

Abstract
The mTOR mediated signaling transduction pathway has been observed to be deregulated in a wide variety of cancer and metabolic diseases. Despite extensive clinical development efforts, the well-known allosteric mTOR inhibitor rapamycin and structurally related rapalogs have failed to show significant single-agent anti-tumor efficacy in most types of cancer. This limited clinical success maybe due to the inability of the rapalogs to maintain a complete blockade mTOR mediated signaling. Therefore numerous efforts have been initiated to develop ATP-competitive mTOR inhibitors that would block both mTORC1 and mTORC2 complex activity. Here we describe our experimental approaches to develop Torin1 using a medium throughput cell-based screening assay and structure-guided drug design.
Keywords: mTOR, mTORC1, mTORC2, PI3K, PIKK, Akt, Rapamycin, Torin1
1. Introduction
mTOR (mammalian Target of Rapamycin) is a serine/threonine protein kinase that is highly conserved across eukaroyotes. It plays a critical role in the regulation of fundamental cellular physiological processes in response to the upstream cellular signals such as growth factors, energy, stress and nutrient to control cell growth, proliferation, and metabolism through two known complexes named mTORC1 and mTORC2 (1). mTORC1 is a primary regulator of protein translation through the phosphorylation of the S6 kinase and the inhibitory 4E-binding proteins (4EBPs). mTORC2 phosphorylates and regulates Akt, a downstream effector of the PI3K signaling pathway that mediates growth and survival signals. Rapamycin, an allosteric inhibitor of mTOR that was originally identified as an immunosuppressant and later as an anti-cancer agent, has greatly facilitated the study of mTOR signaling (2,3). As a key node in the oncogenic phosphoinositide 3-kinase signaling pathway, deregulation of the mTOR kinase has been often observed in human cancers (4,5,6). Inspired by the promising in vitro anti-cancer effect, it is widely believed that rapamycin or related analogs (rapalogs) would be therapeutically effective by blocking the mTORC1’s phosphorylation activity of S6K and 4EBPs, which are key upstream regulators of protein synthesis. However, extensive clinical evaluation has found rapamycin’s in vivo efficacy to be limited to a few rare cancers, such as renal cell carcinoma and mantle cell lymphoma etc (7). This may be partially explained by the recent discovery that rapamycin could not block the function of mTORC1 completely and has little effect on mTORC2 complex in the majority of cell types (4,8). Moreover, the existence of a feedback loop that hyper-activates PI3K when mTORC1 is inhibited (9) and the recent discovery that rapamycin fails to completely inhibit mTORC1 indicates that ATP-competitive small molecule mTOR inhibitors might show broader efficacy.
Structurally, the mTOR catalytic domain exhibits high sequence identity with PI3Ks and PI3K related kinases (PIKK) such as ATR, ATM, DNA-PK and SMG-1. The co-crystal structure of LY294002 with PI3Kγ provides valuable insights that enables structure-guided design of numerous PI3K inhibitors (10). The dual PI3K/mTOR inhibitor PI-103, which like LY294002 uses the morpholine oxygen as the hinge binding moiety provides a molecular-entry point for the development of selective mTOR inhibitors (11,12). For example, PI-103 served as a ‘lead’ compound for the structure-guided development of selective mTOR inhibitors such as KU-0063794 (13) and WYE-354 (14) and other related structures (15) using a rational designed approach. (Fig. 1) Preliminary biochemical analyses indicate that these compounds are, indeed, promising candidates for replacing rapamycin as future anti-cancer agents. Not only do these pharmacological inhibitors have enhanced efficacy in suppressing hyperactive mTOR, but also they are highly specific in targeting mTOR in a large panel of cancer cell lines. More interestingly, these compounds inhibit the proliferation of diseased cells to a greater extent than rapamycin by targeting the rapamycin-insensitive mTOR complex in addition to the rapamycin-sensitive mTOR complex We utilized a complementary approach to develop the mTOR selective inhibitors which consisted of using medium-throughput biochemical screening of an ATP-site directed heterocyclic compound library followed by iterative-chemical synthesis guided by cell-based SAR analysis and molecular modeling.
Fig. 1.

Structures of PI3K and mTOR inhibitors
2. Materials
2.1 Chemical Synthesis materials
All commercially available chemical materials were used directly without further purification.
2.2 Cell Culture and Lysis
Cell culture media: DMEM, 10% fetal bovine serum, penicillin/streptomycin (Invitrogen, Carlsbad, CA)
Centrifuge: RC-5C plus (Sorvall)
2.3 SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
SDS-Polyacrylamide gel: NuPAGE 4–12% Bis Tris Gel 1.5 mm, 15 well (Invitrogen, Carlsbad, CA).
Running buffer (20X): NuPAGE MES SDS running buffer or NuPAGE MOPS SDS running buffer (Invitrogen, Carlsbad, CA).
2.4. Western Blotting for Active S6K1 and Akt
Transfer buffer: 10 mM CAPS, 10% (v/v) ethanol.
Immobilon-P transfer membrane from Millipore, Bedford, MA, and 3 MM Chr chromatography paper from Whatman, Maidstone, UK.
Phosphate-buffered saline with Tween (TBS-T): 0.137 M NaCl, 2.7 mM KCl, 10.0 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4, 0.1% Tween-20.
Blocking buffer: 5% (w/v) nonfat dry milk in PBS-T.
Primary antibody dilution buffer: PBS-T supplemented with 5% (w/v) bovine serum albumin (BSA).
Primary antibodies: Phospho-S6K1 (Thr-389) and phospho-Akt (Ser-473) (Cell Signaling Technology, Danvers, MA).
Secondary antibody: Anti-mouse IgG (for phospho-S6K1) and anti-rabbit IgG (phospho-Akt) conjugated to horse radish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA).
Enhanced chemiluminescent (ECL) reagent: Western Lighting-ECL (Perkin Elmer, Waltham, MA).
2.5. Material in U87MG Model anti-tumor Study
Immunodeficient mice: NCR nude, nu/nu (Taconic Laboratories)
Drug vehicle: 20%: 40%: 40% (v/v) N-methyl-2-pyrrolidone: PEG400: water
3. Methods
3.1 Overview of the Process
As illustrated in the Figure 2, the workflow for developing potent and selective mTOR inhibitors commenced with a medium-throughput biochemical screening of a focused ATP-site directed heterocycle library. Compounds that exhibited activity in this assay were then profiled for selectivity across a panel of approximately 400 kinases using the Ambit KinomeScan methodology from which a class of quinoline-derived compounds appeared to exhibit the greatest selectivity for mTOR. Compounds were then evaluated for their ability to inhibit the downstream target of mTORC1 S6K T389 in a cellular assay utilizing mouse embryonic fibroblasts (MEFs). A key goal of this project was to identify compounds that exhibited selectivity for inhibition of mTOR relative to structurally homologous PIKK family members such as the PI3Ks. The ability of the compounds to inhibit PI3K was evaluated by measuring effects on the activation loop phosphorylation site of Akt T308 in the context of the S473D mutant form of Akt in PC-3 cells. The S473D mutant of Akt was employed because this ‘phospho-mimetic’ mutation abolishes the effects that would result from mTORC2-mediated phosphorylation of this site (8). This is a necessary precaution because most inhibitors in our series are very potent inhibitors of mTORC2 and this would confound our ability to evaluate how potent they are as inhibitors of PI3K. The structure activity relationships derived from these cellular assays was interpreted in the context of a molecular model for the presumed binding mode. A homology model of the 3-dimensional structure of mTOR was constructed using Autodock Vina. This information was then used to inform the design of iterative rounds of compound synthesis. Given the fairly challenging nature of the chemistry required to prepare these compounds, we were never able to prepare focused libraries of derivatives. The most potent and selective compounds were subjected to broader selectivity profiling across a panel of approximately 450 kinases using the Ambit KinomeScan™ technology. The selected compounds chosen from this profiling were then tested in Invitrogen SelectScreen® lipid kinase panel including all of the PI3K isomers and DNA-PK. The most selective compounds were subject to metabolic stability studies using isolated mouse microsomes and then evaluated in mice for their pharmacokinetic and pharmacodynamic properties. Candidate compounds were then tested for anti-tumor efficacy using mouse tumor xenograft models.
Fig. 2.
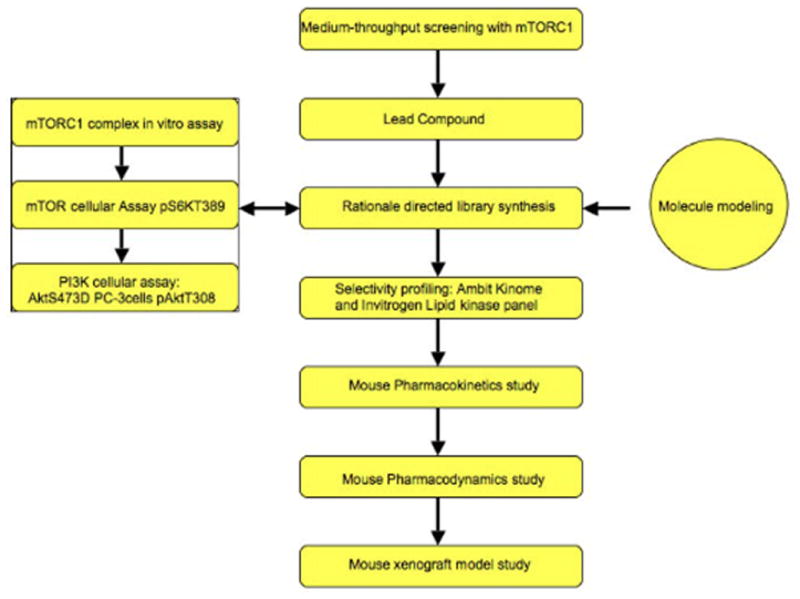
Flowchart for developing mTOR inhibitors
3.2 Chemical Synthesis Methodology
Combinatorial kinase-directed libraries were built from a variety of commercially available heterocycles using both solution and solid phase synthesis (16). Simple and robust chemistries such as nucleophilic aromatic substitution and palladium mediated couplings were used to install diverse elements. For example, disubstituted quinazolines were constructed starting from compound 1 (4,6-dichloroquinazoline) using a nucleophilic substitution reaction to install a particular aniline to the C4 position followed by a Suzuki coupling reaction to install an aromatic side chain to the C6 position of the quinazoline. (Scheme 1)
Rationally designed focused library synthesis was accomplished using the synthetic scheme exemplified in scheme 2 (17). Compound 4 (Ethyl-4,6-dichloroquinoline-3-carboxylate) was subjected nucleophilic addition by particular aryl anilines to afford compound 5. (Scheme 2) Lithium aluminum hydride (LAH) mediated reduction of the ethyl ester furnished the corresponding benzyl alcohol compound 6. Benzylic oxidation with MnO2 followed by Horner-Emmons-Wardsworth olefination generated the cyclized compound 7. Finally, a Suzuki coupling reaction was used to install an additional aryl side chain at C6 position of the quinoline.
Scheme 1.
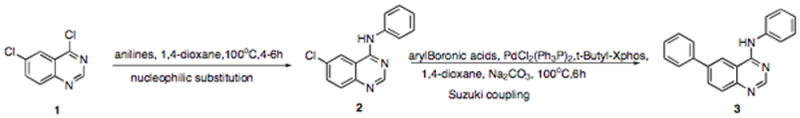
Synthesis of the high-throughput library
Scheme 2.

Synthesis of the focused library
A detailed synthetic protocol is provided below:
-
2.1
To a solution of compound 4 (1 equiv.) in 1,4-dioxane was added aniline (1 equiv.–2 equiv.) at room temperature. The reaction mixture was then heated to 100 °C for 4–6 h and then allowed to cool to room temperature. An aqueous NaOH (1 N) solution was added to neutralize the reaction mixture. The resultant solution was diluted with water and extracted with ethyl acetate. After removal of the solvents under vacuum, the residue was purified by flash column chromatography (hexanes/EtOAc) to afford compound 5. (Note 1)
-
2.2
To a solution of compound 5 (1 equiv.) in THF at 0 °C was added LAH (3 equiv.–5 equiv.) dropwise. After 15–20 min, the solution was warmed to room temperature and stirred for 1–4 h before carefully quenching with methanol and water. Dilution of the mixture with EtOAc and filtration through celite furnished crude 6, which was used in the next step without further purification. (Note 2)
-
2.3
To a solution of compound 6 in CH2Cl2 (1 equiv.) at room temperature was added MnO2 (10 equiv. mass). After 1–4 h, the reaction mixture was filtered through celite. The filtrate was concentrated and placed in a sealed tube and dissolved in dry EtOH. K2CO3 (3 eq.) and triethyl phosphonoacetate were then added sequentially. The resulting mixture was heated to 100 °C for 12–16h before cooling to room temperature. Upon removal of the solvents under vacuum, the residue was diluted with water followed by extraction with ethyl acetate (3X). Purification of the residue by flash column chromatography (Hexanes/EtOAc) provided compound 7. (Note 3)
-
2.4
To a solution of compound 7 in 1,4-dioxane at room temperature was added subsequently PdCl2(Ph3P)2 (0.1 equiv.), t-Bu-Xphos (0.1 equiv.), Na2CO3(3 equiv., 1 N) and boronic acids or pinacol boronic esters. After degassing, the resultant mixture was heated to 100 °C for 6 h before cooling to room temperature and filtering through celite. Upon removal of the solvents, the residue was subjected to column chromatography purification (hexanes/EtOAc) to furnish the desired compounds as exemplified by compound 8. (Note 4) The structure of Torin1 generated following above procedures was shown in Fig. 3.
Fig. 3.

Structure of Torin1
3.3 Biochemical assay of mTORC1 complex
3.3.1. Preparation of human soluble mTORC1 complex
HEK-293T cells that stably expressed N-terminally FLAG-tagged raptor using vesicular stomatitis virus G-pseudotyped MSCV retrovirus was used as a source of mTORC1 complex.
The HEK293T cells were lysed in 50 mM HEPES, pH 7.4, 150 mM NaCl and 0.4% CHAPS at 4°C for 30 min, and the insoluble fraction was removed by centrifugation at 18,000 rpm for 30 min. Supernatants were incubated with FLAG-M2 monoclonal antibody-conjugated agarose for 1 hr and then washed with two column volumes of wash buffer 1 (50 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM DTT and 2 mM ATP and 0.1% CHAPS) and another two column volumes of wash buffer 2 (50 mM HEPES, pH 7.4, 200 mM NaCl, and 0.1% CHAPS).
mTORC1 complex was eluted with 100 μg/ml 3x FLAG peptide in 50 mM HEPES, pH 7.4, 500 mM NaCl and 0.1% CHAPS, then concentrated by centrifugation. Pure mTORC1 was isolated after gel filtration using a tandem Superose 6 10/300 GL column (GE Healthcare) in 50 mM HEPES, pH 7.4 and 150 mM NaCl on an AKTA purifier (GE Healthcare).
3.3.2 In vitro mTORC1 activity assay (Lanthascreen™ time-resolved FRET assay)
mTORC1 (0.1 μg each) was incubated with serially diluted inhibitors (3-fold, 11 points) for 30 min in 5 μL kinase buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2, 4 mM MnCl2, 50 mM KCl) in a 384 well low volume plate (Corning).
The kinase reaction was initiated by the addition of an equal volume of kinase buffer containing 0.8 μM GFP-labeled 4E-BP1 and 100 μM ATP at room temperature. After the kinase reaction for 1 hour, the reaction was stopped by addition of 10 μL of solution containing 20 mM EDTA and 4 nM Tb-labeled anti-phospho 4E-BP1 (T46) antibody (Invitrogen).
After incubation for 30 min, the FRET signal between Tb and GFP within the immune complex was read using Envision plate reader (PerkinElmer). Each data point was duplicated and IC50 values were calculated using Prism4 software (GraphPad).
3.4. Cellular assays of mTOR and PI3K activities
Cell Lysis—Cells were rinsed once with ice-cold PBS and lysed in an ice-cold lysis buffer (40 mM HEPES, pH 7.4, 2 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, and 0.3% CHAPS or 1% Triton X-100, and 1 tablet of EDTA-free protease inhibitors per 25 ml). The soluble fractions of cell lysates were isolated by centrifugation at 13,000 rpm for 10 min in a microcentrifuge. (18)
Cellular IC50 values for mTOR were determined using p53−/− MEFs. Cells were treated with vehicle or increasing concentrations of compound for 1 h and then lysed. Phosphorylation of S6K1 Thr-389 was monitored by immunoblotting using a phospho-specific antibody.
Cellular IC50 values for PI3K were determined using p53 −/−/mLST8 −/− MEFs. Cells were treated with vehicle or increasing concentrations of compound for 1 h and then lysed. Phosphorylation of Akt Thr-308 was monitored by immunoblotting using a phospho-specific anti- body. The summary of Torin1’s biochemical and cellular IC50s was shown in table 1.
Table 1.
Biological evaluation summary of Torin1
| Recombinant mTOR biochemical assay IC50 (nM) | mTORC1 in vitro assay IC50 (nM) | mTOR in cellular assay IC50(nM) | PI3K in cellular assay IC50 (nM) |
|---|---|---|---|
| 4 | 0.29 | 2 | 1800 |
3.5. Molecular modeling
An mTOR homology model was built with Modeller (9v6) based on a published PI3Kγ crystal structure complexed with GDC-0941 (PDB: 3DBS). Compounds were docked into the model with Autodock Vina (19). The presumed proper binding conformation was selected based on the binding modes of structurally related inhibitors whose co-structures had been determined crystallographically. An example of the docking result of Torin1 with mTOR complex was shown in Fig. 4.
Fig. 4.

Pharmacodynamic study of Torin1 (Reproduced from Ref 17)
3.6. Ambit kinome wide and Invitrogen lipid kinase panel selectivity profiling
Ambit kinome wide selectivity profile was performed in Ambit Bioscience with KinomeScan™ technology. Interesting compounds were profiled at a concentration of 10 μM against a diverse panel of 442 kinases by Ambit Biosciences. Scores for primary screen hits are reported as a percent of the DMSO control (% control). For kinases where no score is shown, no measurable binding was detected. The lower the score, the lower the Kd is likely to be, such that scores of zero represent strong hits. Scores are related to the probability of a hit, but are not strictly an affinity measurement. At a screening concentration of 10 μM, a score of less than 10% implies that the false positive probability is less than 20% and the Kd is most likely less than 1 μM. A score between 1–10% implies that the false positive probability is less than 10%, although it is impossible to assign a quantitative affinity from a single-point primary screen. A score of less than 1% implies that the false positive probability is less than 5% and the Kd is most likely less than 1 μM.
Invitrogen lipid kinase panel profile was conducted in LifeScience Inc using SelectScreen® technology by testing IC50 on individual kinase with 10 testing points from a starting 1 μM drug concentration. PIKK family kinase profiling result of Torin1 in Ambit and Invitrogen was shown in Table 2.
Table 2.
Summary of Ambit and Invitrogen selectivity profiles of PIKK-family kinases
| Kinase entry | Ambit score | Invitrogen IC50(nM) | Kinase entry | Ambit score | Invitrogen IC50(nM) |
|---|---|---|---|---|---|
| PIK3C2B | 25 | 549 | PIK3CA(I800L) | 0 | ND |
| PIK3C2G | 11 | ND | PIK3CA(M1043I) | 8.6 | ND |
| PIK3CA | 1.3 | 250 | PIK3CA(Q546K) | 6.8 | ND |
| PIK3CA(C420R) | 0.7 | ND | PIK3CB | 65 | ND |
| PIK3CA(E542K) | 1 | ND | PIK3CD | 51 | 564 |
| PIK3CA(E545A) | 1 | ND | PIK3CG | 1.2 | 171 |
| PIK3CA(E545K) | 0.6 | ND | PIK4CB | 96 | 6680 |
| PIK3CA(H1047L) | 2.6 | ND | DNA-PK | ND | 6.34 |
| PIK3CA(H1047Y) | 12 | ND | mTOR | 0 | 4.32 |
3.7. Pharmacokinetic Studies
The study was performed in Sai Advantium Pharma Limited company (India) with male Swiss Albino mice following single intravenous bolus, oral administration and intraperitoneal injection.
Nine mice were injected 1 mg/kg of Torin1 solution (10% v/v N-methyl pyrrolidone and 50% v/v polyethylene glycol-200 in normal saline) via tail vein. Blood samples were collected at 0, 0.08, 0.25, 0.5, 1, 2, 4, 6 hours. (Note 5)
Nine mice were dosed orally 10 mg/kg of Torin1 suspension (0.5% w/v Na CMC with 0.1% v/v Tween-80 in water). Blood samples were collected at 0, 0.08, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24 hours. (Note 5)
Nine mice were injected 10 mg/kg of Torin1 solution (10% v/v N-methyl pyrrolidone and 20% v/v polyethylene glycol-200 in normal saline and 20% PG in water) via peritoneum. Blood samples were collected at 0, 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 hours. (Note 6)
All samples were processed for analysis by precipitation using acetonitrile and analyzed with a partially validated LC/MS/MS method (LLOQ - 1.138 ng/mL). Pharmacokinetic parameters were calculated using the non-compartmental analysis tool of WinNonlin® Enterprise software (version 5.2). The I.V., P.O. and I.P. studies of Torin1 were summarized in Table 3.
Table 3.
Summary of pharmacokinetic test (reproduced from Ref 17)
| Route | Cmax (ng/mL) | Tmax (h) | AUC (h*ng/mL) | T1/2 (h) | MRT (h) | CL (mL/min/Kg) | Vss (L/Kg) | F (%) |
|---|---|---|---|---|---|---|---|---|
| I.V. | 2757 | ND | 720 | 0.5 | 0.43 | 23.0 | 0.59 | ND |
| P.O. | 223 | 0.25 | 396 | 0.79 | 1.51 | ND | ND | 5.49 |
| I.P. | 5121 | 0.08 | 5718 | 4.52 | ND | ND | ND | ND |
3.8. Pharmacodynamic Studies
Torin1 was formulated by dissolving the compound at a concentration of 25 mg/ml in 100% N-methyl-2-pyrrolidone followed by dilution (1:4) with sterile 50% PEG-400 prior to injection.
Six-week old male C57BL/6 mice were fasted overnight prior to drug treatment. The mice were treated with vehicle (for 10 hr) or Torin1 (20 mg/kg for 2, 6 or 10 hr) by IP injection, and then re-fed 1 h prior to sacrifice (CO2 asphyxiation).
Tissues were collected and frozen on dry ice. The frozen tissue was thawed on ice and lysed by sonication in tissue lysis buffer (50 mM HEPES, pH 7.4, 40 mM NaCL, 2 mM EDTA, 1.5 mM sodium orthovanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, 0.1% SDS, 1.0% sodium deoxycholate and 1.0% Triton, supplemented with protease inhibitor cocktail tablets [Roche]).
The concentration of clear lysate was measured using the Bradford assay and samples were subsequently normalized by protein content and analyzed by SDS-PAGE and immunoblotting. The result of PD study of Torin1 was exemplified in Fig. 5.
Fig. 5.
U87MG tumor xenograft model study of Torin1 (reproduced from Ref 17)
3.9. Animal Xenograft Study (U87 model)
2 × 106 U87MG glioblastoma cells were resuspended in 100 μL of media that had been pre-mixed with matrigel, and was injected subcutaneously in the upper flank region of mice (6 weeks old immunodeficient) that had been anaesthetized with isoflurane. Tumors were allowed to grow to 1 cm3 in size. (Note 6)
Animals were divided into two treatment groups randomly for vehicle and Torin1. Torin1 was formulated as 5 mg/ml in a solution of N-methyl-2-pyrrolidone: PEG400: water (20%:40%:40%). Both vehicle and Torin1 was delivered by IP injection at a dosage of 20 mg/kg (q.d.)
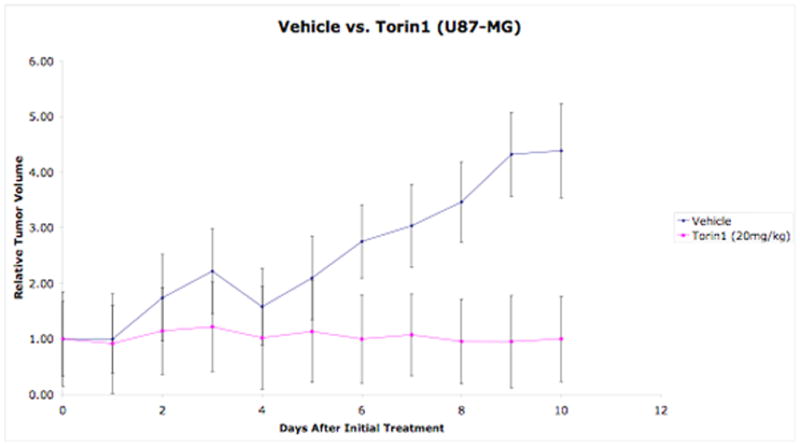
Tumors were measured with calipers in two dimensions every other day. Tumor volumes were estimated with the formula: , where a = short and b = long tumor axes, respectively, in millimeters. The Result of Torin1’s anti-tumor activity was exhibited in Fig. 6.
Acknowledgments
We thank Life Technologies Corporation for SelectScreen® Kinase Profiling Service and Ambit Bioscience for performing KinomeScan™ profiling. We also thank SAI Advantium Pharma Limited Inc. (India) for the pharmacokinetic study.
Footnotes
Some series of compound 5 would require CHCl3/iPrOH (4:1) extraction system due to a poor solubility in EtOAC.
LAH should be added at 0°C to avoid facile over reduction of the benzyl alcohol to the toluene like product. The reaction is typically monitored carefully by LC-MS to avoid over reduction after warming up to room temperature. A 0°C ice-water bath is preferred during the quenching procedure and the methanol should be added drop-wise due to the exothermic reaction. Extra EtOAc would be used to rinse the filtrate to avoid loss of the product in the celite.
Complete removal of the EtOH under vacuum is needed to avoid complications with the purification process.
Some compound 8 analogs are very polar and require reverse phase column chromatography with MeOH/Water or CH3CN/Water solvent system.
All blood samples were collected in microcentrifuge tubes containing K2EDTA as an anticoagulant. Set of three mice at each time point Plasma samples were separated by centrifugation and stored below −70 °C until bioanalysis.
Immunodeficient mice (NCR nude, nu/nu; Taconic Laboratories) were maintained in a pathogen-free facility and were given autoclaved food and water ad libitum. All animal studies were performed according to the official guidelines from the MIT Committee on Animal Care and the American Association of Laboratory Animal Care.
References
- 1.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J antibiotics (Tokyo) 1975;28:727–732. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- 3.Rao RD, Buckner JC, Sarkaria JN. mammalian Target of Rapamycin (mTOR) Inhibitors as Anti-Cancer Agents. Curr Cancer Drug Targets. 2004;4:621–635. doi: 10.2174/1568009043332718. [DOI] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses A, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964–4973. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 6.Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske EP. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–987. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 7.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-insensitive functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 10.Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 11.Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Yamano M, et al. Synthesis and biological evaluation of pyrido[3′,2′:4,5]furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem Lett. 2007;17:2438–2442. doi: 10.1016/j.bmcl.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 12.Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, Willems L, et al. PI-103, a dual inhibitor of class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia. 2008;22:1698–1706. doi: 10.1038/leu.2008.144. [DOI] [PubMed] [Google Scholar]
- 13.García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q, Thoreen CC, Wang J, Sabatini DM, Gray NS. mTOR medicated anti-cancer drug discovery. Drug Discov Today: Therapeutic Strategies. 2009;6:47–55. doi: 10.1016/j.ddstr.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding S, Gray NS, Wu X, Ding Q, Schultz PG. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J Am Chem Soc. 2002;124:1594–1596. doi: 10.1021/ja0170302. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Chang J, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mTOR inhibitor for the treatment of cancer. J Med Chem. 2010 doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Trott O, Olson AJ. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]