

Abstract
OBJECTIVE
Leptin administration is known to directly modulate pancreatic β-cell function in leptin-deficient rodent models. However, human studies examining the effects of leptin administration on β-cell function are lacking. In this study, we examined the effects (16–20 weeks) of leptin replacement on β-cell function in patients with lipodystrophy.
RESEARCH DESIGN AND METHODS
In a prospective, open-label, currently ongoing study, we studied the effects of leptin replacement on β-cell function in 13 patients with congenital or acquired lipodystrophy. Insulin secretory rate (ISR) was calculated by C-peptide deconvolution from plasma glucose and C-peptide levels measured during oral glucose tolerance tests (OGTTs) performed at baseline and after 16–20 weeks of leptin replacement. β-Cell glucose sensitivity and rate sensitivity were assessed by mathematical modeling of OGTT.
RESULTS
There was a significant decrease in triglycerides, free fatty acids, and glycosylated hemoglobin levels (A1C) after leptin therapy. Patients with lipodystrophy have high fasting and glucose-stimulated ISR. However, leptin therapy had no significant effect on fasting ISR, total insulin secretion during OGTT, β-cell glucose sensitivity, rate sensitivity, or insulin clearance.
CONCLUSIONS
In contrast to the suppressive effects of leptin on β-cell function in rodents, 16–20-week treatment with leptin in lipodystrophy patients did not significantly affect insulin secretion or β-cell function in leptin-deficient individuals with lipodystrophy.
Introduction
Leptin, an adipocyte-derived hormone, affects glucose homeostasis in part by directly modulating pancreatic β-cell function (1–3). Systemic leptin infusion acutely decreases insulin secretion and impairs glucose tolerance in rodents (4). Loss of these suppressive actions of leptin in mice with selective absence of leptin receptors in the pancreas leads to fasting hyperinsulinemia and increased responsiveness to glucose- and incretin-stimulated insulin secretion (5–7). These findings have led some to speculate that the diminished responsiveness of pancreatic β-cells to the negative effects of leptin partly contributes to the hyperinsulinemia typically observed in obesity (4,7,8). However, whether leptin negatively affects β-cell function in humans is unclear.
Lipodystrophy, characterized by selective deficiency of adipose tissue, is a hypoleptinemic state associated with hyperinsulinemia, severe insulin resistance, glucose intolerance, and dyslipidemia (1). Leptin replacement in patients with lipodystrophy attenuates insulin resistance, hyperinsulinemia, and hyperglycemia (1,9,10). Although insulin secretion was not assessed in these studies, reductions in insulin levels (fasting and glucose induced) were attributed to improvements in insulin sensitivity (11,12). A solitary study has reported the effects of leptin replacement on insulin secretion in a genetically leptin-deficient adult male (13). In this patient, insulin secretion, but not insulin sensitivity, increased the week after leptin replacement. Except for this single report, we are unaware of studies examining the short-term effects of leptin replacement on β-cell function. To that end, we report the early effects of short-term (16–20 weeks) metreleptin (an analog of human leptin) administration on β-cell function in patients with lipodystrophy.
Research Design and Methods
Study Design and Study Subjects
The study protocol (initiated in July 2000) was approved by the institutional review board of the National Institute of Diabetes and Digestive and Kidney Diseases. Written informed consent was obtained from all subjects or their legal guardian, and assent was obtained from participants under 18 years of age. Patients were from the U.S. and a number of other countries in Europe, Asia, the Middle East, and South America. Study subjects reported here are from an open-label, prospective, currently ongoing study examining the long-term safety and clinical effects of metreleptin treatment in patients with congenital or acquired lipodystrophy (9,11,12). Patients with HIV-associated lipodystrophy were not studied in this protocol. The rationale, inclusion criteria, and study design have been described previously (9,11). Inclusion criteria for leptin replacement included hypoleptinemia (<12 ng/mL), metabolic abnormalities such as hypertriglyceridemia and/or lipoatrophic diabetes, and the ability to adhere to the leptin replacement protocol.
Metreleptin was provided by Amgen (Thousand Oaks, CA) initially and Amylin Pharmaceuticals (San Diego, CA) subsequently (9,11). Leptin therapy was provided as a self-administered once- or twice-daily subcutaneous injection as previously described (9,11). Patients were seen at the Clinical Research Center of the National Institutes of Health every 4–6 months for the first year and then every 6–12 months thereafter. Laboratory data were collected during each visit. The current study examined the early effects of leptin replacement on β-cell function derived from oral glucose tolerance test (OGTT) data. In this report, we include a subgroup of patients (n = 13) who were on stable doses of oral hypoglycemic agents prior to and at 16–20 weeks of leptin therapy. Patients on insulin therapy were excluded in this analysis.
Study Procedures
OGTTs were performed at baseline and after 16–20 weeks of leptin administration. During each visit, after an overnight fast, patients underwent an OGTT with plasma sampling at baseline and 30, 60, 90, 120, and 180 min after glucose ingestion for the measurement of glucose, insulin, and C-peptide concentrations. All oral hypoglycemic agents were held on the day of the OGTT.
Measures of Insulin Secretion and Resistance
The β-cell model used in the current study, describing the relationship between insulin secretion and glucose concentration, has been described (14). Insulin secretory rates (ISRs) were calculated from deconvolution of the plasma C-peptide concentrations. The relationship between insulin secretion and plasma glucose concentrations is modeled as the sum of two components. The first component represents the dependence of insulin secretion on absolute glucose concentration at any time point during the OGTT and is characterized by a dose-response function relating these two variables. The average slope of the dose response is denoted as β-cell glucose sensitivity. The dose response is modulated by various factors (e.g., incretins, neurotransmitters, and degree and duration of hyperglycemia), which are collectively modeled as a “potentiation factor.” The second component represents the dependence of insulin secretion on the rate of change of plasma glucose and depends on the first derivative of plasma glucose concentration against time and a parameter denoted as rate sensitivity. Consequently, this parameter reflects the changes in insulin secretion in response to rapid changes in glucose concentration.
Oral glucose insulin sensitivity (OGIS) and insulin sensitivity index (ISI) were used as surrogate measures of glucose insulin sensitivity/resistance (14,15). OGIS was calculated from OGTT using the 2-h OGIS equation. This method provides an ISI that is an estimate of the glucose clearance during a euglycemic-hyperglycemic clamp and is expressed as milliliters per minute per square meter of body surface area (14). ISI (Matsuda) was calculated as previously described (15).
Statistical Analyses
After testing for normality, data were logarithm transformed where appropriate. Significance of changes in measures of β-cell function (postleptin minus baseline values) was calculated by one-way ANCOVA adjusted for age, value of the dependent variable at baseline, and changes in OGIS. P < 0.05 was considered to represent statistical significance. Data are presented as means ± SD or median (interquartile range [IQR]). The statistical software JMP, Version 8.1 (SAS Institute, Inc., Cary, NC), was used for data analysis.
Results
Clinical Characteristics of Study Subjects
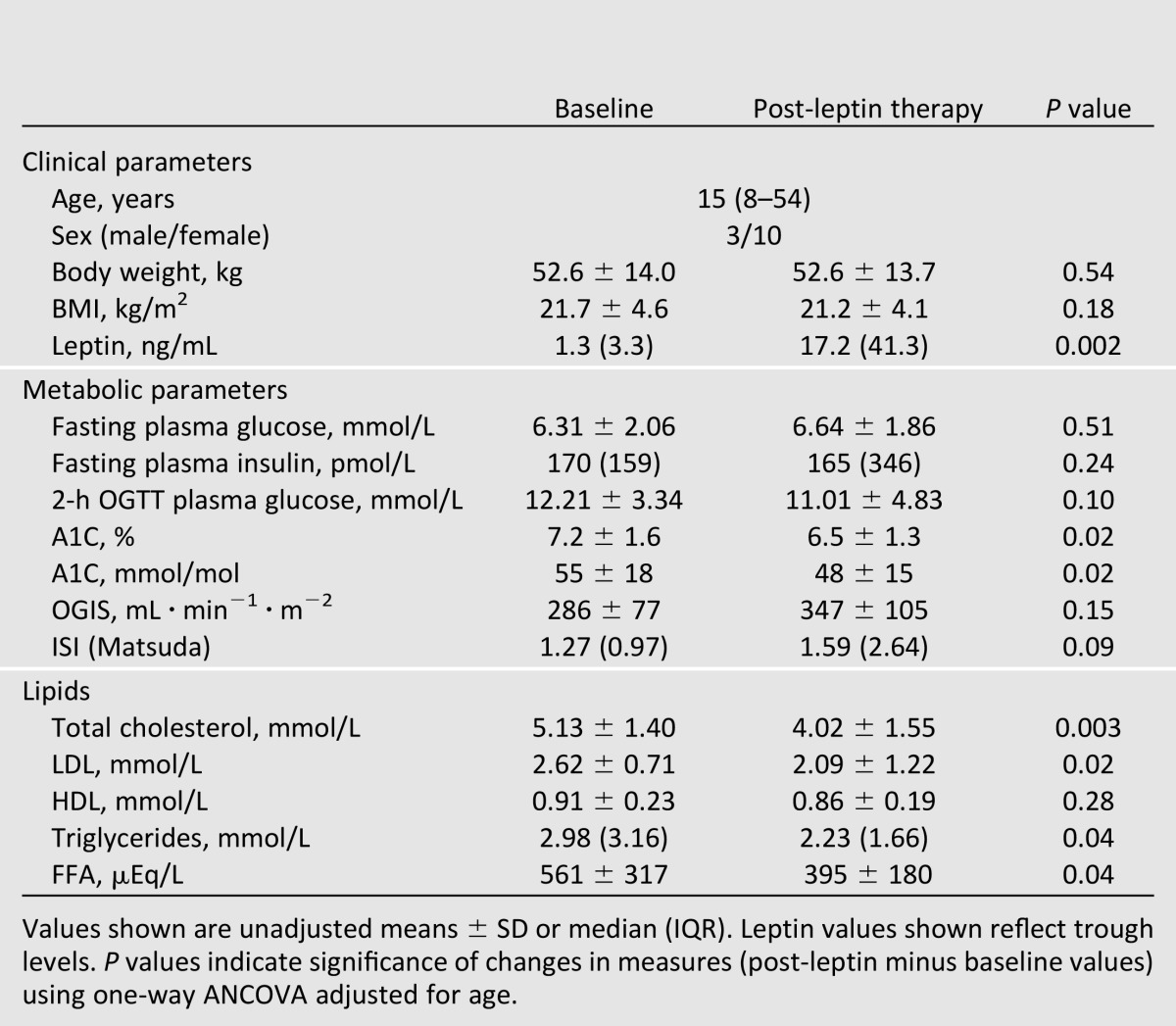
Thirteen patients with lipodystrophy treated with leptin are included in this study (congenital generalized lipodystrophy, n = 5; acquired generalized lipodystrophy, n = 2; familial partial lipodystrophy, n = 5; acquired partial lipodystrophy, n = 1). The median baseline age was 15 years with a range of 8–54 years. Patients were on stable doses of metformin (n = 12) and/or thiazolidinedione (n = 3) prior to and at the end of 16–20 weeks of leptin therapy. Dyslipidemia was treated with stable doses of lipid-lowering agents (fenofibrate, n = 3; omega-3-acid ethyl esters, n = 1) as previously described (9). Prior to leptin therapy, median serum leptin levels (trough) were 1.3 ng/mL (IQR 3.3). Patients with partial lipodystrophy had higher levels of leptin compared with generalized lipodystrophy (mean ± SD 4.1 ± 1.2 vs. 0.7 ± 0.3, P = 0.002). All of our patients were diabetic with an average glycosylated hemoglobin level (A1C) of 7.2% with mean fasting plasma glucose levels <6.6 mmol/L, suggesting that glycemic control was good prior to initiation of leptin. As previously reported, these patients had high levels of triglycerides and free fatty acids (FFAs) (Table 1), and various metabolic parameters were similar in partial and generalized lipodystrophy patients (data not shown).
Table 1.
Clinical characteristics and metabolic parameters before and after leptin replacement in patients with lipodystrophy
Leptin Replacement Therapy
The initial dose of metreleptin was based on weight, sex, and age, and subsequent dose adjustments were based on individual metabolic responses of patients. The average daily dose of metreleptin at the end of 16–20 weeks was 4.19 ± 1.87 mg (mean ± SD). As expected, leptin levels increased significantly with therapy (Table 1).
Effects of Leptin Replacement on Metabolic Parameters, Insulin Sensitivity, and Insulin Secretion
Leptin replacement was associated with a significant decrease in total cholesterol, LDL, triglyceride, and FFA levels with no change in HDL levels (Table 1). After leptin therapy, A1C levels were significantly lower, but fasting glucose and insulin and 2-h post-OGTT values were not different from baseline (Table 1). However, area under the curve for glucose (AUCg) during OGTT was lower after leptin therapy (1,816 ± 731 vs. 2,063 ± 598 mmol ⋅ L ⋅ 180 min, P = 0.02). Surrogate measures of insulin sensitivity, OGIS and ISI, were not significantly modulated, but tended to increase (∼25%) with leptin replacement (Table 1). These changes in insulin sensitivity and the lower glucose AUC partially explain the effects of leptin on A1C.
Changes in parameters of β-cell function after leptin replacement are shown in Fig. 1. Mean basal insulin secretion rate prior to leptin initiation was 324 ± 153 pmol/m2/min (±SD), and basal insulin secretion rate adjusted for age and insulin sensitivity (OGIS) was not significantly different after leptin replacement (345 ± 254 pmol/m2/min, P = 0.10). Similarly, leptin administration had no significant effect on total insulin secretion (post vs. pre: 111 ± 42 vs. 170 ± 123 nmol/m2, P = 0.13). Model-derived β-cell insulin secretion and glucose dose responses obtained from OGTTs administered pre- and post-leptin therapy are shown in Fig. 1C. The slopes of these curves represent the ability of the β-cell to respond to an increment in plasma glucose concentration termed glucose sensitivity. Leptin treatment had no significant effect on glucose sensitivity (post vs. pre): 65 (112) vs. 48 (167) pmol/min/m2/mM, P = 0.39. Likewise, rate sensitivity, a measure of the insulin secretory component that responds to rapid changes in plasma glucose concentrations, was also unaltered with treatment: 206 (1,449) vs. 1,215 (6,915) pmol/min/m2/mM, P = 0.13. Age, duration of diabetes, race, sex, and type of lipodystrophy may potentially modify the effects of leptin. At baseline, age was negatively related to basal ISR (r = −0.72, P = 0.005) and total ISR (r = −0.47, P = 0.09). Changes in measures of β-cell function (post-leptin minus baseline values) were adjusted for age. The median duration of diabetes in this study was 9.5 years (range 4–29). However, duration of diabetes was unrelated to basal ISR (r = −0.14, P = 0.68) or total ISR (r = −0.41, P = 0.18), and the significance of changes in measures of β-cell function after leptin therapy remained unaltered after adjusting for duration of diabetes. The effect of leptin treatment on parameters of β-cell function was not significantly different between partial and generalized lipodystrophy patients (Pgroup × time interaction > 0.5). Thus, these results suggest that leptin replacement in the short-term had no significant effect on insulin secretion or β-cell glucose responsiveness.
Figure 1.

Insulin secretion and β-cell glucose sensitivity in patients with lipodystrophy before and after leptin replacement. Fasting ISR (A), total insulin output after an oral glucose load (B), and model-derived dose response between ISR and plasma glucose concentration (C). Significance of changes in measures of β-cell function (post-leptin minus baseline values) in panels A and B was calculated by one-way ANCOVA adjusted for age, value of the dependent variable at baseline, and changes in OGIS. The ISRs in panel C were compared using a two-way ANOVA. NS, nonsignificant. ○, congenital generalized lipodystrophy; ◇, acquired generalized lipodystrophy; ●, familial partial lipodystrophy; ♦, acquired partial lipodystrophy.
Effects of Leptin Replacement on Insulin Clearance
Basal insulin clearance (fasting ISRs/fasting insulin) and OGTT-insulin clearance (incremental AUCs of ISRs/incremental AUCs of insulin concentrations) rates were measured prior to and after leptin therapy. Before leptin therapy, basal insulin clearance rates were similar in partial (1.99 ± 0.76 L/min/m2) and generalized (1.66 ± 0.33 L/min/m2) lipodystrophy patients. However, OGTT-insulin clearance rates were significantly higher in patients with partial lipodystrophy (1.25 ± 0.33 vs. 0.80 ± 0.25 L/min/m2, P = 0.02). In the overall cohort, leptin treatment had no significant effect on basal (post vs. pre: 1.97 ± 1.04 vs. 1.81 ± 0.57 L/min/m2, P = 0.62) or OGTT-insulin clearance (1.12 ± 0.39 vs. 1.00 ± 0.36 L/min/m2, P = 0.39). The lack of leptin effects on insulin clearance was similar in partial and generalized lipodystrophy patients.
Conclusions
In the current study, leptin replacement therapy had no significant impact on β-cell function in patients with lipodystrophy treated for 16–20 weeks. The effect of leptin on human β-cell function and insulin secretion has not been thoroughly investigated. In that context, our study results suggest that, contrary to the suppressive effects of leptin on insulin secretion consistently observed in animal and in vitro studies, leptin replacement does not attenuate the increased insulin secretion observed in these patients.
Leptin replacement in lipodystrophy patients is associated with reductions in fasting insulin levels and after an oral glucose load after 12 months (11,12). The reduction in insulin levels was attributed to improvements in glycemia and insulin sensitivity. In contrast, in another study, mean serum levels of insulin after an OGTT remained unaltered in patients with congenital generalized lipodystrophy (n = 5) and increased in acquired generalized lipodystrophy (n = 2) despite improvements in insulin sensitivity and glycemia after 4 months of leptin replacement (16). Insulin secretion in one genetically leptin-deficient adult male was deduced by deconvolution of C-peptide levels obtained during a meal (13). In the first week after leptin replacement, the amplitude of C-peptide secretion was higher. However, hepatic extraction of insulin was increased simultaneously, resulting in lower peripheral insulin levels (by ∼20%). After 18 and 24 months of leptin therapy, insulin secretion was reduced along with improvements in insulin sensitivity.
We evaluated β-cell function by using a mathematical model that describes the dynamic relationship between plasma glucose concentration and β-cell insulin secretion. In order to account for changes in insulin sensitivity, we used surrogate indices derived from an OGTT, OGIS and ISI (Matsuda). Patients in this study were hypoleptinemic and severely insulin resistant with hyperinsulinemia (Table 1). All of our patients were diabetic and were on stable doses of insulin sensitizers. Fasting ISRs in these patients were approximately twofold higher than those reported in morbidly obese, insulin-resistant, type 2 diabetic individuals (∼140 pmol/m2/min) (17). This degree of hyperinsulinemia afforded us the opportunity to evaluate the potential inhibitory effects of leptin on insulin secretion. In fact, our sample size (n = 13) was sufficient to provide 80% power with α = 0.05 to detect a 20% decrease in fasting ISR. Nonetheless, we did not observe any significant effects of leptin replacement on fasting insulin secretion or total insulin output during an OGTT (Fig. 1). Glucose sensitivity measures the ability of the β-cell to respond to a glycemic stimulus and is a determinant of insulin secretion (18). β-Cell glucose sensitivity is impaired in individuals with diabetes and glucose intolerance (17,18). In our patients, median values of β-cell glucose sensitivity are in the range typically observed in diabetic individuals (17,18). Even though A1C and FFA levels were significantly better after leptin treatment (Table 1), glucose sensitivity was not significantly improved (Fig. 1).
Our results are inconsistent with findings from studies in cell culture and animals. Leptin receptors are expressed in human islets and mediate the inhibitory actions of leptin on insulin secretion in β-cells in vitro (4,8). Leptin hyperpolarizes β-cells by opening KATP channels, reduces intracellular calcium levels, and accelerates the hydrolysis of cAMP to attenuate glucose- and agonist-induced insulin secretion (8,19). Systemic infusion of leptin in normal or leptin-deficient hyperinsulinemic ob/ob mice, but not leptin receptor–deficient db/db mice, acutely (within minutes) decreases plasma insulin levels (4). In ob/ob mice, leptin administration for 24 h inhibits insulin gene transcription in pancreatic islets (20). These direct insulin-suppressing effects were also observed during short-term (4-week) leptin replacement in ob/ob mice prior to any significant changes in body weight or food intake (21). Consistent with these findings, selective knockout of leptin receptors in the pancreas of mice leads to hyperinsulinemia (5–7).
The reasons for the discrepancy in the results from our study and leptin-deficient rodent models are unclear. It is possible that the hypoleptinemic state and the accompanying significant elevations in FFA and ectopic lipid deposition may have altered the insulin secretory dynamics in human β-cells differently than in rodents. In fact, in obese rodents (due to leptin deficiency or diet induced), leptin treatment reduces pancreatic fat accumulation (22,23). We have previously shown that leptin replacement for 16–20 weeks decreases hepatic steatosis in individuals with lipodystrophy (24,25). Leptin, similar to its effects on hepatic steatosis, may also decrease pancreatic steatosis in humans. However, we are not aware of any human studies that have examined the effects of leptin on pancreatic fat content in patients with non-HIV lipodystrophy. Although hepatic steatosis is frequently observed in individuals with lipodystrophy, postmortem studies in few patients do not report pancreatic steatosis (26–29). Although these findings need further confirmation, it appears that islet amyloidosis may be the characteristic pathologic feature in these patients (26–28). Whether leptin modulates islet amyloidosis in humans is also not known. Thus, species-dependent and tissue-specific antisteatotic effects of leptin may explain the discrepancy in the effects of leptin in rodents and humans.
Leptin inhibits insulin secretion from human islets in vitro (4,8); however, studies examining the acute effects of agonist and/or glucose-stimulated insulin secretion in healthy individuals are necessary to rule out species-dependent in vivo effects of leptin. We also did not observe any significant improvements in insulin sensitivity as measured by OGIS and ISI after leptin replacement (Table 1). Although there was a trend toward higher OGIS and ISI after treatment (P = 0.09), it is possible that a more sensitive method, such as the euglycemic-hyperinsulinemic clamp, would have detected improvements in insulin sensitivity as reported in prior studies (1,24,30). One of the early effects of leptin treatment is an increase in insulin clearance (13). However, 16–20 weeks of leptin replacement did not modulate insulin clearance in this study.
In this study, we chose to report the early effects of leptin replacement on β-cell function. We have previously reported on the long-term effects of leptin therapy (9–11,25). Leptin therapy decreases fasting glucose levels, improves glucose tolerance and insulin sensitivity, reduces A1C, lowers triglycerides, and attenuates steatohepatitis in lipodystrophy (9–11,25). Improvements in insulin sensitivity and changes in ectopic lipid accumulation have been suggested to mediate these effects of leptin. However, a recent study suggests that modest glycemic benefits of long-term leptin therapy extend to patients with insulin receptor mutations (31). Considering that patients with insulin receptor mutations have a fixed defect in insulin sensitivity and do not manifest ectopic lipid accumulation, leptin favorably affects glucose metabolism through other unidentified mechanisms. Future studies examining the short- and long-term effects on β-cell function and pancreatic fat content are warranted to confirm the long-term effects of leptin on pancreatic function.
Our study has several limitations. First, we derived measures of β-cell function by mathematically modeling plasma glucose and C-peptide levels measured during OGTT and used surrogate indices (OGIS and ISI) to assess insulin sensitivity. Although the model is widely used, our findings need to be confirmed using gold-standard techniques such as euglycemic-hyperinsulinemic and hyperglycemic clamp techniques. These techniques will also provide additional information regarding the different phases of insulin secretion. Whether leptin has phase-specific effects on insulin secretion is not known. Nonetheless, the effects of leptin on β-cell glucose sensitivity to first-phase insulin response cannot be adequately assessed during an OGTT. The surrogate measures of insulin sensitivity tended to be significant after leptin therapy. It is likely that glucose clamp technique would have had sufficient sensitivity to detect significant changes in insulin sensitivity after leptin therapy. Second, our study is open-label and nonrandomized and lacks a control treatment arm. Third, because of our study design, we did not examine the initial effects of leptin therapy prior to 16 weeks. Finally, the lack of leptin effects on β-cell function in this group of hypoleptinemic individuals cannot be generalized to normal healthy individuals.
In summary, we report that short-term leptin replacement did not significantly modulate insulin secretion and β-cell function in patients with lipodystrophy. Our study results do not support the notion that hyperinsulinemia in leptin-deficient or leptin-resistant states is partly due to the lack of tonic and direct insulin-suppressive effects of leptin on the pancreas. It is likely that improvements in fasting hyperinsulinemia and β-cell function after prolonged leptin therapy are secondary to changes in insulin sensitivity, glycemia, and dyslipidemia. Further studies in healthy individuals are necessary to confirm whether leptin inhibits insulin secretion.
Article Information
Funding. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. R.M. collected and analyzed data, wrote the manuscript, and approved the final manuscript. R.J.B., J.J., and E.K.C. conducted the study, collected data, and approved the final manuscript. A.M. analyzed data and approved the final manuscript. M.A.W. collected data, wrote the manuscript, and approved the final manuscript. M.C.S. wrote the manuscript and approved the final manuscript. P.G. designed and conducted the study, collected data, wrote the manuscript, and approved the final manuscript. P.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Clinical trial reg. no. NCT00025883, clinicaltrials.gov.
References
- 1.Moon HS, Dalamaga M, Kim SY, et al. Leptin’s role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocr Rev 2013;34:377–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee YH, Magkos F, Mantzoros CS, Kang ES. Effects of leptin and adiponectin on pancreatic β-cell function. Metabolism 2011;60:1664–1672 [DOI] [PubMed] [Google Scholar]
- 3.Marroquí L, Gonzalez A, Ñeco P, et al. Role of leptin in the pancreatic β-cell: effects and signaling pathways. J Mol Endocrinol 2012;49:R9–R17 [DOI] [PubMed] [Google Scholar]
- 4.Kulkarni RN, Wang ZL, Wang RM, et al. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest 1997;100:2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morioka T, Dishinger JF, Reid KR, et al. Enhanced GLP-1- and sulfonylurea-induced insulin secretion in islets lacking leptin signaling. Mol Endocrinol 2012;26:967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morioka T, Asilmaz E, Hu J, et al. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J Clin Invest 2007;117:2860–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray SL, Donald C, Jetha A, Covey SD, Kieffer TJ. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology 2010;151:4178–4186 [DOI] [PubMed] [Google Scholar]
- 8.Seufert J, Kieffer TJ, Leech CA, et al. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: implications for the development of adipogenic diabetes mellitus. J Clin Endocrinol Metab 1999;84:670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan JL, Lutz K, Cochran E, et al. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract 2011;17:922–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010;53:27–35 [DOI] [PubMed] [Google Scholar]
- 11.Javor ED, Cochran EK, Musso C, Young JR, Depaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes 2005;54:1994–2002 [DOI] [PubMed] [Google Scholar]
- 12.Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002;346:570–578 [DOI] [PubMed] [Google Scholar]
- 13.Andreev VP, Paz-Filho G, Wong ML, Licinio J. Deconvolution of insulin secretion, insulin hepatic extraction post-hepatic delivery rates and sensitivity during 24-hour standardized meals: time course of glucose homeostasis in leptin replacement treatment. Horm Metab Res 2009;41:142–151 [DOI] [PubMed] [Google Scholar]
- 14.Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E. Meal and oral glucose tests for assessment of beta-cell function: modeling analysis in normal subjects. Am J Physiol Endocrinol Metab 2002;283:E1159–E1166 [DOI] [PubMed] [Google Scholar]
- 15.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999;22:1462–1470 [DOI] [PubMed] [Google Scholar]
- 16.Ebihara K, Kusakabe T, Hirata M, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab 2007;92:532–541 [DOI] [PubMed] [Google Scholar]
- 17.Nannipieri M, Mari A, Anselmino M, et al. The role of beta-cell function and insulin sensitivity in the remission of type 2 diabetes after gastric bypass surgery. J Clin Endocrinol Metab 2011;96:E1372–E1379 [DOI] [PubMed] [Google Scholar]
- 18.Kanat M, Mari A, Norton L, et al. Distinct β-cell defects in impaired fasting glucose and impaired glucose tolerance. Diabetes 2012;61:447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao AZ, Bornfeldt KE, Beavo JA. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J Clin Invest 1998;102:869–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seufert J, Kieffer TJ, Habener JF. Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc Natl Acad Sci U S A 1999;96:674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995;269:540–543 [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Ravazzola M, Park BH, Bashmakov YK, Orci L, Unger RH. Metabolic mechanisms of failure of intraportally transplanted pancreatic beta-cells in rats: role of lipotoxicity and prevention by leptin. Diabetes 2007;56:2295–2301 [DOI] [PubMed] [Google Scholar]
- 23.Wang MY, Koyama K, Shimabukuro M, Newgard CB, Unger RH. OB-Rb gene transfer to leptin-resistant islets reverses diabetogenic phenotype. Proc Natl Acad Sci U S A 1998;95:714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002;109:1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Javor ED, Ghany MG, Cochran EK, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005;41:753–760 [DOI] [PubMed] [Google Scholar]
- 26.Haque WA, Vuitch F, Garg A. Post-mortem findings in familial partial lipodystrophy, Dunnigan variety. Diabet Med 2002;19:1022–1025 [DOI] [PubMed] [Google Scholar]
- 27.Chandalia M, Garg A, Vuitch F, Nizzi F. Postmortem findings in congenital generalized lipodystrophy. J Clin Endocrinol Metab 1995;80:3077–3081 [DOI] [PubMed] [Google Scholar]
- 28.Garg A, Chandalia M, Vuitch F. Severe islet amyloidosis in congenital generalized lipodystrophy. Diabetes Care 1996;19:28–31 [DOI] [PubMed] [Google Scholar]
- 29.Lüdtke A, Roos GM, van Hettinga M, Horst BA, Worman HJ, Schmidt HH. Post-mortem findings in Dunnigan-type familial partial lipodystrophy. Diabet Med 2010;27:245–246 [DOI] [PubMed] [Google Scholar]
- 30.Lee JH, Chan JL, Sourlas E, Raptopoulos V, Mantzoros CS. Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J Clin Endocrinol Metab 2006;91:2605–2611 [DOI] [PubMed] [Google Scholar]
- 31.Brown RJ, Cochran E, Gorden P. Metreleptin improves blood glucose in patients with insulin receptor mutations. J Clin Endocrinol Metab 2013;98:E1749–E1756 [DOI] [PMC free article] [PubMed] [Google Scholar]