

Abstract
Inflammatory bowel disease (IBD) includes Crohn’s disease and ulcerative colitis. The exact etiology and pathology of IBD remain unknown. Available evidence suggests that an abnormal immune response against the microorganisms in the intestine is responsible for the disease in genetically susceptible individuals. Dysregulation of immune response in the intestine plays a critical role in the pathogenesis of IBD, involving a wide range of molecules including cytokines. On the other hand, besides T helper (Th) 1 and Th2 cell immune responses, other subsets of T cells, namely Th17 and regulatory T cells, are likely associated with disease progression. Studying the interactions between various constituents of the innate and adaptive immune systems will certainly open new horizons of the knowledge about the immunologic mechanisms in IBD.
Keywords: Crohn’s disease, Inflammatory bowel disease, Proinflammatory cytokines, T helper cells, T helper 17 cells, Ulcerative colitis
Core tip: The etiology and pathology of inflammatory bowel disease (IBD) remain elusive, and dysregulation of the mucosal immune response toward commensal bacterial flora together with genetic and environmental factors may play important roles in the pathogenesis of IBD. A better understanding of the mechanisms of immune responses in the intestinal mucosa will provide new insights into the pathogenesis of IBD, and shed some light on targeted immune therapy for this disease.
INTRODUCTION
The etiology and pathogenesis of inflammatory bowel disease (IBD) remain elusive, and accumulating evidence has indicated that sustained intestinal infections, mucosal barrier defects, mucosal immune dysregulation, genetic and environmental factors are involved in the disease process[1-4]. Among these, the dysfunction of the mucosal immune system plays an important role in the pathogenesis of IBD (Figure 1). Among a variety of inflammatory cells in the gut, mucosal CD4+ T cells are thought to play a central role in both the induction and persistence of chronic inflammation by producing proinflammatory cytokines. Previous studies have indicated that T helper (Th) 1-related cytokines [e.g., tumor necrosis factor (TNF), interferon (IFN)-γ, interleukin (IL)-12] as well as Th17-associated cytokines (e.g., IL-17A, IL-21, IL-23) are markedly increased in inflamed mucosa of Crohn’s disease (CD) patients, whereas the cytokine profiles in inflamed areas of ulcerative colitis (UC) patients seem to exhibit increased production of the Th2-associated cytokines such as IL-4 and IL-13[1-3]. These proinflammatory cytokines are potent in vitro stimulators of intestinal mucosal effect or functions, including T cell and macrophage proliferation, adhesion molecule expression, chemokine expression, and secretion of other proinflammatory cytokines.
Figure 1.
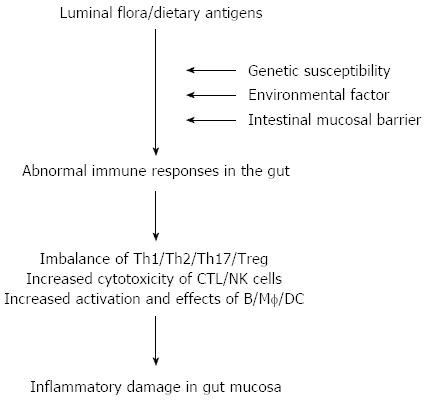
Pathogenesis of inflammatory bowel disease. DC: Dendritic cell; NK: Natural killer; Th: T helper; Treg: T regulator cell; CTL: Cytotoxic T lymphocyte; Mf: Macrophages.
ABNORMAL IMMUNE RESPONSE IN THE INFLAMED MUCOSA OF IBD PATIENTS
Antigen-specific activation of various lymphocytes within the intestinal mucosa by enteric pathogens is an important feature of IBD immunopathology[1-4]. Under physiological conditions, a large number of innate and immune cells are located in the intestinal lamina propria, such as T, B, natural killer (NK), NKT cells, macrophages (Mf), dendritic cells (DCs), mast cells, neutrophils, eosinophils, as well as stromal cells (such as fibroblasts). It is actually surprising that the large lymphoid system in the intestine coexists so peacefully with the external environment, a single epithelial layer away from the luminal microbial flora. However, under inflammatory conditions, a large number of activated immune cells infiltrate into the intestinal mucosa. These immune cells and some stromal cells not only express high levels of adhesion molecules and auxiliary signal molecules (such as CD54, CD62L), but also express high levels of inflammatory mediators and chemokine receptors (such as CCR5, CCR6, and CCR9) and integrin (such as integrin α4β7). Moreover, fibroblasts and capillary endothelial cells in the intestinal mucosa also express high levels of chemokines, selectins (e.g., selectin E) and intracellular adhesion molecule-1 (ICAM-1, or CD54), which further induce intermolecular interactions of leukocytes in the blood circulation to migrate into the intestinal mucosa, and promote local inflammatory response[1-4].
Evidence has demonstrated that CD4+ T cells isolated from inflamed mucosa of CD patients, when stimulated in vitro, are able to produce large amounts of Th1/Th17-associated proinflammatory cytokines (e.g., IFN-γ, TNF, and IL-17A), while in UC inflamed tissue CD4+ T cells and NK T cells secrete high levels of Th2-related cytokines (e.g., IL-4 and IL-13) and Th17-associated proinflammatory cytokines (e.g., IL-17A)[1-4]. The unbalance of pro/antiinflammatory cytokines contributes to intestinal mucosal inflammation. Recent studies have found that some proinflammatory cytokines (e.g., IL-12, IL-18, IL-21, and IL-23) are significantly increased in inflamed mucosa of CD patients[5-7], and that some inhibitory cytokines (e.g., TGF-β, IL-10, IL-25, IL-33, and IL-37) are significantly reduced. Moreover, loss of forkhead proteins (Foxp)3+ regulatory T cells (Treg) and FoxP3-IL-10+ CD4+ cells are also found in the inflamed mucosa of IBD patients, and these events result in not maintaining intestinal mucosal immune tolerance and further promoting local intestinal mucosal immune response, leading to the intestinal mucosal injury[8].
PLEIOTROPIC ROLE OF IL-21 IN IMMUNE RESPONSE
IL-21 is a member of the IL-2 family of cytokines, expressed mainly by CD4+ T cells, including Th1, Th2, and Th17 cells (Figure 2)[9,10]. IL-21 receptor (IL-21R) is structurally related to IL-2R and IL-15R, and is expressed in T, NK, B cells, and DCs[9]. IL-21 exhibits a pleiotropic capacity to regulate T cell differentiation and function, enhances clonal expansion of antigen-activated naive CD4+ and CD8+ T cells, and induces the expression of genes encoding IL-12R, IL-18R, IFN-γ, IL-2Rɑ, and the Th1-associated transcription factor T-bet in activated memory T cells[11,12]. IL-21 is also associated with the Th2-mediated immune response and plays a role in inhibiting the differentiation of naive Th cells into IFN-γ-producing Th1 cells. In synergy with IL-15, Ftl-3 ligand, and stem cell factor, IL-21 promotes human NK cell maturation and activation. It exerts further biological functions in B cells, regulates differentiation and antibody production, including production of all IgG isotypes, and synergizes with anti-CD40 mAb to stimulate B-cell activation, clonal expansion, and maturation (Figure 2)[13-15].
Figure 2.
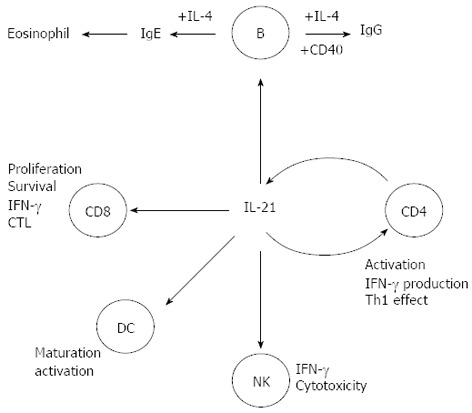
Pleiotropic role of interleukin-21 in immune responses. IFN: Interferon; IL: Interleukin; CD: Crohn’s disease; DC: Dendritic cell; NK: Natural killer; CTL: Cytotoxic T lymphocyte.
In recent years, IL-21 has been found to be produced in excess in the intestine of IBD patients and may be involved in the pathogenesis of human IBD[16-18]. When mucosal T cells from CD patients are activated in vitro with anti-CD3 in the presence of either a neutralizing anti-IL-21 antibody or an IL-21R-IgG fusion protein, the production of both IL-17A and IFN-γ is reduced. These results together with the demonstration that IL-21-deficient mice are resistant against Th1/Th17 cell-driven colitis support the key role of IL-21 in positively regulating Th1 and Th17 cell-associated inflammatory pathways. IL-21 exerts further biological functions that could contribute to its proinflammatory effect in the gut. For example, IL-21 stimulates stromal cells to produce tissue-degrading proteases and enhances secretion of the T-cell chemoattractant macrophage inflammatory protein-3a by intestinal epithelial cells. IL-21 potentates the expression of Th1-related transcription factors and IFN-γ in T and NK cells and the cytotoxic activity of NK cells. IL-21 also inhibits the peripheral differentiation of Tregs and makes CD4+ T cells resistant to Treg-mediated immune suppression. Therefore, through the multiple pathways IL-21 can damage the gut, and neutralizing IL-21 may have a therapeutic potential in the management of IBD[13,18].
We have also investigated expression of IL-21R in inflamed mucosa of IBD patients and evaluated its role in the induction of NK cell cytotoxicity and activation as well as Th17 differentiation[5]. The results have shown that IL-21R-positive cells are significantly increased in inflamed mucosa of IBD patients compared with healthy controls, and IL-21R is mainly expressed in freshly isolated peripheral blood (PB)- and lamina propria (LP)-CD4+, CD8+ T, B, and NK cells. When stimulated with immobilized human IgG and IL-21, PB-NK cells from IBD patients produce higher levels of IFN-γ and TNF than those from controls. IL-21-primed IBD NK cells show a more potent antitumor cytotoxicity to NK-sensitive K562 cells than controls. Moreover, PB-T and LP-T cells from IBD patients produce larger amounts of proinflammatory cytokines (e.g., TNF and IFN-γ) than those from controls when stimulated with IL-21 and anti-CD3. Importantly, IL-21 facilitates IBD CD4+ T cells to differentiate into Th17 cells[5]. In our further study, we have also evaluated the role of anti-TNF mAb (infliximab, IFX) in regulating IL-21 expression and Th17 cell infiltration in the intestinal mucosa of CD patients. Twenty-six CD patients were treated with IFX at weeks 0, 2 and 6. IL-21 and Th17 cells were found to be expressed highly in inflamed mucosa of active CD patients compared with healthy controls. Ten weeks after IFX infusion, CD activity index, erythrocyte sedimentation rate, serum C-reactive protein (CRP) and intestinal mucosal healing were improved markedly in CD patients. Moreover, IL-21 expression and Th17 cell infiltration were also found to be significantly decreased compared with those before IFX therapy[19]. These data indicate that IL-21 plays an important role in the pathogenesis of IBD.
PROINFLAMMATORY ROLE OF IL-23 IN THE PATHOGENESIS OF IBD
Recent advances have also indicated that IL-23, mainly produced by macrophages, is one of the critical cytokines in IBD and is essential for promoting chronic intestinal inflammation[20,21]. IL-23 and IL-12 are members of a small family of proinflammatory heterodimer cytokines, sharing a common p40 subunit covalently linked to a p35 subunit to form IL-12 or to a p19 subunit to form IL-23. IL-12R is comprised of an IL-12Rβ1 and IL-12Rβ2 subunit, whereas the receptor for IL-23 consists of the IL-12Rβ1 subunit and a novel component termed IL-23R[20], which is expressed predominantly on T, NK, and NKT cells and to a smaller extent, on monocytes, macrophages, and DCs. After binding to the IL-23R, IL-23 preferentially induces memory T cell activation. IL-23 exhibits some similar biological activities to IL-12, however, in comparison with IL-12 with profound induction of the Th1 immune response, as well as promotion of cytotoxic, antimicrobial, and anti-tumor responses, IL-23 is found to play a critical role in the maintenance of immune response by controlling T cell memory function and by influencing the proliferation and survival of IL-17-producing Th17 cells[22,23]. Moreover, recent work has also shown that IL-23 could induce naive CD4+ T cells to secrete IL-22, indicating that IL-23 is also associated with the differentiation of naive CD4+ T cells[24].
Previous studies with murine colitis models have demonstrated a requirement for IL-23 in the development of intestinal mucosal inflammation. IL-23p19 subunit knockout results in a decrease of proinflammatory cytokines (e.g., TNF, IFN-γ, IL-6, and IL-17) and the presence of less intestinal mucosal inflammation[25,26]. Moreover, in vivo blockade of IL-23 using anti-IL-23p19 mAb or its inhibitor STA-5326 could inhibit chronic intestinal inflammation in a colitis model and down-regulate a Th1-mediated immune response[26,27]. Elevation of IL-23p19 transcript levels has been observed in inflamed mucosa of IBD patients, and its expression is correlated with the severity of endoscopic lesions[28]. Recent work has demonstrated that myeloid DCs from mesenteric lymphoid nodes of CD patients, when stimulated with exogenous microbial antigens in vitro, produce higher levels of IL-23p19 than UC patients and healthy controls[29].
In order to investigate the pathogenic role of IL-23 in the induction of mucosal inflammation in IBD, we have analyzed IL-23p19 expression in inflamed mucosa of IBD patients and its role in the induction of intestinal epithelial lymphocyte (IEL) and NK cell activation as well as Th17 cell differentiation. Expression of IL-23p19 has been observed to be increased significantly in inflamed mucosa of CD patients compared with that in UC patients and healthy controls. IL-23R cells are increased significantly in PB- and LP-CD4+ and -CD8+ T and NK cells. IL-23 could markedly promote IBD IEL and NK cell activation and cytotoxicity and triggered IBD PB- and LP-T cells to secrete significantly higher levels of IFN-γ, TNF, IL-2, and IL-17A compared with healthy controls. IL-23 promotes IBD PB- or LP-CD4+ T cells to differentiate into Th17 cells. These data indicate that IL-23 plays an important role in the induction of IEL, NK, and T cell activation, proinflammatory cytokine secretion, and Th17 cell differentiation[6]. In two IBD models there is excessive accumulation of short-lived neutrophils and inflammatory monocytes in the intestine. IL-23-driven colitogenic T cell program has been found to regulate upstream hematopoietic stem and progenitor cells (HSPC)[30]. Targeted therapy directed against IL-23 may have a therapeutic role in treatment of IBD.
Additionally, we have also elucidated the further role of IL-23 in the suppression of IL-10 in the IBD intestinal mucosa[7]. IL-10 is an important cytokine in the induction of Th2 response that plays a crucial role in adaptive immunity via the induction of specific antibodies to eliminate the reinvasion of microbes and the absorption of microbial products. IL-10 is also one of the most effective immune regulatory cytokines contributing to maintaining the homeostasis of the body[31,32]. Previous studies indicate that the production of IL-10 in the intestine of IBD patients is suppressed, but the underlying mechanism has not fully understood yet. Therefore, we examined the expression of IL-10, IL-23, and IgA in the surgically removed colon specimens and found that the levels of IgA and IL-10 were significantly lower, and both negatively correlated with IL-23 expression and the infiltration of inflammatory cells in the IBD mucosa. The production of IL-10 by lamina propria mononuclear cells was lower in the IBD group than the control, and these levels could be enhanced by blocking IL-23. The gene transcription of IL-10 was significantly suppressed in CD4+ T cells of IBD mucosa, and this phenomenon could be replicated in vitro by adding IL-23 in the culture of polarized Th2 cells[7]. Thus we conclude that overexpression of IL-23 in the intestinal mucosa weakens the defensive barrier in the gut and disturbs the local immune regulation (IL-10 and Treg) (Figure 3).
Figure 3.
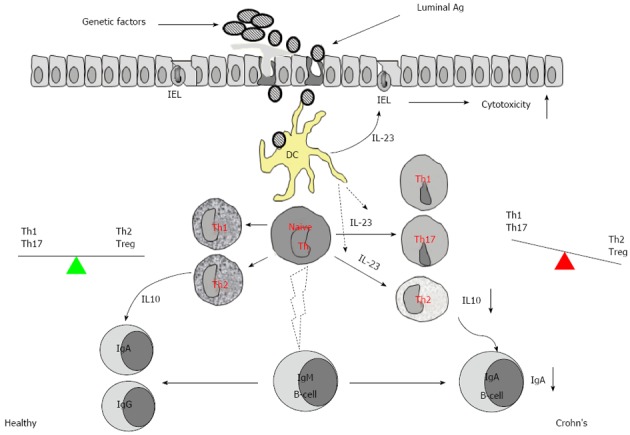
Overexpression of interleukin-23 in inflamed mucosa of patients with inflammatory bowel disease weakens the intestinal defensive barrier and disturbs the immune regulation in intestinal mucosa. IEL: Intestinal epithelial lymphocyte; IL: Interleukin; Th: T helper; Treg: T regulator cell; DC: Dendritic cell.
POTENTIAL ROLE OF IL-25 IN THE DEVELOPMENT OF IBD
IL-25 (also known as IL-17E) is a distinct member of the IL-17 family of cytokines, including IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F. IL-25 shares the receptor IL-17 receptor homolog 1 (IL-17Rh1) (also named the IL-17RB) with IL-17B, although it binds with a much higher affinity. IL-25R is a 56-kDa single transmembrane protein and is expressed in Th2 central memory cells, eosinophils, monocytes, airways smooth muscle cells, fibroblasts, and endothelial cells. Evidence has shown that IL-25 is also involved in the immune responses in gut mucosa[33]. Previous work has demonstrated that IL-25 is constitutively expressed by intestinal mucosal T cells of mouse strains (e.g., BALB/c, C57BL/6 mice) that are resistant to helminth Trichurismuris infection, whereas IL-25-deficient mice on a genetically resistant background fail to develop a Th2-mediated immune response or eradicate Trichuris infection but develop severe infection-induced intestinal inflammation. Moreover, the immunopathology in Trichuris-induced IL-25-deficient mice is also associated with increased expression of IFN-γ and IL-17A in the mesenteric lymph nodes and cecum[33]. Administration of IL-25 could prevent intestinal mucosal inflammation in experimental colitis induced with peptidoglycan, 2,4,6-trinitrobenzenesulphonic acid, or oxazolone in mice. These data indicate that IL-25, which promotes the differentiation and activation of Th2 cells in gut mucosa, plays a critical role in the attenuation of destructive intestinal inflammation[33,34].
IL-25 has been also found to be decreased in the inflamed mucosa of IBD patients and could decrease the synthesis of IL-12 and IL-23 in the CD14+Mf from the inflamed mucosa of patients with CD in vitro[35]. Therefore, IL-25 may be a negative regulator of inflammatory responses in the intestinal mucosa. However, the exact role of IL-25 in the development of IBD remains to be elucidated. Recently, we have also studied the role of IL-25 in the pathogenesis of IBD[36]. The results have demonstrated that IL-25 is significantly decreased in the sera and inflamed mucosa of patients with active IBD compared with controls. The levels of IL-25 in inflamed mucosa and sera are inversely correlated with endoscopic disease activities and CRP, respectively, in IBD. IL-25 could markedly inhibit IBD CD4+ T cells to produce TNF, IFN-γ, and IL-17A but promote IL-10 secretion. IL-25 could suppress the differentiation of IBD CD4+ T cells into Th1 and Th17 cells but did not interfere with Th2 cell differentiation. Importantly, blockade of IL-10 secretion by IBD CD4+ T cells markedly attenuates the inhibitory role of IL-25 in modulating both Th1 and Th17 immune responses (Figure 4). Our study provides evidence that IL-25 is a critical anti-inflammatory cytokine in the pathogenesis of IBD and may be considered as a potential therapeutic agent for human IBD[36].
Figure 4.
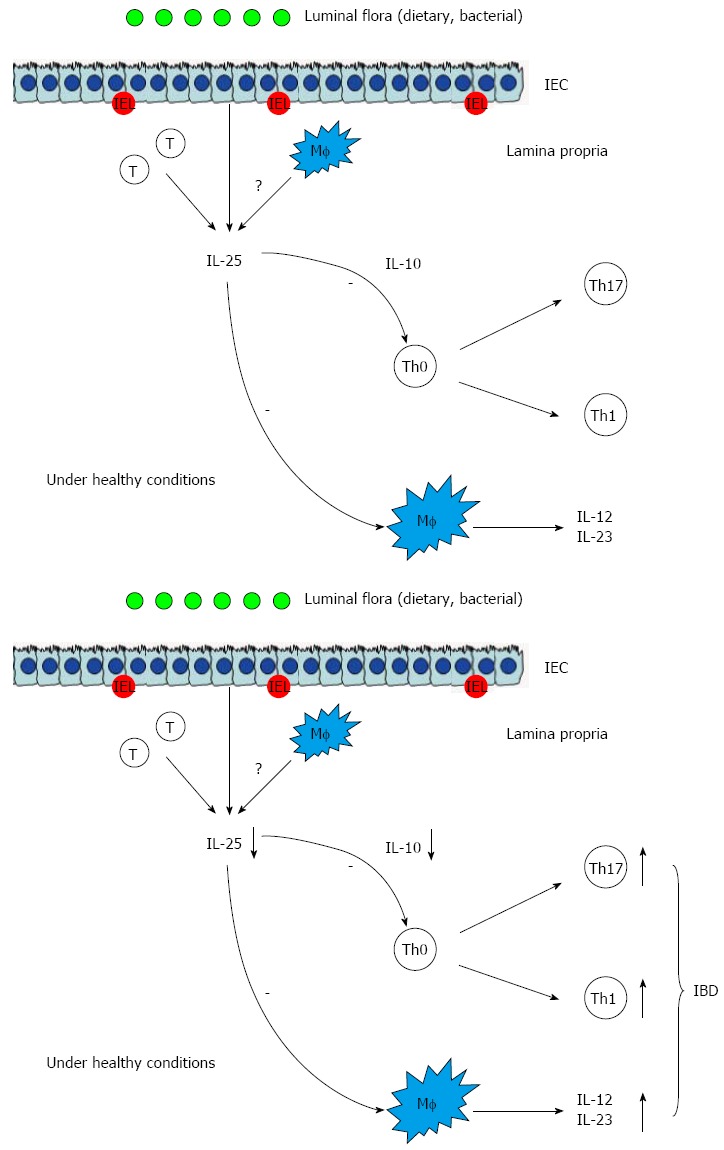
Inhibitory role of interleukin-25 in intestinal mucosa. IEL: Intestinal epithelial lymphocyte; IL: Interleukin; Th: T helper; Treg: T regulator cell; CTL: Cytotoxic T lymphocyte; Mf: Macrophages; IEC: Intestinal epithelial cells; IBD: Inflammatory bowel disease.
POTENTIAL ROLE OF THE TH17/IL-17 AXIS IN THE PATHOGENESIS OF IBD
IL-17 has pleiotropic activities, functions through the adaptive and innate immune system to promote immune response, and plays an important role in immune responses. The identification of the IL-17 family of cytokines as well as the IL-23-mediated expansion of IL-17-producing T cells has uncovered a new subset of Th cells, designated as Th17 cells. Th17 cells originate from naive CD4+ T cells in the presence of TGF-β and IL-6. Th17 cell differentiation does not require IL-17. The amplification and stabilization of Th17 are provided by IL-21 and IL-23. At the same time, the RORγt is identified as the master key regulator and transcription factor of Th17 cell differentiation[37,38]. The IL-17 cytokine family includes six members, IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (or IL-25) and IL-17F, and act in vitro and in vivo as potent proinflammatory cytokines[38]. IL-17A can induce the expression of proinflammatory cytokines (such as IL-6 and TNF), chemokines (such as KC, MCP-1 and MIP-2) and matrix metalloproteases, which mediate tissue infiltration and tissue destruction. It is also involved in the proliferation, maturation and chemotaxis of neutrophils[38].
Evidence has shown that high numbers of Th17 cells are present in the colonic LP of the ileum and colon in conventionallyraised mice, and that these cells are highly infiltrated in inflamed areas of colitic mice[39,40]. Further analysis confirms that commensal gut flora contributes to the expansion of these CD4+ Th17 cells, leading to intestinal mucosal inflammation. In terms of mucosal immunity, the IL-23/IL-17 axis has been observed to play an important role in normal intestinal homeostasis.
To date, IL-17 and other Th17-associated cytokines (e.g., IL-22 and IL-23) have been found to have protective or pathogenic effects dependent on other effective factors in local tissue. Recent work[41] has also demonstrated that most of the transcripts for Th17-related cytokines are increased in IBD patients compared to normal controls, but more abundant in UC than in CD. In contrast, upregulation of IFN-γ mRNA is marked in CD LP CD4+ T cells. Up-regulation of IL-23p19 mRNA is detected in colonic mucosa from both UC and CD patients. The significance of Th17 immunity in UC is further supported by the finding that recombinant IL-23 actually enhances IL-17A production by LP CD4+ T cells in UC, but has a lesser effect on LP CD4+ T cells in CD[42]. IL-17A is protective against dextran sodium sulfate (DSS)-induced colitis and colitis in the T cell transfer model, in which T cells are injected into lymphopoenic mice. However, mice deficient in IL-17F are resistant to DSS-induced colitis. These data suggest that IL-17F, not IL-17A, is pathogenic in the gut. IL-17 may synergize with other inflammatory mediators in the gut. Recent studies have highlighted further potential heterogeneity within Th17 cell populations by demonstrating that some may even secrete IL-10[43], a factor known to inhibit intestinal inflammation. Thus, it is possible that the actions of Th17 cells may differ dependently on other factors that may be present in the local environment. In the normal intestine, the primary function of Th17 cells may be like sentinels which contribute to maintaining epithelial barrier function, whereas in sites of chronic intestinal inflammation, high levels of IL-23 may activate their full pathogenic and antibacterial functions. Recently, Secukinumab (an IL-17A antibody), Brodalumab (an IL-17 receptor antibody) and two small-molecule drugs (Vidofludimus and Tofacitinib) are used in clinical trials for IBD patients, which inhibit IL-17 as part of their overall pharmacological profiles[44].
IL-27 AND IL-35: NEWER MEMBERS OF THE IL-12 FAMILY
The IL-12 family is made up of secreted heterodimers with some overlapping usage between family members. IL-12 (p35/p40) and IL-23 (p19/p40) are the best-known members of the IL-12 family. Other members include IL-27 (EBi3/p28) and IL-35 (EBi3/p35). Like the IL-17 family, however, a critical issue is whether these molecules are pathogenic or protective in the gut. IL-27, produced mostly by myeloid cells, is present at increased concentrations in IBD mucosa. If T cells are taken from mice deficient in the IL-27R and injected into lymphopoenic mice, they induce significantly less colitis than wild-type T cells, clearly suggesting that IL-27 is pathogenic in this model. IL-27R-null mice are also less susceptible to DSS-induced colitis, again suggesting that IL-27 is a proinflammatory mediator. In marked contrast, however, treating mice with established TNBS-induced colitis with IL-27 reduces disease and cytokine production. In humans, IL-27 has also been observed to reduce proinflammatory cytokine production in Mf activated with TNF[45].
MICRORNA AND THE INTESTINAL IMMUNE HOMEOSTASIS
MicroRNA (miR) is an emerging group of short, noncoding RNAs that play an important role in regulating expression of classical genes at the post-transcriptional level. miR regulates cell proliferation, apoptosis, growth, cell differentiation, metabolism and other processes. Recently, miR in intestinal epithelial cells has been found to regulate intestinal mucosal barrier function through its important effect on intestinal epithelial cell proliferation and differentiation. Moreover, differential expression of miR in immune cells within the intestinal mucosa affects the intestinal immune homeostasis[46,47].
In recent years, evidence has suggested that intestinal epithelial miR expression is closely related with the incidence and development of IBD[46]. The expression of miR-192, miR-375 and miR-422b is significantly decreased in inflamed mucosa of patients with active UC, while miR-16, miR-21 and let-7 expression is significantly increased. miR-19b and miR-629 are significantly decreased in patients with active CD inflammation within the intestinal mucosa, while miR-23b, miR-106 and miR-191 are significantly increased. The abnormal miR expression will affect translation process of its corresponding target gene mRNAs, regulate gene expression and thus participate in inflammatory injury of intestinal mucosa in IBD. A recent study by Coskun et al[48] provides the first evidence that miR-20b, miR-98, miR-125b-1*, and let-7e* are deregulated in patients with UC.
We have also investigated the expressions of miR10a in IBD and found that the expression of miR10a is decreased in inflamed intestinal mucosa of IBD patients. Furthermore, we have also found that TNF inhibits miR10a expression, while blockade TNF with anti-TNF mAb markedly enhances miR10a expression in the intestinal mucosa (unpublished data). Our findings, together with previous data showing that miR10a could block intestinal inflammation in mice and reduce the differentiation Th1 and Th17[49], further prove that miR10a is involved in intestinal mucosal inflammatory response, and that targeted therapy may be beneficial for human IBD.
ROLE OF REGULATORY B CELLS
B cells are a source of inhibitory cytokines such as IL-10 and TGF-β. Depending on the signals B cells receive, pro- or antiinflammatory cytokines can be produced, and the shift towards an inflammatory or a protective/suppressive response will be induced. Specific B cell subset found to affect autoreactive responses and suggested to have a regulatory role in autoimmune diseases is B-regulatory cells (Bregs). CD19+CD25+ B cells were the first subset of human B cells previously suggested to have a regulatory role. CD19+CD25+ B cells contribute up to 30% of all peripheral blood B cells in mice and can effectively present peptides to helper T cells. In humans, only B cells expressing high levels of CD25 (BCD25+) seem capable of acting as Bregs with abundant TGF-β production. However, it remains unknown how to identify Bregs with membrane markers or transcription factors. Several signals like B cell receptor (BCR), CD40 and/or Toll-like receptor (TLRs) may include. These different activation signals (alone or combined) were shown by many studies to increase regulatory functions of Bregs[50].
Bacterial molecules have been used to stimulate Bregs. In mice, splenic B cells stimulated ex vivo by bacteria acquire the CD5+ CD1d+ phenotype, which is characterized by high level IL-10 expression and being capable of markedly suppressing the activity of experimental IBD. The protective subset (contributing 1% to 2% of all splenic B cells) is composed of CD5+CD1d(high) B cells activated via the TLR2/4 pathway by bacterial antigens in the gut flora. Investigations into the influence of the gut microbiota on the balance between effector B cells and Bregs may open up new therapeutic possibilities in IBD[51,52].
ROLE AND FUNCTIONAL ALTERATIONS OF THE INNATE IMMUNITY
Innate immunity prevents pathogens from entering and spreading within the body. The intestinal innate immune system involves three lines of defense: the mucus layer, epithelium, and lamina propria. Mucus is the first line of intestinal defense, and the major constitutive proteins are mucins (MUC), with diverse isotypes in different portions of the gastrointestinal tract. CD patients show decreased MUC1 and MUC4 levels in the ileum, while MUC2, MUC5AC, MUC5B, MUC6, and MUC7 are undetectable in lesions. UC patients also show decreased MUC2 expression. As the second line of defense, the intestinal epithelium is composed of a monolayer of fast replicating polarized cells: enterocytes, goblet cells, and enteroendocrine cells. All are bound together through tight junctions that separate the body from intestinal lumen components. The membrane TLRs and cytosolic nucleotide oligomerization domain receptor (NOD) are the most important among intestinal pathogen recognition receptors. NOD2 has also been described as a negative regulator of TLR2-mediated IL-12 secretion. Some NOD2 mutations have been described in CD patients, which are associated with decreased defensin secretion by ileal mucosa Paneth cells.
Lamina propria as a third line of intestinal defense contains innate and immune cells. In IBD patients, DCs promote a robust recognition of bacterial products that might cause an immune responseto commensal bacteria, provoking a loss of intestinal tolerance. Intestinal macrophages from IBD patients have lost the ability to maintain tolerance, mainly through increased surface CD14 content and NF-κB transcription pathway activity, which might induce increased peripheral macrophage recruitment. Increased cytotoxic activity and elevated NK cell counts are also found in IBD patients. Crucial molecules in NK cells, such as IL-15, IL-21 and IL-23, and their cognate receptors, are elevated in the intestinal mucosa of UC patients. Moreover, in IBD the IEL activation increases, resulting in elevated production of IFN-γ, TNF and IL-2, which is associated with increased IL-23[53,54].
Deregulated mucin expression in IBD patients might be due to the cytokine imbalance that characterizes these diseases. These molecules stimulate various transcription factor pathways, such as JAK/STAT and NF-κB, and induce mucin secretion. The combination of TNF and IFN-γ could also decrease claudin-3, claudin-5 and claudin-7 expression, with a marked increase in paracellular permeability in rat colon. Moreover, TNF, IL-6 and IFN-γ increase apoptosis and monolayer permeability in HT-29/B6 cells. These cytokines also inhibit the woundhealing in HT-29/B6 cells. Increased apoptosis and delayed woundhealing of epithelial cells would augment monolayer permeability, and damage the epithelial barrier function[54,55].
CONCLUSION
The exact etiology of IBD is still not completely understood, and increasing data have demonstrated that these conditions occur through an inappropriate immune response to a subset of commensal enteric bacteria in a genetically susceptible host, with disease initiated by environmental triggers. Dysfunction of the mucosal immune system evokes intestinal inflammation through the activation of both innate and acquired immunity in the gut. Among these, T cell activation and Teff/Treg imbalance play an important role in the process of inflammation. Understanding of immunopathogenesis of IBD will help us to find new ideas for diagnosis and treatment.
Footnotes
Supported by Grants from the National Natural Science Foundation of China, No. 81061120521 and No. 81270470; Shanghai Science and Technology Commission, No. 12XD1404000
P- Reviewers: Capasso R, di Sebastiano P, Hewicker-Trautwein M, Neurath MF, Sipos F, Sinagra E, Tsai HH S- Editor: Zhai HH L- Editor: Wang TQ E- Editor: Zhang DN
References
- 1.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 2.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9:599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 3.Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 4.Latella G, Papi C. Crucial steps in the natural history of inflammatory bowel disease. World J Gastroenterol. 2012;18:3790–3799. doi: 10.3748/wjg.v18.i29.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Yang L, Cui Y, Wang X, Guo C, Huang Z, Kan Q, Liu Z, Liu Y. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:1133–1144. doi: 10.1002/ibd.20923. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Yadav PK, Xu X, Su J, Chen C, Tang M, Lin H, Yu J, Qian J, Yang PC, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89:597–606. doi: 10.1189/jlb.0810456. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Feng BS, Yang SB, Chen X, Su J, Yang PC. Interleukin (IL)-23 suppresses IL-10 in inflammatory bowel disease. J Biol Chem. 2012;287:3591–3597. doi: 10.1074/jbc.M111.304949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–859. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat Rev Immunol. 2005;5:688–698. doi: 10.1038/nri1688. [DOI] [PubMed] [Google Scholar]
- 10.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 11.Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 14.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175:7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 15.Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, Ettinger R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179:5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 16.Sarra M, Monteleone I, Stolfi C, Fantini MC, Sileri P, Sica G, Tersigni R, Macdonald TT, Pallone F, Monteleone G. Interferon-gamma-expressing cells are a major source of interleukin-21 in inflammatory bowel diseases. Inflamm Bowel Dis. 2010;16:1332–1339. doi: 10.1002/ibd.21238. [DOI] [PubMed] [Google Scholar]
- 17.Pallone F, Fina D, Caruso R, Monteleone G. Role of IL-21 in inflammatory bowel disease. Expert Rev Clin Immunol. 2010;6:537–541. doi: 10.1586/eci.10.44. [DOI] [PubMed] [Google Scholar]
- 18.MacDonald TT, Biancheri P, Sarra M, Monteleone G. What’s the next best cytokine target in IBD? Inflamm Bowel Dis. 2012;18:2180–2189. doi: 10.1002/ibd.22967. [DOI] [PubMed] [Google Scholar]
- 19.Liu C, Xia X, Wu W, Wu R, Tang M, Chen T, Xu F, Cong Y, Xu X, Liu Z. Anti-tumour necrosis factor therapy enhances mucosal healing through down-regulation of interleukin-21 expression and T helper type 17 cell infiltration in Crohn’s disease. Clin Exp Immunol. 2013;173:102–111. doi: 10.1111/cei.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 21.Croxford AL, Mair F, Becher B. IL-23: one cytokine in control of autoimmunity. Eur J Immunol. 2012;42:2263–2273. doi: 10.1002/eji.201242598. [DOI] [PubMed] [Google Scholar]
- 22.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 24.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 25.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology. 2009;137:1736–1745. doi: 10.1053/j.gastro.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 30.Griseri T, McKenzie BS, Schiering C, Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. 2012;37:1116–1129. doi: 10.1016/j.immuni.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Jarry A, Bossard C, Bou-Hanna C, Masson D, Espaze E, Denis MG, Laboisse CL. Mucosal IL-10 and TGF-beta play crucial roles in preventing LPS-driven, IFN-gamma-mediated epithelial damage in human colon explants. J Clin Invest. 2008;118:1132–1142. doi: 10.1172/JCI32140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caruso R, Sarra M, Stolfi C, Rizzo A, Fina D, Fantini MC, Pallone F, MacDonald TT, Monteleone G. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology. 2009;136:2270–2279. doi: 10.1053/j.gastro.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 36.Su J, Chen T, Ji XY, Liu C, Yadav PK, Wu R, Yang P, Liu Z. IL-25 downregulates Th1/Th17 immune response in an IL-10-dependent manner in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:720–728. doi: 10.1097/MIB.0b013e3182802a76. [DOI] [PubMed] [Google Scholar]
- 37.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 38.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 39.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niess JH, Leithäuser F, Adler G, Reimann J. Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J Immunol. 2008;180:559–568. doi: 10.4049/jimmunol.180.1.559. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, Kitazume MT, Nakazawa A, Sugita A, Koganei K, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57:1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 42.Feng T, Cao AT, Weaver CT, Elson CO, Cong Y. Interleukin-12 converts Foxp3+ regulatory T cells to interferon-γ-producing Foxp3+ T cells that inhibit colitis. Gastroenterology. 2011;140:2031–2043. doi: 10.1053/j.gastro.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 44.Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi: 10.1002/ibd.21677. [DOI] [PubMed] [Google Scholar]
- 45.Cox JH, Kljavin NM, Ramamoorthi N, Diehl L, Batten M, Ghilardi N. IL-27 promotes T cell-dependent colitis through multiple mechanisms. J Exp Med. 2011;208:115–123. doi: 10.1084/jem.20100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:187–193. doi: 10.1002/ibd.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coskun M, Bjerrum JT, Seidelin JB, Nielsen OH. MicroRNAs in inflammatory bowel disease--pathogenesis, diagnostics and therapeutics. World J Gastroenterol. 2012;18:4629–4634. doi: 10.3748/wjg.v18.i34.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coskun M, Bjerrum JT, Seidelin JB, Troelsen JT, Olsen J, Nielsen OH. miR-20b, miR-98, miR-125b-1*, and let-7e* as new potential diagnostic biomarkers in ulcerative colitis. World J Gastroenterol. 2013;19:4289–4299. doi: 10.3748/wjg.v19.i27.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xue X, Feng T, Yao S, Wolf KJ, Liu CG, Liu X, Elson CO, Cong Y. Microbiota downregulates dendritic cell expression of miR-10a, which targets IL-12/IL-23p40. J Immunol. 2011;187:5879–5886. doi: 10.4049/jimmunol.1100535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vadasz Z, Haj T, Kessel A, Toubi E. B-regulatory cells in autoimmunity and immune mediated inflammation. FEBS Lett. 2013;587:2074–2078. doi: 10.1016/j.febslet.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 51.Ochoa-Repáraz J, Mielcarz DW, Haque-Begum S, Kasper LH. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes. 2010;1:103–108. doi: 10.4161/gmic.1.2.11515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berthelot JM, Jamin C, Amrouche K, Le Goff B, Maugars Y, Youinou P. Regulatory B cells play a key role in immune system balance. Joint Bone Spine. 2013;80:18–22. doi: 10.1016/j.jbspin.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 53.Gersemann M, Wehkamp J, Stange EF. Innate immune dysfunction in inflammatory bowel disease. J Intern Med. 2012;271:421–428. doi: 10.1111/j.1365-2796.2012.02515.x. [DOI] [PubMed] [Google Scholar]
- 54.Xia XM, Wang FY, Zhou J, Hu KF, Li SW, Zou BB. CXCR4 antagonist AMD3100 modulates claudin expression and intestinal barrier function in experimental colitis. PLoS One. 2011;6:e27282. doi: 10.1371/journal.pone.0027282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heller F, Fromm A, Gitter AH, Mankertz J, Schulzke JD. Epithelial apoptosis is a prominent feature of the epithelial barrier disturbance in intestinal inflammation: effect of pro-inflammatory interleukin-13 on epithelial cell function. Mucosal Immunol. 2008;1 Suppl 1:S58–S61. doi: 10.1038/mi.2008.46. [DOI] [PubMed] [Google Scholar]