

Abstract
The genetic code is degenerate; thus, protein evolution does not uniquely determine the coding sequence. One of the puzzles in evolutionary genetics is therefore to uncover evolutionary driving forces that result in specific codon choice. In many bacteria, the first 5–10 codons of protein-coding genes are often codons that are less frequently used in the rest of the genome, an effect that has been argued to arise from selection for slowed early elongation to reduce ribosome traffic jams. However, genome analysis across many species has demonstrated that the region shows reduced mRNA folding consistent with pressure for efficient translation initiation. This raises the possibility that unusual codon usage is a side effect of selection for reduced mRNA structure. Here we discriminate between these two competing hypotheses, and show that in bacteria selection favours codons that reduce mRNA folding around the translation start, regardless of whether these codons are frequent or rare. Experiments confirm that primarily mRNA structure, and not codon usage, at the beginning of genes determines the translation rate.
Keywords: codon usage, mRNA structure, translation
Introduction
Which evolutionary constraints shape the genome of an organism? This question has been puzzling researchers for decades (Nirenberg et al, 1966; Sharp and Li, 1987; Trifonov, 1987; Herzel and Grosse, 1997; Karlin, 1998; Itzkovitz and Alon, 2007). Clearly, protein-coding sequences and the segments regulating their expression are the main determinants of genomic sequences in bacteria. The genetic code is degenerate, allowing for diversity to encode one and the same amino acid. Except for tryptophan and methionine, amino acids are encoded by two to six different codons (Nirenberg et al, 1966). Therefore, it is important to understand which mechanisms shape codon choice.
On a genome-wide scale some codons are preferred to others, termed codon usage bias (Grantham et al, 1980; Sharp and Li, 1987). Each genome shows a specific bias, although the origin of it remains unclear (Hershberg and Petrov, 2008). Driving forces for preferentially selecting specific codons (Plotkin and Kudla, 2011) could be the efficiency and accuracy of translation (Trifonov, 1987; Drummond and Wilke, 2008; Cannarozzi et al, 2010; Warnecke and Hurst, 2010; Tuller et al, 2010a; Chu et al, 2011; Gingold and Pilpel, 2011; Shah and Gilchrist, 2011; Chu and von der Haar, 2012), GC content (Lynn et al, 2002), environmental factors (Lynn et al, 2002; Singer and Hickey, 2003), or DNA folding (Herzel et al, 1999). The species-specific codon bias mirrors the amount of the cognate transfer RNAs (tRNAs), i.e., highly used triplets are read by high-abundance tRNAs and rarely used codons pair to low-abundance tRNA (Ikemura, 1981; Dong et al, 1996). Triplets read by major tRNAs are translated faster than the codons read by minor tRNAs (Zhang and Ignatova, 2009). Highly expressed genes thus display a stronger bias towards usage of abundant codons reflecting tRNA availability (Sharp and Li, 1987; Kudla et al, 2009; Tuller et al, 2010b).
Within each gene, rare codons are far from being randomly distributed along the coding mRNA sequences; they tend to cluster and transiently attenuate ribosomal traffic, an effect suggested to synchronize translation with cotranslational folding of multidomain proteins (Komar, 2009; Zhang et al, 2009). Furthermore, rare codons may exert regulatory function under starvation (Elf et al, 2003; Zhang et al, 2010), but may also cause misfolding in highly expressed genes, and thus are avoided (Warnecke and Hurst, 2010).
Genome-wide analysis revealed that codon usage of the protein-coding sequence differs between the beginning of the gene and the rest of the mRNA sequences (Eyre-Walker and Bulmer, 1993; Tuller et al, 2010a). This suggests that there are different evolutionary pressures on codon usage at the beginning of genes than in the rest of the genome. An intriguing hypothesis is that these codons have been selected to reduce elongation speed at the beginning of genes (Tuller et al, 2010a). Such a ‘ramp’ in elongation speed could reduce the likelihood of ribosomal traffic jams along the mRNA, and may be advantageous as such traffic jams may result in premature termination or in protein bursts (Dobrzynski and Bruggeman, 2009). However, other studies have demonstrated that the same region is selected for reduced mRNA folding (Gu et al, 2010; Keller et al, 2012), required for efficient translation initiation (McCarthy and Bokelmann, 1988; de Smit and van Duin, 1990; Kudla et al, 2009; Kertesz et al, 2010).
This finding raises the intriguing possibility that the abnormal codon usage at the beginning of genes is not selected for to reduce elongation speed (‘ramp hypothesis’), but is a side effect of selection for reduced stability (‘structure hypothesis’). To address this fundamental question we analysed 414 bacterial genomes. We found that deviation of codon usage strongly correlates with the suppression of mRNA secondary structure at gene start. Furthermore, we found that rare codons are only selected if they are AU rich, whereas abundant codons are repressed if they are GC rich. Thus, our genome analysis shows that rare codons are not selected because they are rare, but to weaken the mRNA structure. The hypothesis is further corroborated by experimental measurements of the translational efficiency of two Escherichia coli genes with various synonymous starting sequences, underpinning the functional relevance.
Results
Unusual codon usage and reduced folding around the translation start site
To analyse codon usage and mRNA folding energy around the translation start site, we collected a panel of 414 bacterial genomes from the BioCyc database (Keseler et al, 2011). We aligned protein-coding genes with respect to the translation start, and removed the first genes of transcriptional units (TUs), to avoid spurious signals due to misannotated untranslated regions (UTRs). We then used Kullback–Leibler divergence (KLD) to calculate how strongly codon frequency deviates at each position from the overall codon usage (for details, see Materials and methods). For example, in E. coli the first eight codons show higher values of KLD than a null model (Figure 1A, and Supplementary Figure S1a and b), indicating an unusual codon usage in that region, which is consistent with previous findings (Tuller et al, 2010a).
Figure 1.

Unusual codon usage and suppression of mRNA structure at the gene start in bacteria. (A) In E. coli, the frequency of synonymous codons within the first eight codons after translation start deviates from the global codon usage in the genome, as quantified by the KLD (solid line). A null model with SSC (dashed line) shows that the bias due to finite size sampling is significantly lower. (B) Folding energy of E. coli mRNA sequences calculated within a sliding window of 39 nts shows a maximum at translation start site, indicating the suppression of mRNA secondary structure around the start codon. Average folding energy is shown as a solid line, surrounded by the interquartile range in grey. (C) Suppression of mRNA structure around the start codon is largely determined by the global GC content. Insets from bottom to top correspond to KLD profiles calculated for the genomes of Thermoanaerobacter tengcongensis, Bacillus subtilis, E. coli and Aeromonas hydrophila, respectively. (D) Average deviation of codon usage (ΔCU) of the first five codons correlates with the GC content of the organism. The insets are ordered as in C. (E) Deviation from the global codon usage (ΔCU) and suppression of mRNA structure (ΔG) around the start codon are strongly correlated (414 bacterial genomes, correlation coefficient r=0.93).
In addition, we calculated folding energy of 39-nucleotide (nt) long stretches around each position (Hofacker, 2009), which corresponds to the length of a ribosome binding site (Beyer et al, 1994). This allowed us to quantify typical mRNA secondary structure in a position-specific manner. Typical folding profiles showed reduced mRNA stability and less-paired nucleotides around the translation start site (Figure 1B, and Supplementary Figures S1c, d and S2 for E. coli), similar to previous observations (Kudla et al, 2009; Gu et al, 2010).
To compare the deviation of codon usage and reduced mRNA folding between different organisms, we defined two scores: average deviation in the codon usage within the first five codons (ΔCU) and change in folding energy at translation start when compared with typical mRNA folding elsewhere in coding regions (ΔG, see Materials and methods). Clearly, both scores varied strongly among the organisms. The correlation between these scores with genomic features confirmed that suppression of the mRNA structure occurs primarily in GC-rich organisms (Figure 1C; Gu et al, 2010). This is consistent with a stronger pressure to reduce the secondary structure propensity in GC-rich organisms, where mRNA sequences tend to form more stable secondary structures due to the GC content. More surprisingly, we also found unusual codon usage primarily in GC-rich organisms (Figure 1D). Rare codons typically are more slowly elongated, irrespective of GC content (compare Supplementary Figure S3 for an analysis of the tAI scores, a commonly used measure for elongation speed based on tRNA gene copy number (dos Reis et al, 2004)). This suggests that attenuation of elongation occurs primarily in GC-rich organisms. According to the ‘ramp hypothesis’, this would imply that different bacteria would be subject to different pressures to prevent ribosome jamming, and this pressure would be correlated with GC content. Alternatively, codon usage deviates as a side effect of another selective pressure. As the mRNA regions with unusual codon usage and suppressed mRNA structure overlap (Figures 1A and B, and Supplementary Figure S1), the structural constraints required for efficient initiation is one of the prime candidates to exert such a selective pressure. In line with this ‘structure hypothesis’, we find that genomes with suppressed structure show also deviations in codon usage, and the two scores correlate strongly across genomes (r=0.93; Figure 1E, and Supplementary Figures S4 and S5).
AU-rich codons are selected at the beginning of coding regions
Mechanistically, the choice of specific codons could suppress secondary structure by reducing the GC content at the beginning of genes. Indeed, the GC content in E. coli is strongly reduced within the first four to six codons (Figure 2A and Supplementary Figure S6a, dashed line). Furthermore, we observed a strong decrease in the GC content in the third base (GC3 content; Figure 2A and Supplementary Figure S6a, solid line). As synonymous codons differ mainly in their third base, this suggests a preferential selection of codons with A or U at the third position (AU3) to reduce folding propensity. A null model with randomly shuffled synonymous codons (SSC) within each gene (Figure 2A and Supplementary Figure S6a, dotted line) also shows a slight reduction in the GC3 content. This implies that amino acid selection is also biased towards higher AU3 content, albeit the major influence on GC3 is due to the preferential choice of AU3 codons. We also observed a general decrease in the GC content at the first and second nucleotide position (GC1 and GC2, respectively) within the first four to six codons (Figure 2B and Supplementary Figure S6b, solid lines). A similar trend is also detectable with the null models with conserved amino acid sequences (SSC), but almost vanishes if all codons are randomly shuffled (shuffled codons—SC). Taken together, these results suggest that amino acids encoded by AU-rich codons are chosen preferentially at the gene start (Figure 2B and Supplementary Figure S6b).
Figure 2.

GC content at the beginning of genes in E. coli. (A) GC content (dashed line) and GC3 content (solid line) of codons decrease at the beginning of genes inE. coli. Dotted line and grey area shows mean GC3 content±s.d., estimated from the null model (SSC). (B) GC1 and GC2 content are decreased at gene start (solid lines) when compared with a null model with SC. This is primarily due to the choice of amino acids, as the GC2 content is fully determined by the amino acid, and the null model with SSC (dotted line) shows only a small deviation for the GC1 content.
Properties of rare and abundant codons
The reduction of GC content at the beginning of genes might be a consequence of selection of slowly translated and consequently of rare codons. To investigate this, we analysed the properties of rare and abundant codons across many bacterial species. For each genome, we defined subsets of the 15 codons with lowest and highest abundance, and termed these the sets of rare and abundant codons, respectively. With this definition, the set of rare codons typically contained 5% of all codons of an organism, whereas on average 50% belonged to the set of abundant codons (Figure 3A). Intriguingly, we observed that the GC content strongly determines the overall codon distribution. In bacteria with a GC content of 0.5, the abundant codons were about fivefold more abundant in the genome than the rare codons. These differences were more pronounced in bacteria with more extreme GC content, with up to 100-fold difference in frequency (Figure 3B). This dependence is symmetric, i.e., GC-rich and AT-rich genomes show the same disparity.
Figure 3.

Rare and abundant codons. (A) Rare and abundant codons were defined for each genome as the 15 most rare and abundant codons, respectively. The overall frequency for both of these subsets are shown as a box plots for 414 genomes, with median total frequency of ∼0.06 and ∼0.53 for rare and abundant codons, respectively. (B) The ratio of total frequencies of abundant and rare codons is shown as a function of global GC content. Genomes with more extreme GC content show stronger bias in codon usage. (C, D) The normed frequency distribution of rare and abundant codons for different genomes is shown with their GC content indicated by different grey levels. GC content of the genomes increases from left to right. GC-rich organisms tend to have more AU-rich rare codons (C) and GC-rich abundant codons (D), with an inverse relation for AU-rich genomes. Note that E. coli with a GC content of about 0.5 is rather balanced. (E) Average±s.d. of global GC content are shown for organisms grouped according to the number of GC3 codons in the sets for rare and abundant codons. Higher global GC content implies increase of GC3 content for abundant codons and increase of AU3 content for rare codons.
Furthermore, the GC content of an organism is also reflected in the GC content of rare and abundant codons in a particular genome (Figures 3C and D). In AT-rich organisms, such as Thermoanaerobacter tengcongensis, rare codons are GC rich and abundant codons are AU rich. Similarly, in GC-rich organisms, such as Aeromonas hydrophila, AU-rich codons are enriched among the rare codons and abundant codons show high GC content. In contrast, bacteria with intermediate GC content show no particular selection for GC-rich or GC-poor codons. Thus, in organisms with GC content of around 0.5, rare codons are not per se biased towards a particular GC content, and a selective pressure for rare codons is unlikely to strongly influence the GC content. Most importantly, in the context of selection of alternative codons the GC3 content of codons in the sets of rare and abundant codons is primarily determined by the GC content of the organism (Figure 3E). In GC-rich organism, virtually all abundant codons show a G or C at the third position, whereas virtually all rare codons end with an A or a U.
Similarly, when we use tAI scores to classify codons according to their translation elongation speed, we find that in GC-rich organisms slow codons are AU3 rich, whereas fast codons are GC3 rich (Supplementary Figure S7).
Rare codons that reduce GC content are preferentially selected in E. coli
In E. coli, rare codons are enriched at the beginning of genes and abundant codons show a slight decrease (Figure 4 and Supplementary Figure S8). We next asked whether rare codons are selected per se or because of their GC content. We divided the sets of rare and abundant codons in two subsets: those with G or C at the third position (GC3) and those with an A or U (AU3). If there is a selective pressure to increase the frequency of rare codons downstream of the translation start to slow down early elongation (‘ramp hypothesis’), we expected an increase of rare codons irrespective of their GC3 content. In contrast, if codons are chosen in order to weaken the mRNA structure we expected an asymmetry between GC3 and AU3 codons. Within the set of rare codons enriched at the start of genes in E. coli, we detected a clear asymmetry between AU3 and GC3 codons in the set of rare codons (see Figure 4A and Supplementary Figure S8a). Rare AU3 codons are enriched at the beginning of genes, whereas rare GC3 codons are not effected in their frequency, i.e., they remain rare.
Figure 4.

Frequency of extreme codons at beginning of genes in E. coli. Rare codons (A) are enriched and abundant codons (B) are depleted at the beginning of genes in E. coli as shown by the change of total codon frequencies in the left panels. (A) Only rare codons with AU3 (right panel, solid line) are enriched at gene start, whereas rare GC3 codons (right panel, dashed line) are not enriched. (B) Frequency of abundant GC3 codons (right panel, dashed line) is strongly reduced, whereas abundant AU3 codons (right panel, solid line) are even more frequent at gene start. Greyed areas show the corresponding average total frequency±s.d. estimated from the null model SSC.
We also noted a strong asymmetry between GC3 and AU3 abundant codons: abundant GC3 codons are strongly depleted at the beginning of genes, whereas abundant AU3 codons are slightly enriched (Figure 4B and Supplementary Figure S8b). Interestingly, also the null model with SSC shows a drop of GC3 and an increase of AU3 codons (grey lines in Figure 4B and Supplementary Figure S8b). This implies that amino acids preferentially encoded by AU3 codons are enriched and those encoded by GC3 codons are avoided, suggesting a strong evolutionary pressure on GC content that manifests itself in the amino acid sequence. When we used tAI scores to classify codons as slow and fast, we observed an enrichment of slow and a partial depletion of fast codons (Supplementary Figures S9 and S10). Again, in both groups the enrichment depends on the third base of these codons: the frequency of AU3 codons increases, whereas usage of GC3 codons drops. Taken together, our analysis shows that in E. coli rare codons are selected because of their GC content and not because they are rare and slowly translated, supporting the ‘structure hypothesis’.
Widespread selection for low GC content at gene start
We next asked whether the asymmetric enrichment of GC-poor codons at the start of coding sequences is also detectable in other genomes. Bacteria largely differ in their GC content and, therefore, we first grouped them according to the GC3 content in their sets of rare or abundant codons. We observed that abundant codons are depleted only if the fraction of GC3 codons in that subset is above 50%, and rare codons are enriched when they have on average a AU3 content below 50% (Figure 5A and Supplementary Figure S11a). This finding further supports the ‘structure hypothesis’. Under the assumption that the ‘structure hypothesis’ dictates codon usage, we also expect that enrichment of rare codons depends on the GC content of the genome, because (i) mRNAs tend to fold more strongly in GC-rich organisms and (ii) the set of rare codons is enriched of AU-rich codons. Indeed, rare codons are only enriched and abundant codons depleted in genomes with a GC content >0.5 (Figure 5B and Supplementary Figure S11b). When we use tAI scores to classify codons with respect to their elongation speed, we find the same result: slow codons are only enriched at the beginning of genes if they are AU3 rich and fast codons are depleted if they are GC3 rich (Supplementary Figure S12).
Figure 5.

Enrichment of extreme codons and deviation of GC3 content in bacterial genomes. (A, B) Enrichment of codons was assessed by calculating Z-values of fold change for codon frequency at the beginning of genes for rare (red crosses) and abundant (black dots) codons compared with the null model (SSC). (A) Genomes were grouped according to the fraction of GC3 codons in the subset of rare and abundant codons, and mean enrichment±s.d. is shown for these groups. Genomes with GC3-rich abundant codons show a depletion of abundant codons, and genomes with AU3-rich rare codons show an enrichment of rare codons at gene start. (B) Enrichment of extreme codons shown as a function of GC content. Rare codons are only enriched and abundant codons depleted in genomes with GC content larger than about 0.5. (C) The average deviation from the genomic GC3 content for codons 1–5 depends on the global GC content. Virtually all genomes show a reduction in GC3 content at the gene start, and genomes with higher genomic GC content typically show a stronger reduction (correlation coefficient r=−0.66).
In addition, the ‘structure hypothesis’ predicts an asymmetry in GC3 content such that GC3 content at the beginning of genes is always reduced, with stronger depletion for genomes with higher GC content. In the 414 analysed genomes, the GC3 content behaves strongly asymmetric. We observed a reduction in GC3 content proximal to the start codon in most genomes (Figure 5C and Supplementary Figure S13). Importantly, the reduction in the local GC3 content is stronger for genomes with a higher GC content, suggesting that a reduction in the GC content is the main driving force behind the unusual codon bias just downstream of the start codon.
Folding of mRNA at gene start strongly influences translation
We interpreted the suppression of mRNA folding around translation start as a mechanism that facilitates ribosome binding and translation initiation. Moreover, the ‘ramp hypothesis’ predicts that rare codons at the beginning of genes will have an impact on translation efficiency. To discriminate between these two hypotheses, we experimentally dissected the role of the codon usage and mRNA secondary structure on translation efficiency. We selected two genes from E. coli (pykA and ypdE) for which codon choice allows to vary folding energy and codon adaptation independently (Supplementary Figure S14). To investigate the effect of mRNA secondary structure, we chose synonymous codons such that the mRNA structure differs strongly in folding energy (minG and maxG for minimal and maximal folding energy, respectively), but the sequences have similar codon adaptation as the native sequence (wild-type (wt)). Analogously, we studied the effect of codon usage by designing sequences that differ in the codon adaptation but have similar estimated folding energy (minCA and maxCA, for minimal and maximal codon adaptation, respectively). These sequences were fused upstream to a yellow fluorescent protein (Figure 6A), expressed in E. coli, and the translation efficiency was assessed by quantitative measurements of mRNA and YFP fluorescence levels. Alterations in codon usage or secondary structures had no influence on the mRNA abundance as confirmed by qRT–PCR (Supplementary Figure S15). In contrast, the protein yields differed largely (Figures 6B and C, and Supplementary Figure S16). Notably, protein expression between the constructs differed upto 20- and 60-fold for ypdE and pykA, respectively. In line with the structure hypotheses, we found that the constructs with the minimal folding energy (i.e., the strongest secondary structure) showed by far the lowest expression, whereas constructs with the maximal folding energy yielded the highest protein expression (Figures 6B and C). Codon usage also influenced protein expression, but the effects were much weaker and rather inconsistent. For ypdE, the constructs with the minCA and maxCA had a reduced protein expression (Figure 6B). In contrast, pykA showed a slightly enhanced expression for minCA and a decreased expression for maxCA (Figure 6C). The induction of pykA resulted in very high protein levels, which may impair cellular physiology in general. We thus measured the protein levels also in the absence of the inducer β-D-thiogalactoside (IPTG). Although the strong influence of the folding energy on protein level remained, the construct with minCA showed no difference to the wt construct (Figure 6C). Taken together, these results clearly suggest that mRNA structure determines the translation yield, whereas the effects of codon usage are less obvious.
Figure 6.

Influence of synonymous mutations on gene expression. (A) Constructs encoding for the same amino acid sequence, but varying codon usage and folding energy were derived from two different E. coli genes and fused to yellow fluorescent protein (YFP) gene. (B, C) All constructs were expressed in E. coli in triplicates and the fluorescence was measured after induction by flow cytometry. The median of fluorescence distributions was averaged and normalized to wild-type expression. Errors based on the s.d. of the median were calculated by propagating uncertainties. Constructs with varying folding energy but similar codon usage had a pronounced and reproducible effect on translation efficiency. Sequences with strong secondary structure (minG) had weaker expression than wild-type (wt), and constructs with loose structure (maxG) showed higher expression. Effects on gene expression by modifying codon usage, i.e., maximal and minimal adapted, but not changing folding energy, were present but less pronounced and inconsistent (compare minCA and maxCA for usage of codons corresponding to rare and abundant tRNAs, respectively). Moreover, effects of altering codon usage were less pronounced for pykA without gene induction.
Discussion
Clearly, synonymous codons are not chosen randomly and here we present a new aspect that drives the selection of unusual codons. In many genomes, the frequency of synonymous codons at the beginning of genes differs from the overall codon usage in the genome. This has led to the ‘ramp hypothesis’, suggesting that rare codons at the beginning of genes were selected to slow down initial elongation, which may help to avoid traffic jams during translation (Tuller et al, 2010a). However, as the same region has been shown to be under selective pressure for mRNA structure, we investigated whether the unusual codon usage is mainly a side effect of structural constraints (‘structure hypothesis’). Such evolutionary pressure is likely to be strong as secondary structures at the ribosome binding region can reduce or even terminate translation initiation (de Smit and van Duin, 1990; Kudla et al, 2009). Both hypotheses make divergent predictions, most importantly regarding the use of AU-rich and GC-rich codons, allowing us to distinguish between these two hypotheses. Our bioinformatics analysis of many bacterial species with varying GC content showed that codons at the beginning of genes are selected that effectively decrease the propensity of mRNAs to form secondary structure around the ribosome binding site.
We also experimentally show that changing the folding energy while keeping the same codon usage at the beginning of native E. coli genes markedly affects translation efficiency. In contrast, alterations of the codon usage at constant folding energy had mild but inconsistent effects on protein yield. This suggests that translation efficiency is strongly modulated by the folding energy at translation start.
The need to disfavour strong mRNA secondary structure formation around the translation initiation site depends on GC content of the genome: if the GC content is higher, the mRNAs tend to form more stable structures and, therefore, the pressure to avoid these is stronger. As suppression of mRNA folding is primarily determined by decreasing the GC content, we observe an enrichment of AU-rich codons at the beginning of genes. This, however, also explains why rare codons are used more often in this region: with increasing genomic GC content the frequency of AU-rich codons throughout the genome drops, i.e., they become rare. As a consequence, we observe the strong correlation between unusual codon usage and suppression of mRNA folding. However, rare codons at the gene start lead to slower elongation, as codon frequency correlates with tRNA abundance. Indeed, in many organisms elongation speed may be slower at the beginning of genes, such as that in yeast (Ingolia et al, 2009). Our results indicate that this ramp in speed is most likely a consequence of the need to suppress mRNA structure and the resulting use of rare codons. Furthermore, our analysis suggests that the evolutionary pressure to keep the ribosome binding regions free of structure is very strong, and may even impact protein sequence. For example, the N-terminal regions of E. coli proteins show an enrichment of amino acids encoded by codons with A or U at the first or second nucleotide position.
Structural constraints for efficient initiation also extend to higher eukaryotic organisms: shorter genes show less structure at the ribosomal binding sites, which results in higher translation rates (Ding et al, 2012). The role of codon usage in defining mRNA structure might not only be restricted to the translation start but also codon choice along the coding sequences may be shaped by various requirements to favour or disfavour specific secondary structures. For example, mRNA stability, micro-RNA-binding or RNA-binding proteins may require certain structures, which would impact codon choice. Thus, evolution has to solve a multi-dimensional problem: although efficient elongation and error reduction require the usage of abundant codons, certain structural requirements for the mRNA may need infrequent codons (Komar, 2009; Zhang et al, 2009). Thus, codon usage is shaped by many possibly conflicting constraints, which we are just beginning to understand.
Materials and methods
Sequence database
Genome sequences and annotation was collected from EcoCyc database (Keseler et al, 2011) version 13.6 for E. coli, and for the other 414 bacterial genomes from the BioCyc database (Karp et al, 2005) version 13.6, Tier3. We excluded the first gene of each TU, non-chromosomal, non-protein-coding sequences and splicable genes (genes with programmed frame shifts). Furthermore, we removed TUs containing genes of nucleotide length, which are not multiple of 3 or which contained components that were incompletely annotated. Genes present in multiple TUs were assigned to the largest TU. Taxonomy was annotated using the NCBI taxonomy IDs and the Perl module Bio-LITE-Taxonomy-NCBI-0.08.
Null models
We used two null models: (i) SC, where codons were randomly permuted within each gene, and (ii) SSC, which preserves the amino acid sequence by shuffling only synonymous codons within genes. Start and stop codons, codons with sequencing errors (only in the case of SSC), and overlapping sequences were not shuffled.
Calculation of folding energy
We used the Vienna RNA Package, version 1.8.5, available at http://www.tbi.univie.ac.at/%7Eivo/RNA/, to predict free energy of RNA sequences (Hofacker, 2009). Gibbs free energy was calculated within a sliding window of 39 nts that correspond to the approximate number of nucleotides covered by a ribosome (Beyer et al, 1994), and this value was assigned to the nucleotide position of the window centre. Using this method, we defined suppression of mRNA structure around the start codon (ΔG=G0–Gb1) as the difference between average folding energy G0 within a −5 to +5 nt window around gene start. The baseline folding energy Gb1 estimated as the average within a 50-nt window from nucleotide position +150 to +199.
Codon frequencies and KLD
For each set of synonymous codons, we determined the genome-wide frequency qi,j of each codon within this set, where i=1…20 indicates the amino acid and j=1…Si indexes the synonymous codon (where Si is number of synonymous codons). In addition, we defined the position-dependent codon frequencies, pi,j(k), for each codon position k relative to the translation start site (start codon is at k=0). In both cases, pseudocount regularization with a pseudocount of 1 is applied. The position-dependent KLD(k) that quantifies the deviation of the codon usage at each position k is then calculated as:
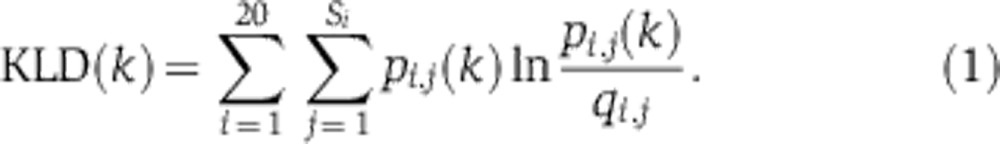 |
Because of finite size effects, the KLD(k) is biased to values larger than 0 even if pi,j and qi,j stem from the same distribution (Herzel and Grosse, 1997; Roulston, 1999). The bias due to this finite size sampling was estimated using the SSC null model.
Enrichment of extreme codons
We define abundant and rare codons as the 15 most abundant and 15 most rare codons in each genome, measured by their codon frequencies. The total frequencies of rare frare and abundant fabund codons are the sum over the codon frequencies in the corresponding set, e.g.
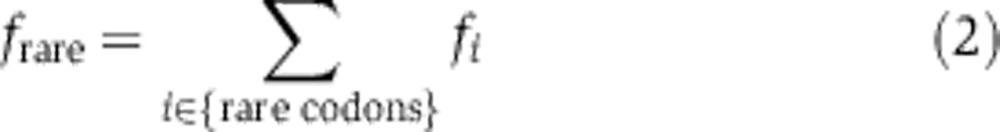 |
where fi denotes the frequency of the ith codon and the same definition applies for abundant codons. For each position k, we defined the fold change fc(k) of the rare codon frequency as:
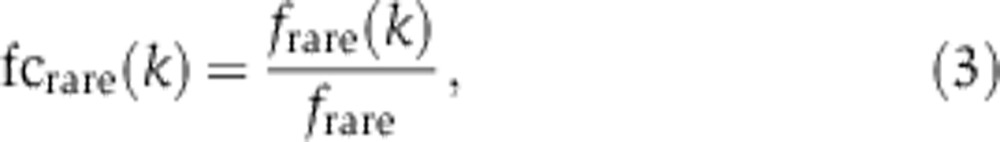 |
and, correspondingly, for abundant codons. Enrichment of rare and abundant codons at the beginning of genes was determined by averaging the fold change from position 1 through 5 (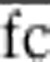 ) and subsequent calculation of Z-values:
) and subsequent calculation of Z-values:
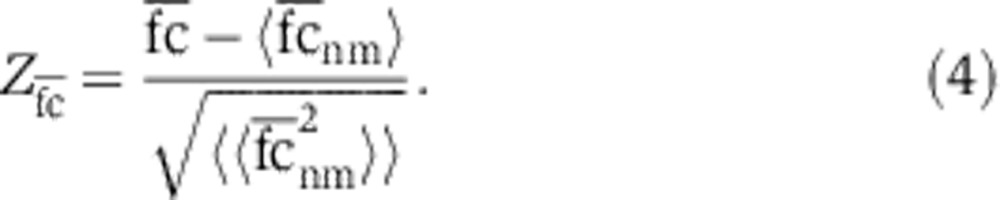 |
where the estimate of the mean 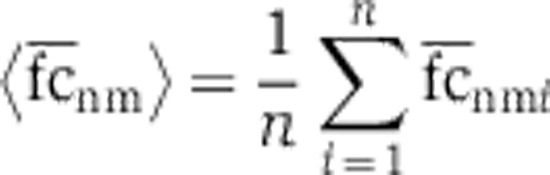 and variance
and variance 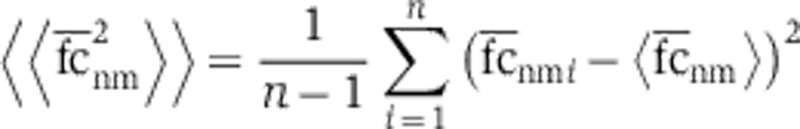 were calculated using n=100 instances of the SSC null model.
were calculated using n=100 instances of the SSC null model.
Enrichment of slow and abundant codons was assessed analogously.
Evaluation of synonymous sequences
For each cytoplasmic protein, we generated in silico all synonymous sequences that differ in the first six codons. Only the beginning of the genes, i.e., 5′-UTR and 31 codons, including ATG, was taken into account. To select appropriate genes to address codon usage and folding energy independently, we judged each synonymous sequence according to the following measures. We calculated the mean folding energies around the translation start (i.e., folding energies of 39-nt long stretches centred at nucleotide positions 0, ±3 and ±6 relative to the translation start). Codon adaptation was estimated by determining the geometric mean of the relative tRNA abundance corresponding to codons from position 1–6 (Dong et al, 1996; Zhang et al, 2009; Zhang and Ignatova, 2009). The folding energy and codon usage profiles for the selected genes ypdE and pykA are shown in Supplementary Figure S14.
Plasmids and strains
We created five constructs (wt, minCA, maxCA, minG and maxG) for each gene with (i) a 50-nt long native 5′-UTR in which additional ATG codons were removed to avoid alternative start sites, (ii) the start codon ATG, (iii) the stretch of variable codons, (iv) a sequence of 24 unaltered codons, followed by three codons coding for glycine (Figure 6A, and Supplementary Tables S1 and S2). All constructs were synthesized de novo (Eurofins MWG Operon) and subcloned into pETDuet-1-yfp vector (a kind gift from IM Axmann) upstream of the yfp sequence. All plasmids carry a T7 promoter inducible by IPTG and an ampicilin resistance for selection. Plasmids were expressed in E. coli BL21(D3) strain.
Growth conditions
Overnight cultures were grown in Luria Bertani broth (LB) containing ampicilin (100 μg/ml) at 37 °C. For measurements of YFP expression, overnight cultures were diluted 1:50 in 20 ml fresh LB medium containing ampicilin. Cells were induced at OD600∼0.5(±0.1) with IPTG at a final concentration of 45 μM. One hour after induction, cell cultures were diluted 1:5 in fresh LB medium containing ampicilin and IPTG, grown for another hour, collected by centrifugation and resuspended in tethering buffer (5 mM K2HPO4, 5 mM KH2PO4, 0.1 mM EDTA, 1 μM L-methionine, 0.1% (v/v) lactic acid (pH 7)). The protein expression was analysed by flow cytometry and mRNA was quantified by qRT–PCR.
Quantification of gene expression
Median expression levels of fluorescent proteins were quantified in a population of approximately 105 cells by flow cytometry on a FACSCalibur (BD Biosciences) equipped with an argon 488-nm laser. A measurement was triggered by forward scatter (fsc) and sideward scatter (ssc) events. FACSCalibur data files were imported for analysis into MATLAB using fca_readfcs.m (developed by Laszlo Balkay, University of Debrecen, downloaded from Matlab Central File Exchange, File ID: 9608). Only non-zero measurements were taken into account for fsc and ssc values between the 10th and 90th percentile. The median value of the autofluorescence background, measured for control cells transformed with an empty pETDuet-1 vector, was subtracted from all values. Measured fluorescence distributions are shown in Supplementary Figure S16.
Total RNA was isolated using the InviTrap Spin Cell RNA Mini Kit (Stratec Molecular) following the protocol for Gram-negative bacteria. The RNA quality was judged by the absorbance ratio A260nm/A280nm. Samples were then treated with DNAseI (Fermentas) for 1 h at 37 °C to remove remaining DNA traces and the enzyme was inactivated by adding 50 mM EDTA and incubated at 65 °C for 10 min.
cDNA was produced from total RNA using Reverse Transcription Kit (Fermentas) with random hexamer primer. For each sample, a control was performed without Revert Aid Transcriptase. mRNA was quantified by qRT–PCR with gene-specific primers on a real-time PCR cycler (7500 Fast Real-Time PCR System, Applied Biosystems) using Fast SYBR Green Master Mix (Applied Biosystems). The level of yfp mRNA was normalized to the level of gapdh mRNA as an internal standard. Two clear outliers were removed from the analysis.
Supplementary Material
Acknowledgments
We thank Naama Barkai and Gong Zhang for stimulating discussions; Melanie Anding for her help with the experiments; Hanspeter Herzel, Debora Marks, Ralf Steuer, Johannes Meisig and Adam Wilkins for commenting on the manuscript. This work was supported by the European Commission (BACTOCOM), Deutsche Forschungsgemeinschaft (DFG, through SFB 618, project A3 and SPP 1395/InKoMBio) and BMBF (FORSYS) to NB and ITN NICHE to ZI.
Author contributions: NB conceived the study; KB and NB designed the computational analysis; ZI, KB and NB designed the experiments; KB carried out the wet lab experiments with PS and RR, and performed the computational experiments. KB and NB wrote the manuscript with input from ZI.
Footnotes
The authors declare that they have no conflict of interest.
References
- Beyer D, Skripkin E, Wadzack J, Nierhaus KH (1994) How the ribosome moves along the mRNA during protein synthesis. J Biol Chem 269: 30713–30717 [PubMed] [Google Scholar]
- Cannarozzi G, Cannarrozzi G, Schraudolph NN, Faty M, von Rohr P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y (2010) A role for codon order in translation dynamics. Cell 141: 355–367 [DOI] [PubMed] [Google Scholar]
- Chu D, Barnes DJ, von der Haar T (2011) The role of tRNA and ribosome competition in coupling the expression of different mRNAs in Saccharomyces cerevisiae. Nucleic Acids Res 39: 6705–6714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D, von der Haar T (2012) The architecture of eukaryotic translation. Nucleic Acids Res 40: 10098–10106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smit MH, van Duin J (1990) Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Natl Acad Sci USA 87: 7668–7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Shah P, Plotkin JB (2012) Weak 5′-mRNA secondary structures in short eukaryotic genes. Genome Biol Evol 4: 1046–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynski M, Bruggeman FJ (2009) Elongation dynamics shape bursty transcription and translation. Proc Natl Acad Sci USA 106: 2583–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Nilsson L, Kurland CG (1996) Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol 260: 649–663 [DOI] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J, Nilsson D, Tenson T, Ehrenberg M (2003) Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300: 1718–1722 [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Bulmer M (1993) Reduced synonymous substitution rate at the start of enterobacterial genes. Nucleic Acids Res 21: 4599–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y (2011) Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M (1980) Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res 8: 1893–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO (2010) A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol 6: e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA (2008) Selection on codon bias. Annu Rev Genet 42: 287–299 [DOI] [PubMed] [Google Scholar]
- Herzel H, Grosse I (1997) Correlations in DNA sequences: The role of protein coding segments. Phys Rev E 55: 800–810 [Google Scholar]
- Herzel H, Weiss O, Trifonov EN (1999) 10-11 bp periodicities in complete genomes reflect protein structure and DNA folding. Bioinformatics 15: 187–193 [DOI] [PubMed] [Google Scholar]
- Hofacker IL (2009) RNA secondary structure analysis using the Vienna RNA package. Curr Protoc Bioinformatics Chapter 12:Unit12.2. [DOI] [PubMed] [Google Scholar]
- Ikemura T (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol 146: 1–21 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Sci N Y 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzkovitz S, Alon U (2007) The genetic code is nearly optimal for allowing additional information within protein-coding sequences. Genome Res 17: 405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S (1998) Global dinucleotide signatures and analysis of genomic heterogeneity. Curr Opin Microbiol 1: 598–610 [DOI] [PubMed] [Google Scholar]
- Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahrén D, Tsoka S, Darzentas N, Kunin V, López-Bigas N (2005) Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res 33: 6083–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller TE, Mis SD, Jia KE, Wilke CO (2012) Reduced mRNA secondary-structure stability near the start codon indicates functional genes in prokaryotes. Genome Biol Evol 4: 80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E (2010) Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muñiz-Rascado L, Bonavides-Martinez C, qPaley S, Krummenacker M, Altman T, Kaipa P, Spaulding A, Pacheco J, Latendresse M, Fulcher C, Sarker M, Shearer AG, Mackie A, Paulsen I, Gunsalus RP et al. (2011) EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res 39: D583–D590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA (2009) A pause for thought along the co-translational folding pathway. Trends Biochem Sci 34: 16–24 [DOI] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Singer GAC, Hickey DA (2002) Synonymous codon usage is subject to selection in thermophilic bacteria. Nucleic Acids Res 30: 4272–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JE, Bokelmann C (1988) Determinants of translational initiation efficiency in the atp operon of Escherichia coli. Mol Microbiol 2: 455–465 [DOI] [PubMed] [Google Scholar]
- Nirenberg M, Caskey T, Marshall R, Brimacombe R, Kellogg D, Doctor B, Hatfield D, Levin J, Rottman F, Pestka S, Wilcox M, Anderson F (1966) The RNA code and protein synthesis. Cold Spring Harb Symp Quant Biol 31: 11–24 [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston MS (1999) Estimating the errors on measured entropy and mutual information. Phys D Nonlinear Phen 125: 285–294 [Google Scholar]
- Shah P, Gilchrist MA (2011) Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc Nat Acad Sci USA 108: 10231–10236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH (1987) The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer GAC, Hickey DA (2003) Thermophilic prokaryotes have characteristic patterns of codon usage, amino acid composition and nucleotide content. Gene 317: 39–47 [DOI] [PubMed] [Google Scholar]
- Trifonov EN (1987) Translation framing code and frame-monitoring mechanism as suggested by the analysis of mRNA and 16 S rRNA nucleotide sequences. J Mol Biol 194: 643–652 [DOI] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y (2010a) An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354 [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E (2010b) Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA 107: 3645–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke T, Hurst LD (2010) GroEL dependency affects codon usage—support for a critical role of misfolding in gene evolution. Mol Syst Biol 6: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Fedyunin I, Miekley O, Valleriani A, Moura A, Ignatova Z (2010) Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res 38: 4778–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Hubalewska M, Ignatova Z (2009) Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat Struc Mol Biol 16: 274–280 [DOI] [PubMed] [Google Scholar]
- Zhang G, Ignatova Z (2009) Generic algorithm to predict the speed of translational elongation: implications for protein biogenesis. PLoS ONE 4: e5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.