% inhibition was identified. A list of experiments, including concentrations used for each condition in the fitness data set, is found in Supplementary Table 5. After growing the pool for about 5–7 generations (typically to saturation), cell pellets were collected (called ‘END’), and the genomic DNA was isolated (Qiagen DNeasy Kit or Qiagen PureGene Kit). We PCR amplified the UPTAGs from the up-pool and DOWNTAGs from the dn-pool as previously described (Deutschbauer et al, 2011). These PCR products were combined and hybridized to an Affymetrix 16K TAG4 array, washed, and scanned as previously described (Pierce et al, 2006). The abundance of each mutant in the ‘END’ sample compared with its abundance in the ‘START’ sample is used to calculate a log2 fitness ratio (END/START), which is called ‘strain fitness’. For most genes, ‘gene fitness’ is defined as the average strain fitness value for all ‘good’ transposon insertions (i.e., within the central 5–80% portion of a gene). We found that the strain fitness values for different insertions within the same gene were well correlated (for all hydrolysate experiments, n=37, R=0.85, mean absolute difference in fitness was only 0.27), and therefore we report only gene fitness values in this paper.
Data normalization and calculation of gene fitness values was done using a modification of our previous method (Oh et al, 2010; Deutschbauer et al, 2011). First, we set the median of strain fitness from each scaffold (main chromosome and five plasmids) to zero. This corrected for differential efficiency of DNA extraction between the main chromosome and five plasmids. In addition, for the main chromosome we used a smooth estimator (loess in R) to remove a small effect of chromosomal position on tag abundance (this was probably due to increased copy number near the origin). Finally, for the main chromosome, we also set the mode of the strain fitness distribution to zero. We did this because the median fitness was generally below the mode (the mode was estimated using the maximum of the kernel density using the density function in R) and because the mode for all proteins matched the mode for hypothetical proteins, which should be less likely to have phenotypes. Data analysis was done using custom R scripts (available upon request). The full data set of conditions tested and fitness values is found in Supplementary Dataset 1.
We found that a putative prophage region (ZMO1920-ZMO1952) contained 18 genes that were often detrimental for fitness (vertical yellow stripe in Figure 2, a little to the right of center); however, they were not specifically detrimental to fitness in hydrolysate and so we did not study them further. In our comparative genome hybridization data, this region appeared to have variable copy number (Supplementary Figure 4H). The positive fitness of these genes could be an artifact—if the prophage increases its copy number when the cell is stressed (Imae and Fukasawa, 1970), then the barcodes in these strains will be amplified and their fitness values will be positive, even though those cells have not increased in abundance.
Modeling hydrolysate fitness using component fitness data
We modeled the average gene fitness in hydrolysate as a linear mixture of gene fitness in ZRMG-rich media and in various stresses, for example, for each gene g:
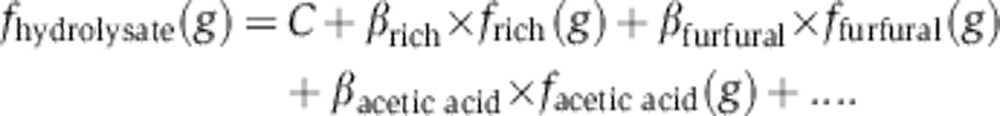 |
where frich is the average gene fitness in rich media across 24 experiments and the parameters C, βrich, βfurfural, βacetic acid, etc., were computed using a standard linear regression (lm in R) to best fit the average gene fitness in hydrolysate across 37 experiments. We started from a model with 37 components and used ANOVA to identify components that did not significantly improve the regression. The results of ANOVA depend on the order of the components; components with the highest concentrations were included first and hence were more likely to be retained. We removed insignificant components and then built a new regression and performed a new ANOVA with fewer components. We repeated this procedure until all components were statistically significant (P<0.05 after Bonferroni correction for multiple testing), resulting in a model with 16 components (Model-16). The regression results are shown in Supplementary Table 7. To test the effect of additional conditions on our model (see Supplementary Table 5 for full condition list), we added them and repeated the ANOVA test.
We also considered adding interaction terms to test for possible synergistic effects of components. We considered adding all terms of the form X × Y, where X and Y are components, and used a P-value cutoff of 10−4 to make up for the large number of terms tested. We identified 20 significant interaction terms. As with the linear regression with individual components, we then used ANOVA to see if these terms were significant when used in combination, again requiring P<10−4. The resulting model contained only three interaction terms. Adding these terms to Model-16 improves the adjusted R2 from 0.880 to 0.893 and alters the predicted fitness of four genes by 0.5 or more. The affected genes were ZMO0975, ZMO1430, ZMO1431, and ZMO1432; also ZMO1429 is in an operon with ZMO1430-ZMO1432 and is near the threshold. All five of these genes were identified as having non-stable insertions, so we are not sure that the improvement of fit for these genes is biologically meaningful.
Yeast pooled fitness experiments
Fitness experiments were performed using a S. cerevisiae homozygous deletion pool. For each pool experiment, 100 μl of our frozen pool aliquot was diluted into 50 ml YPD media, and grown for 6 h to an OD600 of 2.4. Once the pool had recovered, we collected a ‘START’ sample, and then inoculated into various experimental conditions at a starting OD600 of 0.03. Pool experiments were performed in 24-well or 48-well formats and grown in a TECAN Infinite F200 plate reader. Samples were grown in the condition of interest for about seven generations (typically to saturation) and then the ‘END’ sample was collected. Either 1 ml (24-well format) or 2.1 ml (48-well format) was collected for genomic DNA isolation (YeaStar Genomic DNA Kit, Zymo Research). DNA barcode amplification by PCR, Affymetrix hybridization, washing, and scanning of TAG4 arrays was done as previously described (Pierce et al, 2006). Using custom R scripts, we set the median fitness value, as computed using the UPTAG or DNTAG, to zero and then averaged across the measurements for each gene (Supplementary Dataset 1). ‘Gene fitness’ and ‘strain fitness’ values are the same (log2 ratio of END/START) for S. cerevisiae because only one deletion strain exists per gene in the homozygous pool. For most yeast strains, we have both UPTAG and DNTAG measurements of abundance. In addition, the Affymetrix TAG4 array has five replicate probe spots for each UPTAG and DNTAG; therefore, ‘gene fitness’ is an average of multiple measurements (i.e., two different tags and probe replicates). To identify putative S. cerevisiae tolerance genes, we plotted the gene fitness data in YPD-rich media versus gene fitness in YPD-rich media supplemented with batch 1 hydrolysate, and selected genes using the following criterion: fitnesshydrolysate <−1 and fitnesshydrolysate<fitnessrich −1 (Supplementary Figure 13; Supplementary Dataset 2). Gene and GO annotations for S288C were obtained from the SGD database ( http://www.yeastgenome.org/). We used GO annotations to help classify our tolerance gene list into six broad functional groups (Table II).
Identification of promoters for use in Z. mobilis
We developed a series of Gateway (Invitrogen) adapted vectors for overexpression of genes in Z. mobilis. The system is based on pJS71, a broad-host plasmid derived from pBBR1MCS (Skerker and Shapiro, 2000) that we found to be stably maintained in Z. mobilis. Using a fluorescence-based assay based on superfolder GFP (Pédelacq et al, 2005), we tested the relative promoter strengths of one E. coli (Pbad) and one Z. mobilis (Pgap) promoter (Conway et al, 1987; Guzman et al, 1995). Each promoter construct was made using the Gateway system by fusing the promoter of interest to GFP using a pJS71-based destination vector (Supplementary Table 6). These strains were grown in a 10-ml ZRMG media culture with spectinomycin. In the case of Pbad, cultures were induced with increasing concentrations of arabinose. Once saturated, cultures were diluted 1:1000 and allowed to grow until they reached an OD600 of 0.4. Cells were washed twice with 1 × PBS. After washing, 150 μl of resuspended cells was pipetted into a 96-well assay plate (Costar 3603). In addition, a Z. mobilis control strain carrying only the pJS71 plasmid was used as a negative control. Fluorescence (RFU) was measured in a Tecan Safire plate reader. Average fluorescence of the control strain was subtracted from the average of each experimental sample. Relative promoter activity was determined by calculating RFU/OD for all samples and averaged over three biological replicates (Supplementary Figure 14).
Z. mobilis growth experiments
Growth experiments were performed in 24-well and 96-well microplates. Single transposon mutant characterization was performed in 150 μl volume, 96-well format and in 1 ml volume, 24-well format using either a Tecan Sunrise or Tecan Infinite F200 plate reader (Tecan Systems Inc., San Jose, CA). Starting OD600 for all Z. mobilis strains was 0.02. For Z. mobilis transposon mutants, saturated overnight ZRMG cultures were diluted 1:20, grown to OD600 0.5 and then used to inoculate a plate containing 1 × ZRMG with and without 10% batch 1 or 8% batch 2 miscanthus hydrolysate (% v/v). Complementation and overexpression experiments were done using a Tecan Sunrise reader in a 96-well format. Strains were grown overnight in ZRMG+100 μg/ml spectinomycin. For mutants where Pbad controlled expression of the relevant gene, 2% arabinose was added to the media. Once saturated, cultures were diluted back 1:20 and grown to 0.5 OD600. Strains were inoculated at a starting OD600 of 0.1 into plates containing ZRMG, with and without inhibitor. For these experiments, inhibitor concentrations were 10% batch 1 hydrolysate or 1.5 mM methylglyoxal in 150 μl total volume per well. All growth experiments were run for up to 72 h.
Construction of expression clones for complementation and overexpression
We used the Pbad and Pgap promoters for complementation of tolerance gene mutants and for testing the effect of overexpression on growth and fermentation performance. Each promoter construct included an in-frame N-terminal FLAG tag (DDDDYDK), so protein expression could be detected by western blot with anti-FLAG antibodies (Sigma-Aldrich). A series of pENTR clones were generated for putative Z. mobilis tolerance genes (Supplementary Table 6). We used Gateway cloning to generate Pbad-ZMOxxxx or Pgap-ZMOxxxx expression clones, where ZMOxxxx is the systematic gene name (Supplementary Table 6). These plasmids were electroporated into wild-type Z. mobilis to examine the effects of overexpression on cell growth and ethanol production. For electroporation, competent cells were made from an overnight wild-type culture grown to 0.8–1.0 OD600. Cells were harvested by centrifugation and resuspended twice in ice-cold sterile Milli-Q water (EMD Millipore, Billerica, MA). Cells were then washed twice with ice-cold 10% glycerol and frozen in 80 μl aliquots at −80 °C. To perform the electroporation, 40 μl of cells was thawed on ice and 1 μg of DNA was added, along with 2.5 μg of Type One Restriction Inhibitor (Epicentre). The transformation mixture was pipetted into an electroporation cuvette and cells were electroporated using the following settings: 1600 V, 200 ohms, and 25 μF. Cells were recovered in ZRMG media for 6 h, shaking at 30 °C. After recovery, 10 μl or 100 μl of recovered cells was spread on selective plates and incubated at 30 °C for at least 2 days. A single colony for each overexpression strain was archived at −80 °C (Supplementary Table 6). The same constructs were also used for complementation studies, except that they were electroporated into the corresponding mutant (ZMOxxxx::TN5) strain. Overexpression and complementation experiments were performed with at least three biological replicates, using freshly streaked single colonies from ZRMG+spectinomycin plates.
Western blot analysis of overexpression strains
The relative expression of each clone used for overexpression was examined by western blot analysis (Supplementary Figure 15). Strains containing expression plasmids were grown in ZRMG+100 μg/ml spectinomycin+2% arabinose. After 24 h, cultures were diluted 1:20, grown to 0.5 OD600, and 1 ml was harvested. Cells were resuspended in freshly made 1 × SDS–PAGE sample buffer (Bio-Rad) containing β-mercaptoethanol and boiled for 5 min. All samples were stored at −80°C until used. Protein samples were separated by SDS–PAGE (using precast 2–40% gradient gels, Bio-Rad), transferred at 4°C onto a PVDF membrane and then blocked overnight in PBST+5% non-fat instant dry milk. Once blocked, the membrane was washed briefly in PBS+3% non-fat milk and subsequently incubated with the primary antibody, 1:5000 dilution of anti-FLAG (Sigma F3165), for 1 h, shaking at room temperature. After thoroughly washing with PBST, the membrane was incubated with the secondary antibody, 1:1000 goat anti-mouse horseradish peroxidase (Thermo Scientific), again for 1 h with shaking. The membrane was washed with PBST and incubated with ECL reagent (1:1 mixture, Thermo Scientific) for 5 min. After allowing to drip dry briefly, the blot was imaged using a Fujifilm LAS-4000 imager in chemiluminescence mode.
Fermentation experiments
The fermentation capability of mutants was measured during aerobic growth in rich media with and without hydrolysate (Figures 1 and 6; Supplementary Figure 16). Starter cultures were grown in 1 × ZRMG+100 μg/ml spectinomycin and induced with 2% arabinose. After 24 h, cultures were diluted 1:20 and grown to 0.5 OD600. Fermentations were started at 0.1 OD600 and set up in 10 ml tubes, shaking 200 r.p.m. at 30°C. Fermentation media contained 1 × ZRMG, 2% arabinose, and spectinomycin for control conditions. To test the effects of hydrolysate on fermentation, the media was supplemented with 8% (v/v) batch 2 hydrolysate. Samples were collected every 3 h for 50 h. At each time point, OD600 was measured and supernatant was removed for HPLC analysis of glucose and ethanol concentrations. Vials were stored at 4°C until processed. For HPLC analysis of the supernatant, samples were analyzed at 55°C on a Rezex RFQ fast acid column (Phenomenex, Torrance, CA) and compounds were eluted with 0.005 M sulfuric acid at a flow rate of 1 ml/min. Ethanol and glucose were detected by RID. Specific ethanol productivity (g/l/h/OD600) was calculated during the time period (t1 to t2) when glucose was being consumed, using the formula (ethanolt2−ethanolt1)/(t2−t1)/(OD600t1+OD600t2)/2). The following intervals were used: WT+pJS71 (0–15 h), WT+pJS71 in hydrolysate (0–27 h), WT+Pbad-ZMO1875 in hydrolysate (0–18 h). Fermentation experiments for WT+pJS71 and WT+Pbad-ZMO1875 were performed using four biological replicates and average productivity values are reported. Fermentation experiments using WT+Pbad-ZMO1722, WT+Pbad-ZMO0760, and WT+Pbad-ZMO0100) were performed in duplicate.
Supplementary Material
Supplementary figures S1-18, Supplementary tables S1-8
Composition of plant hydrolysates and synthetic hydrolysate mixtures determined by GC/MS and LC-RID.
List of mutants in our Z. mobilis barcoded transposon pools.
List of putative essential genes in Z. mobilis.
List of 54 amino acid synthesis genes in Z. mobilis.
List of experiments used to compile fitness datasets for Z. mobilis and S. cerevisiae.
Strains, Plasmids, Primers used in this study.
Linear regression model results.
Transposon stability results.
Complete gene fitness datasets for Z. mobilis and S. cerevisiae.
Gene fitness data for 44 Z. mobilis hydrolysate tolerance genes identified in this study.
Gene fitness data for 99 S. cerevisiae tolerance genes identified in this study.
Acknowledgments
We thank the Stanford Genome Technology Center for help with sequencing and for the yeast deletion collection. We thank Rebecca Arundale (UIUC) for providing the samples of miscanthus and switchgrass. This work was funded by the Energy Biosciences Institute grant OO7G02.
Author contributions: JMS, JSM, KMW, AMD, and DRT generated the transposon library and performed pool experiments. DL performed single mutant, overexpression, complementation, and fermentation studies. MNP analyzed fitness data and modeled hydrolysate fitness. SB, ABI, and VDM generated and analyzed plant hydrolysates and performed glucose and ethanol measurements. CHW and PH developed the arabinose inducible expression plasmid. TH and APA managed the project and helped plan experiments. JMS, DL, MNP, SB, and APA wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Agrawal M, Wang Y, Chen RR (2012) Engineering efficient xylose metabolism into an acetic acid-tolerant Zymomonas mobilis strain by introducing adaptation-induced mutations. Biotechnol Lett 34: 1825–1832 [DOI] [PubMed] [Google Scholar]
- Alic N, Higgins VJ, Dawes IW (2001) Identification of a Saccharomyces cerevisiae gene that is required for G1 arrest in response to the lipid oxidation product linoleic acid hydroperoxide. Mol Biol Cell 12: 1801–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SA, Clark W, McCaffery JM, Cai Z, Lanctot A, Slininger PJ, Liu ZL, Gorsich SW (2010) Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol Biofuels 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida JR, Modig T, Petersson A, Hahn-Hagerdal B, Lidén G, Gorwa-Grauslund M-F (2007) Increased tolerance and conversion of inhibitors in lignocellulosic hydrolysates by Saccharomyces cerevisiae. J Chem Technol Biotechnol 82: 340–349 [Google Scholar]
- Amin G, Van den Eynde E, Verachtert H (1983) Determination of by-products formed during the ethanolic fermentation, using batch and immobilized cell systems of Zymomonas mobilis and Saccharomyces bayanus. Eur J Appl Microbiol Biotechnol 18: 1–5 [Google Scholar]
- Angert ER (2012) DNA replication and genomic architecture of very large bacteria. Annu Rev Microbiol 66: 197–212 [DOI] [PubMed] [Google Scholar]
- Bitoun JP, Wu G, Ding H (2008) Escherichia coli FtnA acts as an iron buffer for re-assembly of iron–sulfur clusters in response to hydrogen peroxide stress. BioMetals 21: 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimi P, Hitz W (2012) Xylose utilizing Zymomonas mobilis with improved ethanol production in biomass hydrolysate medium. WO Patent 2012/082711 [Google Scholar]
- Cameron JM, Janer A, Levandovsky V, MacKay N, Rouault TA, Tong WH, Ogilvie I, Shoubridge EA, Robinson BH (2011) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-Oxoacid dehydrogenase enzymes. Am J Hum Genet 89: 486–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoensuk K, Irie A, Lertwattanasakul N, Sootsuwan K, Thanonkeo P, Yamada M (2011) Physiological importance of cytochrome peroxidase in ethanologenic thermotolerant Zymomonas mobilis. J Mol Microbiol Biotechnol 20: 70–82 [DOI] [PubMed] [Google Scholar]
- Chen M (2010) irrE, an exogenous gene from Deinococcus radiodurans, improves the growth of and ethanol production by a Zymomonas mobilis strain under ethanol and acid stresses. J Microbiol Biotechnol 20: 1156–1162 [DOI] [PubMed] [Google Scholar]
- Clark TA, Mackie KL (1984) Fermentation inhibitors in wood hydrolysates derived from the softwood Pinus radiata. J Chem Tech Biotechnol 34: 101–110 [Google Scholar]
- Cokol M, Chua HN, Tasan M, Mutlu B, Weinstein ZB, Suzuki Y, Nergiz ME, Costanzo M, Baryshnikova A, Giaever G, Nislow C, Myers CL, Andrews BJ, Boone C, Roth FP (2011) Systematic exploration of synergistic drug pairs. Mol Syst Biol 7: 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway T, Sewell GW, Ingram LO (1987) Glyceraldehyde-3-phosphate dehydrogenase gene from Zymomonas mobilis: cloning, sequencing, and identification of promoter region. J Bacteriol 169: 5653–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer A, Price MN, Wetmore KM, Shao W, Baumohl JK, Xu Z, Nguyen M, Tamse R, Davis RW, Arkin AP (2011) Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet 7: e1002385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djaman O (2004) Repair of oxidized iron-sulfur clusters in Escherichia coli. J Biol Chem 279: 44590–44599 [DOI] [PubMed] [Google Scholar]
- Drobna E, Gazdag Z, Culakova H, Dzugasova V, Gbelska Y, Pesti M, Subik J (2012) Overexpression of the YAP1, PDE2, and STB3 genes enhances the tolerance of yeast to oxidative stress induced by 7-chlorotetrazolo[5,1-c]benzo[1,2,4]triazine. FEMS Yeast Res 8: 958–968 [DOI] [PubMed] [Google Scholar]
- Freedberg WB, Kistler WS, Lin EC (1971) Lethal synthesis of methylglyoxal by Escherichia coli during unregulated glycerol metabolism. J Bacteriol 108: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitomi K, Sanda T, Hasunuma T, Kondo A (2012) Deletion of the PHO13 gene in Saccharomyces cerevisiae improves ethanol production from lignocellulosic hydrolysate in the presence of acetic and formic acids, and furfural. Bioresour Technol 111: 161–166 [DOI] [PubMed] [Google Scholar]
- Gao L, Xia L (2012) Sm-like protein enhanced tolerance of recombinant Saccharomyces cerevisiae to inhibitors in hemicellulosic hydrolysate. Bioresour Technol 124: 504–507 [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW (2004) Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci USA 101: 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BR, Lawrence SJ, Boulton CA, Box WG, Graham NS, Linforth RST, Smart KA (2008) The oxidative stress response of a lager brewing yeast strain during industrial propagation and fermentation. FEMS Yeast Res 8: 574–585 [DOI] [PubMed] [Google Scholar]
- Gibson BR, Lawrence SJ, Leclaire JPR, Powell CD, Smart KA (2007) Yeast responses to stresses associated with industrial brewery handling. FEMS Microbiol Rev 31: 535–569 [DOI] [PubMed] [Google Scholar]
- Goodman AE, Rogers PL, Skotnicki ML (1982) Minimal medium for isolation of auxotrophic Zymomonas mutants. Appl Environ Microbiol 44: 496–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsich SW, Dien BS, Nichols NN, Slininger PJ, Liu ZL, Skory CD (2006) Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 71: 339–349 [DOI] [PubMed] [Google Scholar]
- Griese M, Lange C, Soppa J (2011) Ploidy in cyanobacteria. FEMS Microbiol Lett 323: 124–131 [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heer D, Sauer U (2008) Identification of furfural as a key toxin in lignocellulosic hydrolysates and evolution of a tolerant yeast strain. Microb Biotechnol 1: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J, Nelson E, Tilman D, Polasky S, Tiffany D (2006) Environmental, economic, and energetic costs and benefits of biodiesel and ethanol biofuels. Proc Natl Acad Sci USA 103: 11206–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer ME, Ericson E, Davis RW, Nislow C, Koller D, Giaever G (2010) Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol 11: R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueso G, Aparicio-Sanchis R, Montesinos C, Lorenz S, Murguía JR, Serrano R (2012) A novel role for protein kinase Gcn2 in yeast tolerance to intracellular acid stress. Biochem J 441: 255–264 [DOI] [PubMed] [Google Scholar]
- Huynen MA, Spronk CAEM, Gabaldón T, Snel B (2005) Combining data from genomes, Y2H and 3D structure indicates that BolA is a reductase interacting with a glutaredoxin. FEBS Lett 579: 591–596 [DOI] [PubMed] [Google Scholar]
- Imae Y, Fukasawa T (1970) Regional replication of the bacterial chromosome induced by derepression of prophage lambda. J Mol Biol 54: 585–597 [DOI] [PubMed] [Google Scholar]
- Jansen G, Lee AY, Epp E, Fredette Ael, Surprenant J, Harcus D, Scott M, Tan E, Nishimura T, Whiteway M, Hallett M, Thomas DY (2009) Chemogenomic profiling predicts antifungal synergies. Mol Syst Biol 5: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakody LN, Hayashi N, Kitagaki H (2011) Identification of glycolaldehyde as the key inhibitor of bioethanol fermentation by yeast and genome-wide analysis of its toxicity. Biotechnol Lett 33: 285–292 [DOI] [PubMed] [Google Scholar]
- Jeppsson M, Johansson B, Jensen PR, Hahn-Hägerdal B, Gorwa-Grauslund MF (2003) The level of glucose-6-phosphate dehydrogenase activity strongly influences xylose fermentation and inhibitor sensitivity in recombinant Saccharomyces cerevisiae strains. Yeast 20: 1263–1272 [DOI] [PubMed] [Google Scholar]
- Kawahata M, Masaki K, Fujii T, Iefuji H (2006) Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res 6: 924–936 [DOI] [PubMed] [Google Scholar]
- Keller FA, Bates D, Ruiz R, Nguyen Q (1998) Yeast adaptation on softwood prehydrolysate. Appl Biochem Biotechnol 70-72: 137–148 [DOI] [PubMed] [Google Scholar]
- Klein-Marcuschamer D, Oleskowicz-Popiel P, Simmons BA, Blanch HW (2010) Technoeconomic analysis of biofuels: a wiki-based platform for lignocellulosic biorefineries. Biomass Bioenergy 34: 1914–1921 [Google Scholar]
- Klein-Marcuschamer D, Oleskowicz-Popiel P, Simmons BA, Blanch HW (2011) The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnol Bioeng 109: 1083–1087 [DOI] [PubMed] [Google Scholar]
- Klinke H, Olsson L, Thomsen A, Ahring B (2003) Potential inhibitors from wet oxidation of wheat straw and their effect on ethanol production of Saccharomyces cerevisiae: wet oxidation and fermentation by yeast. Biotechnol Bioeng 81: 738–747 [DOI] [PubMed] [Google Scholar]
- Klinke HB, Thomsen AB, Ahring BK (2004) Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl Microbiol Biotechnol 66: 10–26 [DOI] [PubMed] [Google Scholar]
- Koch B, Nybroe O (2006) Initial characterization of a bolA homologue from Pseudomonas fluorescens indicates different roles for BolA-like proteins in P. fluorescens and Escherichia coli. FEMS Microbiol Lett 262: 48–56 [DOI] [PubMed] [Google Scholar]
- Koppram R, Albers E, Olsson L (2012) Evolutionary engineering strategies to enhance tolerance of xylose utilizing recombinant yeast to inhibitors derived from spruce biomass. Biotechnol Biofuels 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumanovics A, Chen OS, Li L, Bagley D, Adkins EM, Lin H, Dingra NN, Outten CE, Keller G, Winge D, Ward DM, Kaplan J (2008) Identification of FRA1 and FRA2 as genes involved in regulating the yeast iron regulon in response to decreased mitochondrial iron-sulfur cluster synthesis. J Biol Chem 283: 10276–10286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Murthy GS (2011) Impact of pretreatment and downstream processing technologies on economics and energy in cellulosic ethanol production. Biotechnol Biofuels 4: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langridge GC, Phan M-D, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK (2009) Simultaneous assay of every Salmonella typhi gene using one million transposon mutants. Genome Res 19: 2308–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen RA, Wilson MM, Guss AM, Metcalf WW (2002) Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch Microbiol 178: 193–201 [DOI] [PubMed] [Google Scholar]
- Larsson S, Nilvebrant NO, Jonsson LJ (2001) Effect of overexpression of Saccharomyces cerevisiae Pad1p on the resistance to phenylacrylic acids and lignocellulose hydrolysates under aerobic and oxygen-limited conditions. Appl Microbiol Biotechnol 57: 167–174 [DOI] [PubMed] [Google Scholar]
- Li H, Outten CE (2012) Monothiol CGFS glutaredoxins and BolA-like proteins: [2Fe-2S] binding partners in iron homeostasis. Biochemistry 51: 4377–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZL (2011) Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl Microbiol Biotechnol 90: 809–825 [DOI] [PubMed] [Google Scholar]
- Ma M, Liu ZL (2010) Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genomics 11: 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín C, Almazán O, Marcet M, Jönsson LJ (2007a) A study of three strategies for improving the fermentability of sugarcane bagasse hydrolysates for fuel ethanol production. Int Sugar J 109: 33–39 [Google Scholar]
- Martín C, Marcet M, Almazán O, Jönsson LJ (2007b) Adaptation of a recombinant xylose-utilizing Saccharomyces cerevisiae strain to a sugarcane bagasse hydrolysate with high content of fermentation inhibitors. Bioresour Technol 98: 1767–1773 [DOI] [PubMed] [Google Scholar]
- Masters CI, Smith MD, Gutman PD, Minton KW (1991) Heterozygosity and instability of amplified chromosomal insertions in the radioresistant bacterium Deinococcus radiodurans. J Bacteriol 173: 6110–6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergaert P, Uchiumi T, Alunni B, Evanno G, Cheron A, Catrice O, Mausset A-E, Barloy-Hubler F, Galibert F, Kondorosi A, Kondorosi E (2006) Eukaryotic control on bacterial cell cycle and differentiation in the Rhizobium-legume symbiosis. Proc Natl Acad Sci USA 103: 5230–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, Jarboe LR, Turner PC, Pharkya P, Yomano LP, York SW, Nunn D, Shanmugam KT, Ingram LO (2009a) Furfural inhibits growth by limiting sulfur assimilation in ethanologenic Escherichia coli strain LY180. Appl Environ Microbiol 75: 6132–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, Jarboe LR, Yomano LP, York SW, Shanmugam KT, Ingram LO (2009b) Silencing of NADPH-dependent oxidoreductase genes (yqhD and dkgA) in furfural-resistant ethanologenic Escherichia coli. Appl Environ Microbiol 75: 4315–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, Turner PC, Jarboe LR, Ingram LO (2010) Genetic changes that increase 5-hydroxymethyl furfural resistance in ethanol-producing Escherichia coli LY180. Biotechnol Lett 32: 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Imlay J (2012) Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch Biochem Biophys 525: 145–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Rios E, de la Rosa-Morales F, Mendoza-Hernandez G, Rodriguez-Zavala JS, Celis H, Zarco-Zavala M, Garcia-Trejo JJ (2010) A novel 11-kDa inhibitory subunit in the F1F0 ATP synthase of Paracoccus denitrificans and related alpha-proteobacteria. FASEB J 24: 599–608 [DOI] [PubMed] [Google Scholar]
- Nakata T, Miyafuji H, Saka S (2006) Bioethanol from cellulose with supercritical water treatment followed by enzymatic hydrolysis. Appl Biochem Biotechnol 129-132: 476–485 [DOI] [PubMed] [Google Scholar]
- Nieves IU, Geddes CC, Miller EN, Mullinnix MT, Hoffman RW, Fu Z, Tong Z, Ingram LO (2011) Effect of reduced sulfur compounds on the fermentation of phosphoric acid pretreated sugarcane bagasse by ethanologenic Escherichia coli. Bioresour Technol 102: 5145–5152 [DOI] [PubMed] [Google Scholar]
- Nilvebrant N-O, Reimann A, Larsson S, Jönsson LJ (2001) Detoxification of lignocellulose hydrolysates with ion-exchange resins. Appl Biochem Biotechnol 91-93: 35–50 [DOI] [PubMed] [Google Scholar]
- Nisamedtinov I, Kevvai K, Orumets K, Arike L, Sarand I, Korhola M, Paalme T (2011) Metabolic changes underlying the higher accumulation of glutathione in Saccharomyces cerevisiae mutants. Appl Microbiol Biotechnol 89: 1029–1037 [DOI] [PubMed] [Google Scholar]
- Oh J, Fung E, Price MN, Dehal PS, Davis RW, Giaever G, Nislow C, Arkin AP, Deutschbauer A (2010) A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic Acids Res 38: e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani N, Tomita M, Itaya M (2010) An extreme thermophile, Thermus thermophilus, is a polyploid bacterium. J Bacteriol 192: 5499–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva JM, Ballesteros I, Negro MJ, Manzanares P, Cabañas A, Ballesteros M (2004) Effect of binary combinations of selected toxic compounds on growth and fermentation of Kluyveromyces marxianus. Biotechnol Prog 20: 715–720 [DOI] [PubMed] [Google Scholar]
- Ozyamak E, Black SS, Walker CA, Maclean MJ, Bartlett W, Miller S, Booth IR (2010) The critical role of S-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol Microbiol 78: 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist E, Grage H, Meinander NQ, Hahn-Hägerdal B (1999) Main and interaction effects of acetic acid, furfural, and p-hydroxybenzoic acid on growth and ethanol productivity of yeasts. Biotechnol Bioeng 63: 46–55 [DOI] [PubMed] [Google Scholar]
- Palmqvist E, Hahn-Hägerdal B (2000a) Fermentation of lignocellulosic hydrolysates. II: Inhibitors and mechanisms of inhibition. Bioresour Technol 74: 25–33 [Google Scholar]
- Palmqvist E, Hahn-Hägerdal B (2000b) Fermentation of lignocellulosic hydrolysates. I: inhibition and detoxification. Bioresour Technol 74: 17–24 [Google Scholar]
- Parawira W, Tekere M (2011) Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: review. Crit Rev Biotechnol 31: 20–31 [DOI] [PubMed] [Google Scholar]
- Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS (2005) Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24: 79–88 [DOI] [PubMed] [Google Scholar]
- Pereira FB, Guimarães PM, Gomes DG, Mira NP, Teixeira MC, Sá-Correia I, Domingues L (2011) Identification of candidate genes for yeast engineering to improve bioethanol production in very high gravity and lignocellulosic biomass industrial fermentations. Biotechnol Biofuels 4: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson A, Almeida J, Modig T, Karhumaa K (2006) A 5-hydroxymethyl furfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast 23: 455–464 [DOI] [PubMed] [Google Scholar]
- Pierce SE, Fung EL, Jaramillo DF, Chu AM, Davis RW, Nislow C, Giaever G (2006) A unique and universal molecular barcode array. Nat Methods 3: 601–603 [DOI] [PubMed] [Google Scholar]
- Roe AJ, O’Byrne C, McLaggan D, Booth IR (2002) Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiology 148: 2215–2222 [DOI] [PubMed] [Google Scholar]
- Rouhier N, Couturier J, Johnson MK, Jacquot J-P (2010) Glutaredoxins: roles in iron homeostasis. Trends Biochem Sci 35: 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda T, Hasunuma T, Matsuda F, Kondo A (2011) Repeated-batch fermentation of lignocellulosic hydrolysate to ethanol using a hybrid Saccharomyces cerevisiae strain metabolically engineered for tolerance to acetic and formic acids. Bioresour Technol 102: 7917–7924 [DOI] [PubMed] [Google Scholar]
- Sandoval NR, Kim JYH, Glebes TY, Reeder PJ, Aucoin HR, Warner JR, Gill RT (2012) Strategy for directing combinatorial genome engineering in Escherichia coli. Proc Natl Acad Sci USA 109: 10540–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval NR, Mills TY, Zhang M, Gill RT (2011) Elucidating acetate tolerance in E. coli using a genome-wide approach. Metab Eng 13: 214–224 [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA 98: 12712–12717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbach MS, Keating DH, Tremaine M, Marner WD, Zhang Y, Bothfeld W, Higbee A, Grass JA, Cotten C, Reed JL, da Costa Sousa L, Jin M, Balan V, Ellinger J, Dale B, Kiley PJ, Landick R (2012) Complex physiology and compound stress responses during fermentation of alkali-pretreated corn stover hydrolysate by an Escherichia coli ethanologen. Appl Environ Microbiol 78: 3442–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakamuri P, Zhang B, Johnson MK (2012) Monothiol glutaredoxins function in storing and transporting [Fe2S2] clusters assembled on IscU scaffold proteins. J Am Chem Soc 134: 15213–15216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerker JM, Shapiro L (2000) Identification and cell cycle control of a novel pilus system in Caulobacter crescentus. EMBO J 19: 3223–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, Roth FP, Giaever G, Nislow C (2009) Quantitative phenotyping via deep barcode sequencing. Genome Res 19: 1836–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville C, Youngs H, Taylor C, Davis SC, Long SP (2010) Feedstocks for Lignocellulosic Biofuels. Science 329: 790–792 [DOI] [PubMed] [Google Scholar]
- Sommer MO, Church GM, Dantas G (2010) A functional metagenomic approach for expanding the synthetic biology toolbox for biomass conversion. Mol Syst Biol 6: 360 ; DOI: 10.1038/msb.2010.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoutenburg RM, Perrotta JA, Nakas JP (2011) Overcoming inhibitors in a hemicellulosic hydrolysate: improving fermentability by feedstock detoxification and adaptation of Pichia stipitis. J Ind Microbiol Biotechnol 38: 1939–1945 [DOI] [PubMed] [Google Scholar]
- Sundström L, Larsson S, Jönsson LJ (2010) Identification of Saccharomyces cerevisiae genes involved in the resistance to phenolic fermentation inhibitors. Appl Biochem Biotechnol 161: 106–115 [DOI] [PubMed] [Google Scholar]
- Tao L, Aden A, Elander RT, Pallapolu VR, Lee YY, Garlock RJ, Balan V, Dale BE, Kim Y, Mosier NS, Ladisch MR, Falls M, Holtzapple MT, Sierra R, Shi J, Ebrik MA, Redmond T, Yang B, Wyman CE, Hames B et al. (2011) Process and technoeconomic analysis of leading pretreatment technologies for lignocellulosic ethanol production using switchgrass. Bioresour Technol 102: 11105–11114 [DOI] [PubMed] [Google Scholar]
- Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344: 109–116 [PMC free article] [PubMed] [Google Scholar]
- Turnbull AL, Surette MG (2010) Cysteine biosynthesis, oxidative stress and antibiotic resistance in Salmonella typhimurium. Res Microbiol 161: 643–650 [DOI] [PubMed] [Google Scholar]
- US Department of Energy. (2011) US Billion-Ton Update: Biomass Supply for a Bioenergy and Bioproducts Industry. In: Perlack RD, Stokes BJ (Leads), ORNL/TM-2011/224. Oak Ridge National Laboratory: Oak Ridge, TN. 227 p
- Van Opijnen T, Bodi KL, Camilli A (2009) Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6: 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veal EA, Ross SJ, Malakasi P, Peacock E, Morgan BA (2003) Ybp1 is required for the hydrogen peroxide-induced oxidation of the Yap1 transcription factor. J Biol Chem 278: 30896–30904 [DOI] [PubMed] [Google Scholar]
- Vicari KJ, Tallam SS, Shatova T, Koh KJ, Scarlata CJ, Humbird D, Wolfrum EJ, Beckham GT (2012) Uncertainty in techno-economic estimates of cellulosic ethanol production due to experimental measurement uncertainty. Biotechnol Biofuels 5: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J, Reeder P, Karimpour-Fard A, Woodruff L, Gill R (2010) Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol 28: 856–862 [DOI] [PubMed] [Google Scholar]
- Wheals A, Basso L, Alves D (1999) Fuel ethanol after 25 years. Trends Biotechnol 17: 482–487 [DOI] [PubMed] [Google Scholar]
- Willems P, Wanschers B, Esseling J, Szklarczyk R, Kudla U, Duarte I, Forkink M, Nooteboom M, Swarts H, Gloerich J, Nijtmans L, Koopman WJH, Huynen M (2012) BOLA1 is an aerobic protein that prevents mitochondrial morphology changes induced by glutathione depletion. Antioxid Redox Signal 18: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood K, Nishida S, Sontag ED, Cluzel P (2012) Mechanism-independent method for predicting response to multidrug combinations in bacteria. Proc Natl Acad Sci USA 109: 12254–12259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman CE, Dale BE, Elander RT, Holtzapple M, Ladisch MR, Lee YY (2005) Coordinated development of leading biomass pretreatment technologies. Bioresour Technol 96: 1959–1966 [DOI] [PubMed] [Google Scholar]
- Yang S, Pappas KM, Hauser LJ, Land ML, Chen G-L, Hurst GB, Pan C, Kouvelis VN, Typas MA, Pelletier DA, Klingeman DM, Chang Y-J, Samatova NF, Brown SD (2009a) Improved genome annotation for Zymomonas mobilis. Nat Biotechnol 27: 893–894 [DOI] [PubMed] [Google Scholar]
- Yang S, Tschaplinski TJ, Engle NL, Carroll SL, Martin SL, Davison BH, Palumbo AV, Rodriguez M Jr, Brown SD (2009b) Transcriptomic and metabolomic profiling of Zymomonas mobilis during aerobic and anaerobic fermentations. BMC Genomics 10: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Land ML, Klingeman DM, Pelletier DA, Lu T-YS, Martin SL, Guo H-B, Smith JC, Brown SD (2010a) Paradigm for industrial strain improvement identifies sodium acetate tolerance loci in Zymomonas mobilis and Saccharomyces cerevisiae. Proc Natl Acad Sci USA 107: 10395–10400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Pelletier DA, Lu T-YS, Brown SD (2010b) The Zymomonas mobilis regulator hfq contributes to tolerance against multiple lignocellulosic pretreatment inhibitors. BMC Microbiol 10: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W (2002) Repair of nitric oxide-modified ferredoxin [2Fe-2S] cluster by cysteine desulfurase (IscS). J Biol Chem 277: 12868–12873 [DOI] [PubMed] [Google Scholar]
- Youngs H, Somerville C (2012) Development of feedstocks for cellulosic biofuels. F1000 Biol Rep 4: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaldivar J, Ingram L (1999) Effect of organic acids on the growth and fermentation of ethanologenic Escherichia coli LY01. Biotechnol Bioeng 66: 203–210 [DOI] [PubMed] [Google Scholar]
- Zaldivar J, Martinez A, Ingram L (1999) Effect of selected aldehydes on the growth and fermentation of ethanologenic Escherichia coli. Biotechnol Bioeng 65: 24–33 [DOI] [PubMed] [Google Scholar]
- Zaldivar J, Martinez A, Ingram LO (2000) Effect of alcohol compounds found in hemicellulose hydrolysate on the growth and fermentation of ethanologenic Escherichia coli. Biotechnol Bioeng 68: 524–530 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1-18, Supplementary tables S1-8
Composition of plant hydrolysates and synthetic hydrolysate mixtures determined by GC/MS and LC-RID.
List of mutants in our Z. mobilis barcoded transposon pools.
List of putative essential genes in Z. mobilis.
List of 54 amino acid synthesis genes in Z. mobilis.
List of experiments used to compile fitness datasets for Z. mobilis and S. cerevisiae.
Strains, Plasmids, Primers used in this study.
Linear regression model results.
Transposon stability results.
Complete gene fitness datasets for Z. mobilis and S. cerevisiae.
Gene fitness data for 44 Z. mobilis hydrolysate tolerance genes identified in this study.
Gene fitness data for 99 S. cerevisiae tolerance genes identified in this study.