

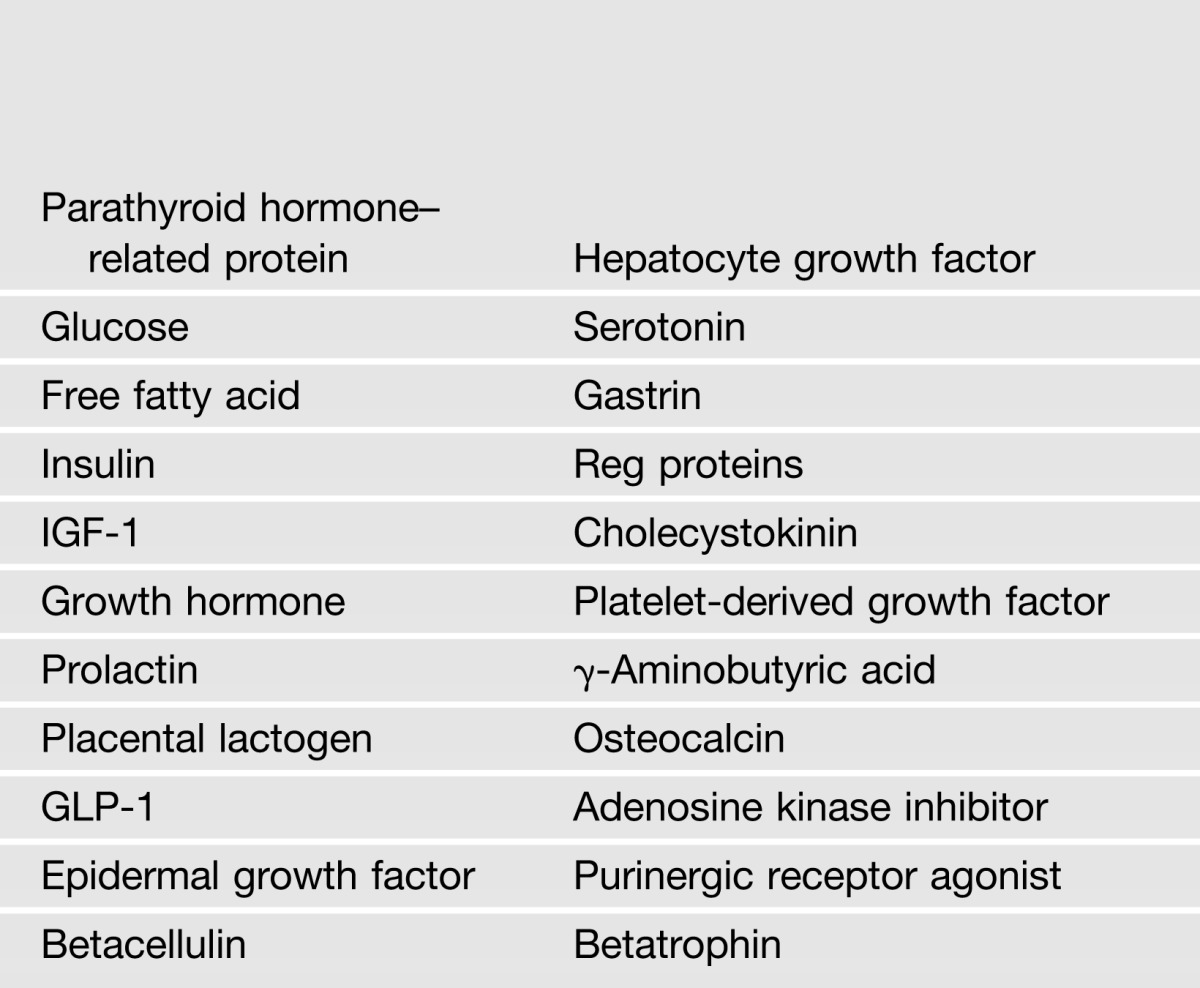
The purpose of studying pancreatic β-cells in nonhuman species is to learn how to protect and regenerate the supply of residual endogenous β-cells in the face of the immunologic attack of type 1 diabetes, and the glucolipotoxic, inflammatory, endoplasmic reticulum- and oxidative stress–inducing environment of type 2 diabetes. β-Cell replication was unimaginable 20 years ago, when many believed that β-cells were irrevocably, terminally differentiated or senescent, and could never reenter the cell cycle. Enormous progress has been made over the past two decades. There are now many, many agonists that induce β-cells to replicate and expand. The agonists have included nutrients, growth factors, intracellular signaling molecules, small molecules discovered in high-throughput screens, proteins discovered in similar screens, or transgenic mouse models. Examples of these agonists are shown in Table 1, some of which are derived from work in the author’s group. Importantly, most of the β-cell replication and expansion resulting from these agonists has been observed in β-cells from mice, rats, pigs, and zebra fish. Surprisingly few induce human β-cells to replicate at therapeutically meaningful rates. Demonstration of human β-cell replication is an unspoken “elephant in the room” that is avoided in most reports.
Table 1.
Example of small molecules, growth factors, hormones, and nutrients that induce robust β-cell replication in rodents, but not in adult humans
One recent case in point is a report by Yi et al. (1) that attracted considerable attention both in the diabetes research community and in the lay media. Very briefly, Yi et al. described a novel β-cell mitogen. By infusing an insulin receptor antagonist, S961, Yi et al. induced insulin resistance, and found that an mRNA-encoding angiopoietin-like 8, or lipasin, which the investigators renamed “betatrophin,” was induced in liver and adipocytes. When they administered a betatrophin-encoding plasmid to generate systemic expression in normal mice, endogenous β-cell replication was dramatically enhanced. Whether this remarkable proliferation was a direct effect on β-cells or an indirect effect mediated by another factor(s) generated in other tissues is uncertain. Importantly, as with so many of its predecessors, the Yi et al. study was confined to mice. No betatrophin effect on human islets was included in the report, although tools are readily available to conduct these experiments: plasmids expressing both mouse and human betatrophin, human cadaveric islets, and many methods to assess proliferation. So while the work is lovely and innovative, the elephant in the room in the article by Yi et al. is the obvious question: “Would betatrophin make human β-cells replicate?” Put another way, “Will this be another bitter ending—bitter-trophin—to what might otherwise have been a groundbreaking advance in diabetes?”
This is the question that is asked in the article by Jiao et al. in this issue (2). The new study uses immunodeficient NOD-Scid mice to study the effects of betatrophin on transplanted human islets in vivo. In brief, the investigators reproduced observations of Yi et al. that the insulin receptor antagonist, S961, induces betatrophin mRNA expression in liver, and induces proliferation in both endogenous as well as transplanted mouse β-cells. The principal finding in Jiao et al., however, is that despite induction of vigorous replication in mouse β-cells, endogenous betatrophin fails to induce replication in transplanted human β-cells. Thus, betatrophin may simply be the most recent example of a mouse β-cell mitogen that fails to induce human β-cell replication.
This may be a hasty interpretation, as unanswered questions remain. Failure to observe human β-cell replication could result from the heterologous species model employed in these experiments. Perhaps mouse betatrophin cannot activate human betatrophin receptors, or mouse and human betatrophin undergo different posttranslational processing. Or, perhaps the 1-week duration of these experiments was not sufficient to observe induction of human β-cell proliferation (although it was long enough to observe proliferation in simultaneously transplanted mouse β-cells). And betatrophin was not actually measured, so perhaps it really was not elevated systemically. Moreover, there was no positive control for human β-cell replication, so perhaps the human islets that were selected were unhealthy. Thus, the final chapter of the betatrophin story has not been written, but at present, the plot does not point toward a promising conclusion.
More important than any single mitogenic factor, there is a larger and more important message for β-cell biology in the pair of articles by Yi et al. (1) and Jiao et al. (2). It is the elephant in the room, an elephant whose size increases with each new report of rodent β-cell replication. Since the principal goal of human β-cell regeneration research is to identify strategies to induce human β-cells to replicate, it follows that addressing this question should be an integral component of reports describing novel approaches to β-cell replication. The tools are available: 1) human islets can be obtained from the Integrated Islet Distribution Program (http://iidp.coh.org) in the U.S. as well as from similar sources in other countries, and these can be studied in vitro or in vivo; 2) simple screening assays for human β-cell proliferation (BrdU, Edu, PCNA, Ki67, PHH3, etc.) currently exist; and 3) small molecules and nutrients are testable in these systems, and larger proteins can be made recombinantly or expressed in viruses and examined for proliferation. Since human β-cell proliferation is the goal, and since the tools are available directly or collaboratively, it seems reasonable to require that these kinds of studies are included in any important report of a novel agent that claims to induce β-cell replication. Adopting this requirement should be the foundation of a new baseline, or the “new normal,” in this line of research.
Investigators in the field will recognize that the story does not end with these kinds of measures: Additional technical end points must be addressed. These include evaluation of DNA damage and repair, cell death, β-cell function in vitro or in vivo, and the actual demonstration of increases in human β-cell numbers using in vitro or in vivo models. Clearly, the latter has proven to be a particular challenge.
There are also more profound questions. First, how much human β-cell proliferation is “enough” to be therapeutically relevant? Is a rate of 2–3%, as has been reported in native neonatal human β-cells (3–6), all that is required? Second, if it is possible to drive proliferation at rates of 1–10%, as has been reported in some studies, why is it that the other 90–99% of β-cells refuse to replicate? Is it senescence? Terminal differentiation? Excessive cell cycle inhibitory tone? Repressive DNA methylation and histone marks on critical unidentified target loci? Loss of receptors for key growth factors or transporters for key nutrients? Or loss of key mitogenic signaling molecules? Inquiring minds want to know. Third, what attributes of neonatal and juvenile human β-cells that permit replication are lost in adult human β-cells? Fourth, do we still need to study mouse, rat, and zebra fish models? The answer to this is an unequivocal “yes,” because at the present time these models provide more rapid screens and mechanism-of-action information that is unattainable in human β-cells. So while these smaller species are indeed critically important, let’s not forget to give the elephant equal time.
Article Information
Funding. This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grants U01-DK-089538 and R01-DK-55023, and JDRF grants 1-2011-603 and 17-2011-598.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Footnotes
See accompanying article, p. 1283.
References
- 1.Yi P, Park J-S, Melton DA. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell 2013;153:747–758 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Jiao Y, Le Lay J, Yu M, Naji A, Kaestner KH. Elevated mouse hepatic betatrophin expression does not increase human β-cell replication in the transplant setting. Diabetes 2014;63:1283–1288 [DOI] [PMC free article] [PubMed]
- 3.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 2000;49:1325–1333 [DOI] [PubMed] [Google Scholar]
- 4.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008;57:1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in human islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011;54:572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012;97:3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]