

Abstract
The efficacy of DNA-damaging anticancer drugs is highly influenced by cellular DNA repair capacity, and by inhibiting the relevant DNA repair pathway, efficacy of alkylating agents may be increased. Therefore, combining DNA repair inhibitors with anticancer agents that selectively target tumour tissue should improve cancer treatment. The objective of this study was to test the hypothesis that co-treatment of cancer cells with acylfulvene (AF, alkylating agent) and UCN-01 (DNA repair inhibitor) would improve drug efficacy and promote the persistence of DNA adducts. Previous data regarding the relative susceptibility of repair proficient versus deficient cells toward an AF analog suggests that corresponding adducts are repaired by nuclear excision repair (NER), a cellular process that has been shown to be prevented with UCN-01. In this study, cells were co-treated with non-toxic levels of UCN-01 together with increasing doses of AF. The efficacy of AF was assessed by measuring cytotoxicity and DNA adducts. In addition, cells were co-treated with non-toxic levels of methoxyamine, a known base excision repair (BER) inhibitor, to determine if inhibiting BER also promotes cytotoxicity of AF. DNA-adducts were measured in a sensitive and precise manner by using stable isotope-labeled mass spectrometry analysis. The data obtained in this study demonstrate for the first time that pharmacological inhibition of the NER pathway of DNA repair leads to the persistence of AF-specific adducts and promotes AF cytotoxicity.
Introduction
Alkylating agents that work by producing DNA damage, causing cell death directly or following DNA replication, continue to be a useful and effective strategy for anticancer therapy. However, drug resistance and toxicity to healthy tissue can be major limiting problems.1 Acylfulvenes (AFs), including the unsubstituted analog acylfulvene (AF, see Figure 1), are a class of experimental anticancer alkylating agents that are selectively toxic towards cancer tissue compared to normal tissue. In addition, AFs have better therapeutic indices than the more ubiquitously toxic natural product illudin S, from which the AFs are derived.2,3,4 Extensive data suggests that a contributing factor to the selective toxicity of AFs is a greater extent of reductase-mediated bioactivation in cancer cells. Thus, AFs are bioactivated by prostaglandin reductase 1 (PTGR1) resulting in an activated intermediate that can react with DNA, and sensitive cancer cells have higher PTGR1 activity and/or expression levels.4,5,6 While selective bioactivation appears to enhance selectivity, drug resistance may arise from the DNA damage response.7,8. Potentially overcoming repair-induced resistance requires an understanding of how the toxic effects of DNA adducts are avoided and devising strategies for interfering with their repair. In the case of AFs, it has been demonstrated that the AF analog HMAF, and its natural product precursor illudin, are more toxic in NER-deficient cells.9,10 DNA repair is a target for cancer treatment, and co-treatment of cancer cells with a DNA repair inhibitor and a selective alkylating agent could improve efficacy.1
Figure 1.

Structures of acylfulvene, its analog hydroxymethylacylfulvene, and its natural product precursor illudin S; AF-DNA adducts formed in HT29 cells: 3-acylfulvene-adenine, 3-acylfulvene-guanine and 7-acylfulvene-guanine.
It has been demonstrated for certain anticancer drugs that by specifically inhibiting a relevant DNA repair pathway, the efficacy of certain anticancer drugs can be increased.11,12,13 In this context, two major pathways include nuclear excision repair (NER) and base excision repair (BER) .14 For example, cisplatin adducts are repaired by NER, and when used in combination with the NER inhibitor UCN-01 (7-hydroxystaurosporing, Figure 2), cisplatin cytotoxicity was observed to be enhanced in lung epithelial cells.15,16 There are many examples of enhancing drug toxicity by inhibiting BER,17 for example combining methoxyamime (MX, Figure 2) with temozolomide to treat ovarian cancer.18
Figure 2.

Structures of the inhibitors UCN-01 (7-hydroxystaurosporine) and methoxyamine used in this study. IC50 values are for HT29 cells.
NER is involved in repairing bulky alkylation adducts (i.e. cisplatin and benzo(a) pyrene adducts) .19 After damage recognition by one of two mechanisms involving either transcriptional stalling or by the damage sensor XPC-RAD23B in global genome repair, multiple protein effectors are recruited and act on the damaged DNA. The abnormal strand is separated from the normal strand and xeroderma pigmentosum group A (XPA) isolates the damaged segment on the strand to be cut. Subsequently, 25–30 bases around the bulky adduct are excised by xeroderma pigmentosum group G (XPG) on the 3 side and by a heterodimeric protein, xeroderma pigmentosum group F (XPF) - excision repair cross-complementation group 1 (ERCC1), on the 5 side. Afterwards the gap is filled by the action of polymerases.1,12,14
Yang and co-workers demonstrated that the small molecule UCN-01 interferes with the NER signaling pathway and prevents ERCC1 from binding XPA, thereby preventing recognition and 5 -side incision during NER.1,16,20 The prevention of ERCC1-XPA binding is of further interest since ERCC1 has been demonstrated to be elevated in tumor tissue compared to normal tissue.21 In addition, UCN-01 is also known to be a protein kinase inhibitor that can abrogate the G2 checkpoint.22 While DNA alkylation may normally activate checkpoints and promote an opportunity for repair, UCN-01 appears to allow cells to bypass this process. Thus, a complete picture of how UCN-01 acts on cells is not fully understood yet, however it is clearly an effective means for reducing the protective effects of NER on cells.
In BER, specific small adducts such as oxidized bases are detected and removed, forming an abasic site to which AP endonucleases bind and initiate ligation. Methoxyamine (MX) binds to abasic sites, inhibiting AP endonucleases and thereby preventing BER function. An assumption underlying the success of these strategies, both in the case of NER and BER inhibition, is that inhibiting a drug-specific repair function promotes retention of drug-induced adduct levels, however, to the best of our knowledge this model has not been demonstrated by quantifying DNA adducts during treatment. Furthermore, in the case of UCN-01, data regarding how DNA adducts are impacted by co-treatment with an alkylating agent are expected to provide further insight regarding the influence of UCN-01 on NER function.
It appears that the DNA adduct resulting from the AF analog hydroxymethylacylfulvene (HMAF) ,9,10 are exclusively repaired by NER (Figure 1) . A very similar repair pathway specificity was observed for illudin S, from which the AFs are derived.9,10,23,24 This repair-specific relationship has been established primarily by comparing drug toxicity in various cell lines lacking a specific repair factor. On the basis of these data, it was concluded that the HMAF-induced DNA damage is repaired by NER and not by BER, and furthermore, that the repair process is selective for actively transcribed strands of DNA. Thus, removal of the adducts is selectively initiated by the transcription-coupled NER sub-pathway and largely ignored by global genome NER.9,10,23,24 However, the impact of inherent cellular repair deficiency on adduct levels was not examined in these studies. Furthermore, these studies were performed in human fibroblasts, and the phenomenon has not been examined further in rapidly growing cancer cell lines.
DNA adducts arising from AF in drug-sensitive cells has been characterized previously, providing a drug-specific chemical marker for investigating toxicity pathways.2,3,4 Thus, the major adduct results from minor groove alkylation, predominantly at the 3-position of adenine (Figure 1) . Two minor adducts result from reactions at the 3- and 7-positions of guanine (Figure 1) .3 On the basis of data regarding AF-DNA alkylation and repair, we hypothesized that by inhibiting NER with UCN-01, more AF adducts would persist in DNA, and the drug would be rendered more cytotoxic. However, in addition to repair-induced removal, AFs also spontaneously depurinate due to the instability of the corresponding nucleoside adduct.3 Upon depurination, an abasic site is formed and accumulation of abasic sites can be a precursor for toxicity.25 In general BER is effective in abasic site repair, but can be inhibited by the small molecule MX. If the resulting abasic sites are toxic to the cells, the combination of MX with AF would be anticipated to result in an accumulation of unrepaired abasic sites and be more toxic than drug alone.
In this study, the influence of DNA repair inhibitors on drug-induced alkylation of DNA in cancer cells was evaluated by combining the DNA-alkylating anticancer agent AF with NER inhibitor UCN-01 and comparing to the results of the same approach with BER inhibitor MX. We evaluated the impact of co-treatment on drug cytotoxicity to the colon cancer cell line HT29. Significantly, the impact of repair inhibition on drug-specific adduct levels and persistence were demonstrated. Thus, AF-adducts on genomic DNA, as well as excreted depurinated adducts, were measured by isotope labeling liquid chromatography-mass spectrometry. The data demonstrate the principle that AF cytotoxicity can be enhanced by co-treatment with an NER-specific inhibitor and that excreted depurinated drug adducts are an effective marker for magnitude of DNA damage.
Material and Methods
Chemical and Reagents
DMSO, methoxyamine (MX) and UCN-01 (7-hydroxystaurosporine) were purchased from Sigma-Aldrich (Buchs, Switzerland) . AF26,27, N3-AF-adenine (3-AF-Ade) and deuterated-N3-AF-adenine (3-AF-Ade-d3) 2 were prepared as previously described. Pure deionized water was obtained from a Milli-Q Integral water purification system (Millipore Corporation, Billerica, MA) .
Cell culture
HT-29 cells from the German Cell Culture Collection (Braunschweig, Germany) were provided by Professor Christophe Lacroix (ETH Zurich) . The cell line was grown in RPMI 1640 medium with glutaMAX (Invitrogen, Switzerland) containing 10% bovine calf serum, and 1% penicillin/streptomycin. The cells were incubated in a humidified incubator containing 5% CO2 at 37 °C.
Cell viability
Cells were seeded in a 96-well plate at a density of 1.0*103 cells/well and were allowed to attach overnight. Experiments were initiated by replacing the growth media with media containing the repair inhibitor MX (5 mM in 0.1% DMSO, final concentration) or UCN-01 (20 nM in 0.1% DMSO, final concentration) or with DMSO as control (0.1%, final concentration) . After one hour treatment with MX or UCN-01, AF (0.1% DMSO, final concentration) was added to the wells. The total final DMSO concentration in all wells was 0.2%. After 48 h AF treatment, metabolic capacity of viable cells as an indicator of cell viability was measured with CellTiter 96® AQueous One Solution cell proliferation assay (Promega Corporation, Madison, WI) . Absorbance at 490 nm was measured 4 h after adding 20 μL MTS solution to each well. Regression analysis and IC50 calculations were performed with Sigma Plot (version 12.2) .
DNA adducts
Cells were seeded in a 6-well plate at a density of 1.0*105 cells/well and were allowed to attach overnight. As in the cell viability measurements, medium was replaced with 2 mL fresh medium containing 0.1% DMSO (solvent control), 5 mM MX or 20 nM UCN-01. After 1 h, 2 μL stock solution of acylfulvene was added to the medium resulting in the following final concentrations: 50, 100, 250, 500, 750 or 1000 nM AF (total DMSO concentration was 0.2%) . Cells were incubated for 48 h; thereafter DNA of intact cells was isolated (according to procedure mentioned below) and medium was collected and stored at -80 °C. In addition to incubation of cells with AF for 48 h, cells were also incubated for 24 h with AF and medium subsequently was replaced with medium lacking AF to allow the repair of damage DNA. In this case cells were incubated with MX (5 mM), UCN-01 (20 nM) or DMSO, together with 500 nM AF for 24 h. Thereafter medium was replaced with fresh medium containing MX or UCN-01, but lacking AF. Samples were taken after 3, 8, 24, 48 and 72 h after adding AF and MX, UCN-01, or DMSO. DNA of intact cells was isolated according to the procedure mentioned below and medium was collected and stored at −80 °C.
Isolation of DNA and sample preparation
DNA was extracted from the incubated cells by using the Wizard® SV Genomic DNA Purification System (Promega, Switzerland) according to the protocol reported by us before.4 In short, cells were washed once with PBS and lysed by adding 500 μL lysis buffer to the 6-well plate. The lysis buffer was transferred to the Wizard® SV mini-column assembly. The assembly was centrifuged for 2 min at 13,000g. Thereafter the column was washed 4 times with 650 μL wash solution to remove any contaminants (1 min, 13,000 g) . After washing the column, the binding matrix was dried by centrifuging it for 2 min at 13,000 g, and the column was transferred to a new 1.5 mL eppendorf tube containing 2 μL of RNAse. Nuclease-free water (250 μL) was added to the column and after 2 min at 20 °C the assembly was centrifuged for 5 min at 13,000 g. The DNA concentration of the resulting solution was determined with a NanoDrop™ 1000 spectrophotometer (Thermo Scientific), and the solution was stored at 4 °C for up to 72 h.
Depurinated adducts were obtained from the DNA by neutral thermal hydrolysis according to a slightly modified version of the procedure reported by Neels et al.2 To the purified DNA solution an equal volume of water was added plus 3 μL 3-AF-Ade-d3 (200 nM in 50% methanol/water (v/v) ) as internal standard. The solution was heated at 90 °C for 1 h. The resulting sample was dried by rotary vacuum centrifugation and the solid was extracted 3 times with 300 μL methanol. The combined fractions were filtered through an in-line 0.45 μm nylon syringe filter (Millipore) . The methanol was removed by rotary vacuum centrifugation and the resulting solids were dissolved in 20 μL 50% methanol/50% water (v/v) for quantitative LC-MS analysis.
Cell medium sample preparation
Cell medium samples were obtained after incubating cells with AF and UCN-01/MX/DMSO in order to determine the amount of depurinated adducts excreted by the cells. 3 μL 3-AF-Ade-d3 (200 nM in 50% methanol/water (v/v) ) as internal standard was added to the medium samples (2 mL each) . Samples were concentrated by off-line solid-phase extraction on a 50 mg Sep-Pak C18 cartridge (Waters) . The cartridge was first activated with 1 mL methanol followed with 1 mL of 80% acetonitrile/20% water and equilibrated with 2 x 1 mL water. The medium samples (2 mL each) were loaded on the activated cartridges, and upon loading, the cartridge was washed with 1 mL water followed by 0.5 mL of 5% acetonitrile in water. Finally, the depurinated adducts, 3-AF-Ade, N3-AF-gua, N7-AF-gua and the internal standard, 3-AF-Ade-d3, were eluted with 1 mL of 80% acetonitrile/20% water. Solvent was removed by rotary vacuum centrifugation and dried samples were reconstituted for LC-MS analysis in 20 μL of 50% methanol/50% milliQ water.
Quantitative analysis of DNA adducts by LC-ESI-MS/MS
The LC-MS/MS equipment consisted of a nanoAcquity UPLC system (Waters) connected to a tandem quadrupole mass spectrometer (Thermo LCQ Vantage) with an electrospray ionization interface (ESI) . Quantitative analyses were carried out in selected reaction monitoring (SRM) mode, and MS ionization parameters were optimized by tuning with 1 μM AF-Ade solution in 50% acetonitrile and 0.1 % formic acid in water. The ESI source was set in positive ion mode with the following parameters: capillary temperature, 300 °C; voltage, 3.5 kV; sheath gas pressure, 10; Q2 CID gas pressure, 1.5 mTorr; collision gas, argon; scan width, m/z 0.100; scan time, 0.050 s; collision energy, 17 V; Q1 peak width, 0.70 amu; Q3 peak width, 0.70 amu. Mass transitions monitored were m/z 336.2 to 201.1 for 3-AF-ade, m/z 339.2 to 204.1 for 3-AF-ade-d3, and m/z 352.0 to 201.0 for N3-AF-gua and N7-AF-gua. Xcalibur software (Thermo) was used for data acquisition and processing. Chromatography was performed with a Phenomenex Synergi Polar-RP 80 Å column (150 × 0.5 mm, 4 μm particle size) . The HPLC flow rate was 10 μL / min and the mobile phases were 3% acetonitrile and 0.1% formic acid in H2O (v/v) (mobile phase A) and 0.1% formic acid in acetonitrile (v/v) (mobile phase B) . The following gradient was used: 0 min – 0% B, 20 min – 90% B, 30 min – 90% B, 32 min – 0% B, 40 min – 0% B.
In case of the cell medium samples, a divert valve was used before the MS entrance. By the sample work-up procedure, phenol red present in the medium also was preconcentrated and thereby suppressed ionization. Therefore the first 9 min were directed to waste and after 9 min, the divert valve was switched and the LC-flow was directed to the MS for adduct detection.
LOD and LOQ were determined on the basis of a signal to noise ratio of 3 and 10, respectively. LOD was 0.3 fmol (~1 adduct per 108 nucleotides for 10 μg DNA) and LOQ was 1 fmol (3 adducts per 108 nucleotides for 10 μg DNA) .
Statistical evaluation
All data are expressed as means ± standard error of the mean (SEM) . All the results were analyzed for significant differences using the Student t-test, with p < 0.05 considered as significantly different.
Results
Cell viability
The impact of NER vs BER inhibition on AF cytotoxicity was assessed by treating HT29 colon cancer cells with AF either alone or in combination with UCN-01, to inhibit NER, or MX, to inhibit BER. The measured IC50-values for UCN01 in HT29 cells was 150 nM and for MX 50 mM (Figure 2, inset) . Concentrations of UCN-01 and MX used in this study were 20 nM and 5 mM, respectively. The rationale for using these concentrations was based on both empirically measured IC50 values in the present test system, indicating that the inhibitors would be non-toxic to the cells, as well as consistency with results reported by others.28,29 Cells were incubated for 48 h in the presence of both repair inhibitor and AF, and thereafter the cell viability was measured (Figure 3) . In the case of treatment with AF alone, no cytotoxicity was observed up to 30 nM drug concentration. At higher concentrations, a decrease in cell viability was observed, with an IC50 of 155±25 nM. In the presence of MX, no change in the AF cytotoxicity profile was observed, and the IC50 (133±13 nM) was not significantly different compared to AF alone (p = 0.3) . On the other hand, co-treatment with UCN-01 resulted in a shift of the AF cytotoxicity curve such that for this combination, cells were two-fold more sensitive to AF than without UCN-01. The IC50 in this case (64±7 nM) was significantly different compared to treatment with AF alone (p = 0.003) .
Figure 3.
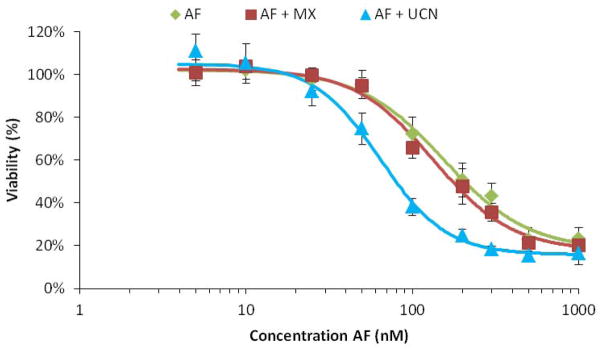
Dose-response curves of HT29 cells with acylfulvene (green diamonds) and with combined treatment of acylfulvene with methoxyamine (red squares) and acylfulvene with UCN- 01 (blue triangles) . Cells were incubated for 48h and viability was measured using the MTS assay (mean ± SEM, n=3) .
DNA adduct formation versus acylfulvene concentration
To determine how DNA repair inhibitors influence levels of drug-induced DNA adducts, the amount of genomic AF-adducts in co-treated cells was quantitatively evaluated by stable isotope dilution mass spectrometry. Following 48 h treatment of cells with increasing concentrations of AF, DNA was isolated and adduct levels were evaluated. In all samples, the major N3-AF-ade adduct and the two minor N3-AF-gua and N7-AF-gua adducts were detected (Figure 4) . At a drug concentration of 50 nM AF alone, AF-induced adduct formation was just above the LOQ, which was 3 adducts per 108 nucleotides for 10 μg DNA. With increasing AF concentration, more DNA adducts were detected, up to almost 100 N3-AF-ade adducts per 107 nucleotides for the incubation of the HT29 cells with 1 μM AF alone.
Figure 4.

Formed (a) N3-adenine-AF-DNA adducts, (b) N3-guanine-AF-DNA adducts, and (c) N7-guanine-AF-DNA adducts versus acylfulvene concentration in HT29 cells. For the combined treatment 5 mM MX was used and 20 nM UCN-01 (mean ± SEM, n=5) . Asterisks denote statistically significant differences towards AF (p<0.05) .
When cells were co-treated with AF and UCN-01, the levels of the major AF-adduct found on the intact DNA approximately doubled. Thus, in the case of N3-AF-ade, the AF-adduct was for all concentrations significantly higher compared to the situation lacking the NER inhibitor (Figure 4a) . In the case of the N3-AF-gua and N7-AF-gua adducts, adduct levels in DNA were not significantly higher for the lower concentrations (50–250 nM), but were significantly increased for 500 nM AF and higher (Figure 4b and 4c) . At the lower concentrations, the lack of an observed change was most likely because the AF-gua adducts detected were almost at the LOD.
When cells were co-treated with MX and AF, there was also an increase of AF adducts detected with increasing concentrations of AF, however for each AF concentration, adducts were slightly higher, but statistically insignificant, compared to treatment with AF alone.
Spontaneous depurination of AF adducts continuously occurs due to the instability of the corresponding nucleoside adduct.3 To determine the relationship between the AF adducts found on genomic DNA and the amount of depurinated adducts excreted by cells, the cell medium was assessed for excreted depurinated adducts (Figure 5) . All three spontaneously depurinating adducts were detected in the medium. With increasing AF concentration, as observed for AF adducts in DNA and mentioned above, more depurinated adducts also were detected in cell medium. Despite the concentration-associated increases, however, there were no significant differences in excreted adduct levels between the cells treated with AF alone or in the presence of a DNA repair inhibitor. In the cases of N3-AF-ade and N7-AF-gua, adduct levels were slightly higher for the UCN-01-AF co-treated cells compared to AF alone, but the difference was not significant.
Figure 5.

Depurinating adducts found in medium after 48h incubation with acylfulvene versus concentration: (a) N3-adenine-AF, (b) N3-guanine-AF, and (c) N7-guanine-AF. For the combined treatment, 5 mM MX was used and 20 nM UCN-01 (mean ± SEM, n=3) . No significant differences were observed between AF treated samples and AF+MX or AF+ UCN-01.
Repair of DNA adducts in the presence of repair inhibitors
In the absence of DNA repair inhibitors, HT29 cells are proficient in NER and BER. The effectiveness of repair was assessed by first alkylating the DNA and subsequently testing the ability of the cells to repair the damage in the absence of the alkylating agent. Thus, cells were challenged for 24 h with a combination of AF and repair inhibitor. Afterwards, medium was replaced with fresh medium lacking AF, but still containing the original concentration of MX or UCN-01. In this way, cells had the potential to repair damaged DNA and recover. Adduct levels were monitored at various time points to determine the rates of formation and repair of AF adducts (up to 72 h) .
Already after 3 h, all three adducts were detected on the DNA and these increased over time (Figure 6) . Whereas in the presence of NER inhibitor UCN-01 the detected AF adducts increased at a constant rate, in the presence of BER inhibitor MX or no inhibitor, the AF adducts were constant after 8 h treatment up to 24 h. Therefore N3-AF-ade and N3-AF-gua adducts after 24 h were slightly higher in the UCN-01-treated cells compared to in those treated with MX or no inhibitor.
Figure 6.

Formation and repair of DNA adducts versus time. Cells were incubated for 24h with 500 nM acylfulvene with or without repair inhbitors and medium was replaced after 24h with fresh medium lacking acylfulvene to measure the repair. (a) N3-Adenine-AF-DNA adducts, (b) N3-guanine-AF-DNA adducts, and (c) N7-guanine-AF-DNA adducts. For the combined treatment, 5 mM MX was used and 20 nM UCN-01 (mean ± SEM, n=3) . Asterisks denote statistically significant differences towards AF (p<0.05) .
When changing the medium to fresh medium lacking AF, a decrease in adducts on the DNA was detected in the case of treatment with AF alone, and with MX plus AF. In the case of N3-AF-ade, the adduct levels dropped from around 70 adducts per 107 nucleotides to 20 adducts per 107 nucleotides. This decrease, observed in both MX-treated samples and in the control samples, suggests active repair of the damaged DNA strand. The rates of repair of N3- or N7-AF-gua adducts were much slower than for the N3-AF-ade. The repair of N3-AF-gua adducts was around 1 adduct per 107 nucleotides per 48 h and repair of N7-AF-gua adduct was 3 adducts per 107 nucleotides per 48 h. In the case of UCN-01-treated cells, however, there was little change in the amount of adducts detected on genomic DNA. N3-AF-ade levels decreased from 90 adducts per 107 nucleotides to approximately 80 adducts per 107 nucleotides in 48 h, which is most probably due to spontaneous depurination of adducts rather than repair; the amount of N3- and N7-AF-gua adducts were approximately constant during the 48h recovery time.
The rate of disappearance of AF adducts in UCN-01-treated samples was significantly different than control cells. Assuming that in this situation adduct release is due to depurination, and that the process follows first order kinetics, the half-life for spontaneous depurination of 3-AF-ade from genomic cellular DNA under conditions of NER inhibition was approximately 12 days, which is long compared to the depurination half-life of about 1 day in NER-proficient cells, and dramatically slower than the half-life of 8.5 h from naked DNA.3 These data provide for the first time an estimate of the rate of depurination of AF adducts in cells.
In addition to depurination, adducts may be transported from nucleus to cytoplasm, and also exported from the cell to the external culture medium. Therefore, levels of excreted depurinated adducts also were measured as a function of time (Figure 7) . This experiment was performed in a manner similar to that described above, involving increasing concentrations of AF, but with removal of the drug after 24 h to allow cells to recover. The cumulative amount of adducts per time point were integrated to determine the total amount of depurinated adducts in the cell medium. For the first 8 h, hardly any depurinated adducts were detected in the cell medium, but at 24 h, a significantly larger amount was detected and all three adducts were excreted by the cells (Figure 7) . After 24 h, the total amount of depurinated adducts for both the control and MX-treated samples hardly increased over time, since the amount of AF adducts on the DNA decreased (Figure 6) . In contrast, the depurinated adducts found in the medium from the UCN-01-treated cells increased over time at a constant rate, which is in agreement with the constant increase of AF adducts found on the DNA from UCN-01-treated cells.
Figure 7.

Depurinating adducts found in medium after incubation with 500 nM acylfulvene versus time. Acylfulvene was added the first 24h and thereafter medium was refreshed without acylfulvene but with inhibitors. (a) N3-adenine-AF, (b) N3-guanine-AF, and (c) N7-guanine-AF. For the combined treatment, 5 mM MX was used and 20 nM UCN-01 (mean ± SEM, n=3) . Asterisks denote statistically significant differences towards AF (p<0.05) .
Discussion
AFs are bioactivated by reductase enzymes (i.e., PTGR1) after entering cells and the bioactivated form can alkylate DNA.30 Due to this alkylation, AFs cause cells to undergo apoptosis, and they effectively target fast-dividing cells like tumor cells. However, when the AF-DNA adducts are formed, the cells can repair this damage via the NER pathway, and increased repair has been observed in tumor cells making the drug less effective.10 Therefore a co-treatment of AF with an NER repair inhibitor could improve the efficacy of the drug. Before being repaired, AF adducts may also spontaneously depurinate from the strand due to the instability of the corresponding nucleoside adduct.3 This depurination results in an abasic site, which may be repaired by both NER and by BER.31 In addition to the initially formed AF adducts, also the accumulation of abasic sites may promote entry into apoptosis. A standing question concerns whether the AF adducts or the accumulation of abasic sites are more significant contributors to AF-induced cytotoxicity. In this study, the repair inhibitors MX and UCN-01 were combined with AF. MX inhibits BER by binding to abasic sites and blocking endonuclease-mediated ligation. UCN-01 inhibits NER by preventing ERCC1 from binding to XPA and thereby preventing dual incision. Experiments involving co-treatment of cells with AF plus the inhibitors under varying treatment protocols were performed to better understand the relationship between AF-specific adducts, abasic site accumulation, and DNA repair in AF cytotoxicity.
The only cytotoxicity value reported previously for AF in HT29 cells was an IC50 of 379 nM.32 This value was slightly higher compared to the IC50 measured in this study (155 nM) . However, in the earlier study by Kelner et al, the IC50 of 379 nM was determined after 2 h treatment, and with a colony-forming assay. In this study, cells were incubated for 48 h and it is common that with increasing time, the IC50 may be expected to diminish.33 During the co-treatment of AF with MX, there was no change in cytotoxicity, suggesting that the formation of abasic sites is not a relevant mechanism of cytotoxicity. When NER was inhibited, the cells were two-fold more sensitive towards AF, indicating that when the cell cannot remove the AF-adduct, cytotoxicity is promoted. This observation is in agreement with previous studies where it was demonstrated that NER-deficient cells were more sensitive to AF, suggesting that the associated DNA damage is an NER substrate.10,23,24 Furthermore, in a previous screening study, UCN-01 was amongst a group of miscellaneous compounds that showed evidence of a synergistic effect with HMAF.34 These data are consistent with the notion that when treating cancer cells with a combination of UCN-01 and AF, the concentration of cytotoxic drug can be lower to achieve the same effect as when treating solely with AF. Translated to therapeutic applications, this property might result in fewer side-effects.13
UCN-01 has been used in combination with carboplatin, a platinum-based anticancer drug, in a phase I study to treat advanced solid tumours.35 There are two modes of action known for UCN- 01: (1) it inhibits the binding of ERCC1 and XPA and thereby inhibits the NER repair pathway1,16,20 and (2) it can abrogate the G2 checkpoint, therefore not permitting repair of the DNA adducts.22 With regard to AF-induced DNA alkylation, the amount of AF adducts found on genomic DNA would be expected to increase with UCN-01 co-treatment, as was observed in this study. Since UCN-01 can inhibit the human checkpoint kinase 1 and thereby promote the initiation of DNA replication36, DNA yields were also determined after 48 h incubation with AF and at different time points with 500 nM AF (Figure 8) . There was no significant difference in the DNA yield in samples treated with UCN-01 compared to samples treated with AF. For this reason it was concluded that at the concentration used in this study, UCN-01 acted as an NER inhibitor. Nonetheless, it has been suggested that the checkpoint pathways play an important role in regulating the NER processes, and therefore their contributions cannot be strictly separated.37 The influence of repair inhibitors on cytotoxicity has been demonstrated in other inhibitor-drug cases before, but the corresponding effect on DNA adducts, to best of our knowledge, has not been addressed. In the case of NER inhibition, DNA-adduct levels are expected to reflect whether repair inhibition takes place before or after the incision. If UCN-01 inhibited the function of polymerases that act after the incision, it would not be possible to measure adducts on genomic DNA as we were able to do. Thus, AF adduct levels were higher in UCN-01-treated samples compared to those treated with only AF, suggesting therefore that the dual-incision of the NER-pathway was inhibited.16,38 The amount of adducts on the DNA in UCN-01-treated cells was approximately two-times higher compared to cells without UCN-01 treatment, in agreement with the two-fold shift in cytotoxicity. At the IC50 dose, the total amount of adducts was around 6 adducts per 107 nucleotides, which was for all three treatment situations similar. This pattern is in agreement with previous DNA binding studies comparing cisplatin and illudin S,23 as well as illudin S and AF4, where it was shown that on an adduct-per-adduct basis, each drug was equally lethal.
Figure 8.

DNA yields of HT29 cells incubated with AF (blue, diamonds), AF + MX (red, squares) or AF + UCN-01 (green, triangles) for (a) different concentration AF (48h) or for (b) collected at different time points (mean ± SEM, n=3) . Asterisks denote statistically significant differences towards AF (p<0.05) .
MX-treated vs untreated cells demonstrated no differences in AF adduct levels or cytotoxicity. The lack of an influence of MX suggests that inhibiting the AP endonuclease did not improve the effectiveness of AF treatment. As expected also, levels of the depurinated adducts in the medium were not significantly different, i.e. the same amount of AF adducts were attached to the DNA and therefore also the same amount of adducts depurinated (the depurination rate constant did not change) .
Whether cells were co-treated with MX or no inhibitor, they were able to almost fully remove the AF adducts from the intact DNA (Figure 6) . The yields of DNA isolated from cells were consistent between these cases, indicating that the amount of cells proliferating and going into apoptosis was constant, and the cells were recovering. In addition the rate of depurination, measured by the amount of depurinated adducts found in the cell medium, diminished, i.e. less adducts were attached to the DNA, so fewer depurinated adducts were excreted. In contrast, for the UCN-01-treated cells the AF adducts attached to the DNA were constant over time and the amount of depurinated adducts was also constant over time. This trend indicated that there was no detectable repair in these cells. In addition the DNA yield was decreased, indicating that the cells were dying due to the AF-adducts attached to the DNA.
Conclusion
In this study, we demonstrated that by inhibiting the NER pathway with a non-toxic amount of the small molecule UCN-01, the concentration of the anticancer drug AF required to kill HT29 cancer cells was reduced by two-fold. In addition, data obtained here for AF suggest that if DNA adducts are susceptible to depurination, depurinated adducts measured in cell medium may be a convenient indicator of DNA bound levels. This study is a proof of biochemical principle that the treatment of cancer cells with both AF and UCN-01 can be a future therapeutic strategy.
Acknowledgments
Funding support We acknowledge the US National Cancer Institute (R01 CA123007) and the Swiss National Science Foundation (136247) for support of this research.
Abbreviations
- MX
methoxyamine
- UCN-01
7-hydroxystaurosporine
- BER
base excision repair
- NER
nucleotide excision repair
- AF
acylfulvene
- ade
adenine
- gua
guanine
- ERCC1
excision repair cross-complementing rodent repair deficiency, complementation group 1
- XPA
xeroderma pigmentosum group A
- PTGR1
prostaglandin reductase 1
- LOD
limit of detection
- LOQ
limit of quantitation
References
- 1.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 2.Neels JF, Gong J, Yu X, Sturla SJ. Quantitative correlation of drug bioactivation and deoxyadenosine alkylation by acylfulvene. Chem Res Toxicol. 2007;20:1513–1519. doi: 10.1021/tx7001756. [DOI] [PubMed] [Google Scholar]
- 3.Gong JC, Vaidyanathan VG, Yu X, Kensler TW, Peterson LA, Sturla SJ. Depurinating acylfulvene-DNA adducts: Characterizing cellular chemical reactions of a selective antitumor agent. J Am Chem Soc. 2007;129:2101–2111. doi: 10.1021/ja0665951. [DOI] [PubMed] [Google Scholar]
- 4.Pietsch KE, van Midwoud PM, Villalta PM, Sturla SJ. Quantification of acylfulvene- and illudin S-DNA adducts in cells with variable bioactivation capacities. Chem Res Toxicol. 2012;26:146–155. doi: 10.1021/tx300430r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dick RA, Yu X, Kensler TW. NADPH alkenal/one oxidoreductase activity determines sensitivity of cancer cells to the chemotherapeutic alkylating agent irofulven. Clin Cancer Res. 2004;10:1492–1499. doi: 10.1158/1078-0432.ccr-03-0162. [DOI] [PubMed] [Google Scholar]
- 6.Yu X, Erzinger MM, Pietsch KE, Cervoni-Curet FN, Whang J, Niederhuber J, Sturla SJ. Up-Regulation of Human Prostaglandin Reductase 1 Improves the Efficacy of Hydroxymethylacylfulvene, an Antitumor Chemotherapeutic Agent. J Pharmacol Exp Ther. 2012;343:426–433. doi: 10.1124/jpet.112.195768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaney SG, Sancar A. DNA repair: Enzymatic mechanisms and relevance to drug response. J Natl Cancer Inst. 1996;88:1346–1360. doi: 10.1093/jnci/88.19.1346. [DOI] [PubMed] [Google Scholar]
- 8.Woynarowska BA, Woynarowski JM, Herzig MCS, Roberts K, Higdon AL, MacDonald JR. Differential cytotoxicity and induction of apoptosis in tumor and normal cells by hydroxymethylacylfulvene (HMAF) Biochem Pharmacol. 2000;59:1217–1226. doi: 10.1016/s0006-2952(00)00254-9. [DOI] [PubMed] [Google Scholar]
- 9.Koeppel F, Poindessous V, Lazar V, Raymond E, Sarasin A, Larsen AK. Irofulven cytotoxicity depends on transcription-coupled nucleotide excision repair and is correlated with XPG expression in solid tumor cells. Clin Cancer Res. 2004;10:5604–5613. doi: 10.1158/1078-0432.CCR-04-0442. [DOI] [PubMed] [Google Scholar]
- 10.Jaspers NGJ, Raams A, Kelner MJ, Ng JMY, Yamashita YM, Takeda S, McMorris TC, Hoeijmakers JHJ. Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair. 2002;1:1027–1038. doi: 10.1016/s1568-7864(02)00166-0. [DOI] [PubMed] [Google Scholar]
- 11.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 12.Pallis AG, Karamouzis MV. DNA repair pathways and their implication in cancer treatment. Cancer Metastasis Rev. 2010;29:677–685. doi: 10.1007/s10555-010-9258-8. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Perez I. DNA repair inhibitors in cancer treatment. Clin Transl Oncol. 2006;8:642–646. doi: 10.1007/s12094-006-0034-8. [DOI] [PubMed] [Google Scholar]
- 14.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ljungman M. Targeting the DNA damage response in cancer. Chem Rev. 2009;109:2929–2950. doi: 10.1021/cr900047g. [DOI] [PubMed] [Google Scholar]
- 16.Jiang H, Yang LY. Cell cycle checkpoint abrogator UCN-01 inhibits DNA repair: Association with attenuation of the interaction of XPA and ERCC1 nucleotide excision repair proteins. Cancer Res. 1999;59:4529–4534. [PubMed] [Google Scholar]
- 17.Reed AM, Fishel ML, Kelley MR. Small-molecule inhibitors of proteins involved in base excision repair potentiate the anti-tumorigenic effect of existing chemotherapeutics and irradiation. Future Oncol. 2009;5:713–726. doi: 10.2217/FON.09.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 19.Rosell R, Lord RVN, Taron M, Reguart N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer. 2002;38:217–227. doi: 10.1016/s0169-5002(02)00224-6. [DOI] [PubMed] [Google Scholar]
- 20.Jiang H, Li L, Yang LY. 7-Hydroxystaurosporine (UCN-01) inhibits nucleotide excision repair (NER) : Attenuation of ERCC1-XPA interaction. P Am Assoc Canc Res. 1999;40:402. [PubMed] [Google Scholar]
- 21.McNeil EM, Melton DW. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012;40:9990–10004. doi: 10.1093/nar/gks818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy thinking beyond the cell cycle. Mol Interv. 2011;11:133–140. doi: 10.1124/mi.11.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelner MJ, McMorris TC, Estes L, Rutherford M, Montoya M, Goldstein J, Samson K, Starr R, Taetle R. Characterization of illudin-S sensitivity in DNA repair-deficient chinese-hamster cells - unusually high-sensitivity of ERCC2 and ERCC3 DNA helicase-deficient mutants in comparison to other chemotherapeutic-agents. Biochem Pharmacol. 1994;48:403–409. doi: 10.1016/0006-2952(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 24.Tanasova M, Sturla SJ. Chemistry and biology of acylfulvenes: sesquiterpene-derived antitumor agents. Chem Rev. 2012;112:3578–3610. doi: 10.1021/cr2001367. [DOI] [PubMed] [Google Scholar]
- 25.Mendez F, Goldman JD, Bases RE. Abasic sites in DNA of HeLa cells induced by lucanthone. Cancer Invest. 2002;20:983–991. doi: 10.1081/cnv-120005914. [DOI] [PubMed] [Google Scholar]
- 26.McMorris TC, Kelner MJ, Wang W, Diaz MA, Estes LA, Taetle R. Acylfulvenes, a new class of potent antitumor agents. Experientia. 1996;52:75–80. doi: 10.1007/BF01922420. [DOI] [PubMed] [Google Scholar]
- 27.Pietsch KE, Neels JF, Yu X, Gong JC, Sturla SJ. Chemical and enzymatic reductive activation of acylfulvene to isomeric cytotoxic reactive intermediates. Chem Res Toxicol. 2011;24:2044–2054. doi: 10.1021/tx200401u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan UPF, Lee JFY, Wang SH, Leung KL, Chen GG. Induction of colon cancer cell death by 7-hydroxystaurosporine (UCN-01) is associated with increased p38 MAPK and decreased Bcl-X-L. Anti-Cancer Drugs. 2003;14:761–766. doi: 10.1097/00001813-200310000-00012. [DOI] [PubMed] [Google Scholar]
- 29.Yan L, Bulgar A, Miao YL, Mahajan V, Donze JR, Gerson SL, Liu LL. Combined treatment with temozolomide and methoxyamine: Blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase II alpha. Clin Cancer Res. 2007;13:1532–1539. doi: 10.1158/1078-0432.CCR-06-1595. [DOI] [PubMed] [Google Scholar]
- 30.Liu XD, Pietsch KE, Sturla SJ. Susceptibility of the antioxidant selenoenyzmes thioredoxin reductase and glutathione peroxidase to alkylation-mediated inhibition by anticancer acylfulvenes. Chem Res Toxicol. 2011;24:726–736. doi: 10.1021/tx2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres-Ramos CA, Johnson RE, Prakash L, Prakash S. Evidence for the involvement of nucleotide excision repair in the removal of abasic sites in yeast. Mol Cell Biol. 2000;20:3522–3528. doi: 10.1128/mcb.20.10.3522-3528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelner MJ, McMorris TC, Montoya MA, Estes L, Uglik SF, Rutherford M, Samson KM, Bagnell RD, Taetle R. Characterization of MGI 114 (HMAF) histiospecific toxicity in human tumor cell lines. Cancer Chemother Pharmacol. 1999;44:235–240. doi: 10.1007/s002800050972. [DOI] [PubMed] [Google Scholar]
- 33.Riss TL, Moravec RA. Use of multiple assay endpoints to investigate the effects of incubation time, dose of toxin, and plating density in cell-based cytotoxicity assays. Assay Drug Dev Technol. 2004;2:51–62. doi: 10.1089/154065804322966315. [DOI] [PubMed] [Google Scholar]
- 34.Kelner MJ, McMorris TC, Rojas RJ, Estes LA, Suthipinijtham P. Synergy of irofulven in combination with various anti-metabolites, enzyme inhibitors, and miscellaneous agents in MV522 lung carcinoma cells: marked interaction with gemcitabine and 5-fluorouracil. Invest New Drugs. 2008;26:407–415. doi: 10.1007/s10637-008-9113-8. [DOI] [PubMed] [Google Scholar]
- 35.Edelman MJ, Bauer KS, Smith R, Bisacia S, Dancey J. Phase I and pharmacokinetic study of 7-hydroxystaurosporine and carboplatin in advanced solid tumors. Clin Cancer Res. 2007;13:2667–2674. doi: 10.1158/1078-0432.CCR-06-1832. [DOI] [PubMed] [Google Scholar]
- 36.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Musich PR, Zou Y. Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol Biol. 2011;2:138–145. [PMC free article] [PubMed] [Google Scholar]
- 38.Yamauchi T, Keating MJ, Plunkett W. UCN-01 (7-hydroxystaurosporine) inhibits DNA repair and increases cytotoxicity in normal lymphocytes and chronic lymphocytic leukemia lymphocytes. Mol Cancer Ther. 2002;1:287–294. [PubMed] [Google Scholar]