

Abstract
Systems Toxicology is the integration of classical toxicology with quantitative analysis of large networks of molecular and functional changes occurring across multiple levels of biological organization. Society demands increasingly close scrutiny of the potential health risks associated with exposure to chemicals present in our everyday life, leading to an increasing need for more predictive and accurate risk-assessment approaches. Developing such approaches requires a detailed mechanistic understanding of the ways in which xenobiotic substances perturb biological systems and lead to adverse outcomes. Thus, Systems Toxicology approaches offer modern strategies for gaining such mechanistic knowledge by combining advanced analytical and computational tools. Furthermore, Systems Toxicology is a means for the identification and application of biomarkers for improved safety assessments. In Systems Toxicology, quantitative systems-wide molecular changes in the context of an exposure are measured, and a causal chain of molecular events linking exposures with adverse outcomes (i.e., functional and apical end points) is deciphered. Mathematical models are then built to describe these processes in a quantitative manner. The integrated data analysis leads to the identification of how biological networks are perturbed by the exposure and enables the development of predictive mathematical models of toxicological processes. This perspective integrates current knowledge regarding bioanalytical approaches, computational analysis, and the potential for improved risk assessment.
1. Introduction
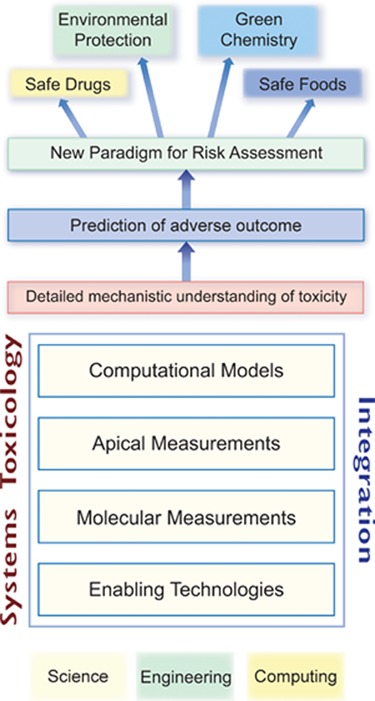
Assessing the potential risk that a substance poses to human health or the environment involves a combined quantitative analysis of both exposure and hazard, including the estimation of associated uncertainties. For decades, the evaluation of toxicological hazards in the context of regulatory risk assessment has relied heavily on animal experimentation, often described in internationally recognized test guidelines (e.g., OECD). Animal experiments are typically designed either to indicate the dose at which substance-induced pathological effects, other than cancer, are first observed after a defined treatment duration (e.g., lowest observed adverse effect level, LOAEL) or to infer the dose below which no effect is expected to occur (e.g., no observed adverse effect level, NOAEL). Then, through a process of extrapolation using empirically derived uncertainty factors, the assessor can arrive at various hazard reference values (e.g., a derived no effect level, DNEL) useful for an ecological or human health risk assessment. Throughout this process, information about the mechanistic basis of the observed adverse effects is often considered nice to have rather than necessary to have. Despite its shortcomings, over the years this traditional approach to toxicity assessment has been generally considered as offering an acceptable level of protection to our health and the environment. However, we are arguably positioned to shift toward a new paradigm for toxicity assessment because of the limitations of this process in addressing increasing demands imposed by modern society together with the opportunities offered by considerable developments in biomedical research tools to do better. The possibilities of improved safety from applying Systems Toxicology (Figure 1) approaches provide a scientific impetus to bring about change.1,2
Figure 1.

What is Systems Toxicology? Systems Toxicology is aimed at decoding the toxicological blueprint of active substances that interact with living systems. It resides at the intersection of Systems Biology with Toxicology and Chemistry. It integrates classic toxicology approaches with network models and quantitative measurements of molecular and functional changes occurring across multiple levels of biological organization. The multidisciplinary Systems Toxicology approach combines principles of chemistry, computer science, engineering, mathematics, and physics with high-content experimental data obtained at the molecular, cellular, organ, organism, and population levels to characterize and evaluate interactions between potential hazards and the components of a biological system. It is aimed at developing a detailed mechanistic as well as quantitative and dynamic understanding of toxicological processes, permitting prediction and accurate simulation of complex (emergent) adverse outcomes. Thereby, the approach provides a basis for translation between model systems (in vivo and in vitro) and study systems (e.g., human, ecosystem). Systems Toxicology, therefore, has an ultimate potential for extrapolating from early and highly sensitive quantifiable molecular and cellular events to medium- and long-term outcomes at the organism level, and its application could be part of a new paradigm for risk assessment. Artwork by Samantha J. Elmhurst (www.livingart.org.uk).
There are numerous regulatory drivers promoting the development and application of innovative methodology for risk assessment, many of which have particular emphasis on alternative in vitro and computational methods that avoid or limit the use of animals.3 Since 1986, the European Union (EU) has had legislation in place covering the use of animals for scientific purposes, in particular Directive 2010/63/EU,4 which came into full effect in January 2013, restricting the use of animals for assessing the toxicological properties of substances for either regulatory or research purposes to cases where it can be clearly demonstrated that no alternative method is available. The EU Cosmetics Regulation5 bans the marketing of cosmetics products in the EU that have had any ingredient tested on an animal since 11 March 2013, irrespective of the fact that full replacement methods are not yet available for complex health effects such as chronic systemic toxicity, reproductive toxicity, and cancer. Legislation dealing with risk management in other sectors, such as the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH),6 the Plant Protection Products Regulation,7 and the Biocides Regulation,8 still allows the use of animal testing to satisfy information requirements; however, the use of alternative methods is strongly encouraged and, in some cases, obligatory if available and accepted by regulatory authorities. Thus, never before has there been such demand for animal-free solutions to toxicological testing and assessments that are fit for the purpose of regulatory decision making.
Considering the diversity of chemical exposures under real-world conditions, there is currently no mechanism within the framework of EU legislation to facilitate the systematic, comprehensive, and integrated assessment of the combined effects of chemicals resulting from different routes and times of exposure. Although risk assessment for mixtures is typically driven by exposure considerations, a key question is whether certain combinations of chemicals have independent, additive, or even synergistic toxic effects. However, mechanism-based mixture toxicology is severely hindered because the mechanism(s) or mode(s) of action of most chemicals is unknown or poorly described, there are no established criteria for characterizing mechanisms or modes of action, and there is no agreed terminology or formal description thereof.9 In addition, although assessing the combined action of co-acting chemicals may be practically possible when they have the same basic mechanism of action (e.g., act on the same molecular target), the problem becomes much more difficult when considering chemicals that impact the same biological system but act through different mechanisms. These issues have emerged prominently, for example, in the contemporary dialogue regarding endocrine-disrupting chemicals (EDCs) because there is much debate regarding what constitutes the endocrine system and criteria to identify EDCs. Tackling issues such as non-monotonic dose–response and the existence of thresholds is severely hampered by an over-reliance on empirical evidence derived primarily from animal experiments, a lack of emphasis on understanding the fundamentals of endocrine system dynamics, and the limitations of analytical and computational tools suitable to characterize and model the problem at hand.
In the context of chemical risk assessment, the application of the new tools of experimental and computational Systems Toxicology is anticipated to occur first for the large numbers of environmental chemicals that have little or no toxicological data in the public domain. In this manner, there can be a prioritization of chemicals to evaluate for endocrine-disrupting activity (see http://www.epa.gov/endo/pubs/edsp21_work_plan_summary%20_overview_final.pdf). This approach also will be used to prioritize chemicals under the U.S. Toxic Substances Control Act (TSCA) (see http://www.epa.gov/agriculture/lsca.html) once the methodologies are capable of deciphering modes of action beyond those related to the endocrine system. For drug development, motivators for the application of systems tools include identification and characterization of disease-related therapeutic targets and uncovering chemical liabilities early in the drug-discovery pipeline, thus avoiding costly failures once a candidate molecule reaches clinical trials.10 Drug safety objectives are being addressed, for example, by a large public–private partnership, the eTOX project in the European Union (see http://www.etoxproject.eu), under the Innovative Medicines Initiative (see http://www.imi.europa.eu/). The aim is to develop a drug safety database for the pharmaceutical industry and novel tools to predict better the toxicological profiles of molecules in early stages of the drug-development pipeline. Another application on the horizon is the assessment of alternative chemicals designed and produced within the framework of green or sustainable chemistry. The aim in this case is to minimize the use and generation of hazardous substances. Without an understanding of the toxicological liability of proposed alternatives, the push for greener chemicals is limited to using chemical engineering tools to reduce the use of known hazardous precursors, maximize atom economy in synthetic processes, and increase the energy efficiency of reactions. The U.S. National Academy of Science has recently undertaken a project to provide a framework for evaluating safer substitutes (see studies in progress at http://www8.nationalacademies.org/cp/projectview.aspx?key=49569), including, in particular, Systems Toxicology. Together with regulatory drivers, an overarching expectation is that Systems Toxicology approaches, applied in a systematic and rigorous manner, will provide more sound information on which to judge how chemicals cause biological perturbations, moving knowledge beyond knowing only what phenotypes are altered. The resulting understanding of biological responses to chemical exposure and biomolecular interactions is expected to reduce uncertainties in species extrapolations, in high-to-low dose extrapolation, and to shed light on life stage and other susceptibility factors such as genetics and pre-existing diseases.
When paired with advances in hazard detection and biomonitoring strategies,11 Systems Toxicology holds the long-term promise, at least in part, to address these challenges. It will enable the gradual shift from toxicological assessment using solely apical end points toward understanding the biological pathways perturbed by active substances. Systems Toxicology is expected, therefore, to create knowledge regarding both the dynamic interactions between biomolecular components of a complex biological system and how perturbing these interactions with active substances alters homeostasis and leads to adverse reactions and disease. This knowledge, represented as adverse outcome pathways (AOPs),12 is then expected to enable our system-level understanding of exposure to active substances and to lead to more effective risk assessment.
Systems Toxicology is rooted in the premise that morphological and functional changes in cellular, tissue, organ, and organism levels are caused by, and cause, changes at the genomic, proteomic, and metabolomic level. A molecular system can be represented initially by high-level static biological network (BN) models that employ basic graph representations to map the molecular entities (nodes) and their interactions (edges). Although these basic BN models serve to map the underlying molecular wiring of a biological system, they are too simplistic in nature to describe a toxicological pathway and its dynamic nature. The description of a toxicological pathway requires the development of more sophisticated computable BNs by incorporating mechanistic information through the encoding of causal relationships between the nodes. Such models enable the computational analysis and contextualization of experimental data, for instance, for the quantification of network perturbation, which is further described in this perspective. Ultimately, the description of dynamic toxicological processes requires the development of even more sophisticated executable BN models by incorporating the mathematical description of the dynamic behavior of these causal relationships. Such BNs are capable of simulating changes in state of the molecular network caused by external perturbation or modification of boundary conditions. To be useful in current risk-assessment frameworks, however, mathematical models need to be multiscale or able to traverse the different levels of biological organization ultimately to relate the dynamics at the BN level to apical adverse outcomes on which regulations are based. The most direct causal path, traversing multiple levels of organization and linking exposure, binding of the toxicants to biomolecules, intermediate key events, and apical adverse outcome, is what characterizes AOPs (Figure 2).12
Figure 2.

Steps that define the Systems Toxicology paradigm, from biological network models to dynamic adverse outcome pathway (AOP) models. The development of dynamic AOP models enabling the simulation of the population-level effects of an exposure is the ultimate goal of Systems Toxicology. This development follows three broad steps of maturity from top to bottom. The first level consists of the development of causal computable biological network models that link the system’s interaction of a toxicant with the organ-level responses. Such models can be used to quantify the biological impact of an exposure in the context of quantifiable end points such as histology or physiological measurements. In a second step, as more mechanistic knowledge derived from quantitative measurements accumulates, dynamic models linking the exposure with the organ-level responses can be developed. Ultimately, the third level of maturity is reached when the link between the exposure and the population outcome can be represented by mathematical models that enable the simulation of population-level effects of an exposure. Blue arrows denote causal links, which are mainly derived from correlative studies. Artwork by Samantha J. Elmhurst (www.livingart.org.uk).
AOPs are tools for describing the entire toxicological process from the molecular level, triggered by an active substance, to an adverse outcome of regulatory concern, typically observed at a tissue, organ, organism, and potentially population level. Although not traditionally associated with Systems Toxicology, an AOP is, in fact, a model that discretizes a process into key events (KE) (nodes) and the causal relationship between them (edges). An AOP13 is developed first on the basis of a qualitative model that captures KEs and their high-level relationships. Gradually, as detailed mechanistic knowledge accumulates, they will capture the causal relationships between the KEs and the underlying molecular events. Even qualitative AOP models serve a very important purpose. For example, such AOPs can be used to map quantitative information for visualization purposes or to compare and identify common features and intersections between AOPs. As AOPs are developed to include more detailed causal and quantitative information, they are expected to become computable and eventually executable, making them quantitative, sensitive, and predictive of outcome for risk assessment.
The implementation and practice of Systems Toxicology requires several experimental approaches to generate data relevant to the question at hand. First, the exposure needs to be characterized both in terms of composition, dose, and duration and in terms of biomarkers of exposure at the subject level. The next step is to examine the effect of these exposures in biological model systems such as cellular and organotypic cultures relevant to the exposure and the observed toxicological effects. The results obtained from these in vitro experiments can then further guide the selection of relevant models of human diseases and environmental health. All of these studies will, however, yield quantitative phenotypic readouts in the context of quantitative system-wide molecular measurements. The third step is to integrate the data from these large-scale experiments, to analyze them to decipher the biological mechanisms by which adverse effects arise from exposure, and to build a functional model thereof. A new framework for risk assessment should therefore integrate this mechanistic knowledge. This perspective, derived from the presentations and discussions held at a recent symposium on Systems Toxicology,98 offers an integrated view on this trajectory from experimentation to computational analysis and risk assessment.
2. The Exposome: A Systems Approach to Understanding Exposure
Xenobiotic exposures are acknowledged to play an overwhelmingly important role in common chronic diseases such as cancer, diabetes, cardiovascular, and neurodegenerative diseases, which constitute the major health burden in economically developed countries.14 It has been estimated that 90% of cancer deaths and half of heart disease mortality cannot be explained by genetic factors, suggesting an environmental origin.15 Sequencing the human genome has permitted identification of individual susceptibility to disease through genome-wide association studies, but so far these have explained comparatively little of the variability in chronic disease prevalence.16 In contrast, there has been a relative paucity of comparable tools for exposure assessment17,18 and exposure estimates (e.g., in epidemiology studies) have relied primarily on questionnaires, geographic information, and a few targeted measurements. In environmental toxicology, exposure assessment is commonly based on quantifying chemical concentrations in the exposure medium (e.g., water or soil), although there is increasing recognition that quantification of intraorganism concentrations provides a better means to link exposure to toxicological effect.19
Remarkable advances in analytical sensitivity and capacity, including omics technologies, have now made environment-wide association study (EWAS) feasible,20−,22 permitting detection of associations between biomarkers and human health outcomes on a much larger scale than previously possible. Wild,17 therefore, proposed the concept of the exposome, representing all environmental exposures (including diet, lifestyle, and infections) from conception onward, as a complement to the genome in studies of disease etiology. In this concept, the body’s internal environment is subject to exposure to biologically active chemicals. Exposures are therefore not only restricted to chemicals (toxicants) entering the body from air, water, or food but also include endogenous chemicals produced by inflammation, oxidative stress, lipid peroxidation, infections, gut flora, and other natural processes. This internal chemical environment continually fluctuates during life because of changes in external and internal sources, aging, infections, life-style, stress, psychosocial factors, and pre-existing diseases.22
A challenge lies in choosing on which of the thousands of measurable biologically active chemicals to focus. Paraphrasing Abraham Lincoln, Stephen Rappaport has suggested, “We cannot measure everything all the time, but we can measure most things some of the time, and we can measure some things most of the time.”98 Thus, the primary purpose of biomarker-based exposure monitoring should be to identify risk factors for use in epidemiological studies rather than to provide exhaustive personalized exposomes that cannot be interpreted at an individual level. Even a partial exposome characterization can lead to major public health benefits, for example, one or more cross-sectional exposure measurements at different time points in a prospective cohort study.18 Because the serum exposome is enormously complex, an EWAS focuses on measuring network features, such as N-linked glycoproteins or reactive electrophiles stabilized as adducts of human serum albumin.23 In this way, potential biomarkers can be identified for subsequent use in targeted epidemiological studies. The issue of timing of exposure assessment may have to be tailored to specific hypotheses by examining exposure in cohorts of different ages.18
The term biomarker can relate to any interaction between an external environmental agent and a biological system such as the human body. In the context of risk assessment, biomarkers of interest have been defined in terms of whether they relate quantitatively to a particular exposure, its effect in a biological system, or to the way in which the system reacts.24 These biomarkers are distinguished as follows:
-
•
Biomarker of exposure: an exogenous substance or its metabolite or the product of an interaction between a xenobiotic agent and some target molecule or cell that is measured in a compartment within an organism;
-
•
Biomarker of effect: a measurable biochemical, physiological, behavioral or other alteration within an organism that, depending upon the magnitude, can be recognized as associated with an established or possible health impairment or disease;
-
•
Biomarker of susceptibility: an indicator of an inherent or acquired ability of an organism to respond to the challenge of exposure to a specific xenobiotic substance.
However, the boundaries between exposure and effect are fluid and it is therefore helpful to describe the measurement of xenobiotics and their metabolites in biological systems as biological monitoring and to restrict the term biomarker to consequences of functional or structural change (e.g., protein and DNA adducts, chromosome aberrations, urinary proteins, and enzymes) that represent an integration of exposure and various biological processes. For example, chemical-induced DNA adducts vary at the individual level because of carcinogen metabolism, DNA repair, and cell turnover, at least partially as a result of interindividual genetic variation. Therefore, such biomarkers may not strictly correlate with exposure but are more likely to be associated with disease outcome, for example, cancer risk, because they reflect the consequences of exposure on a pathway relevant to carcinogenesis.18
In assessing biomarkers, particularly omics-based biomarkers, it is therefore important to separate those generated by environmental exposures ultimately leading to disease (causal pathway) from those that are manifestations of disease (reactive pathway). This process of biomarker identification and validation can be separated into two main phases.23 In the discovery phase, EWAS is used in an untargeted, data-driven way with a focus on small molecules and proteins. The main objective is to identify discriminating features that are consistent across studies, ideally those with a prospective cohort design. The exact chemical nature of the discriminating feature has to be identified, a process that may involve searching for matches in existing databases as well as additional experiments using a battery of different analytical techniques. In the second phase, candidate biomarkers are investigated in targeted, knowledge-driven follow-up studies to establish causality, prevention, diagnosis, prognosis, and treatment of diseases. An example of such a stepwise approach to the identification of biomarkers is the screening of several thousand small nonpolar molecules in serum to discover a molecular biomarker of susceptibility to colorectal cancer linked to low serum GTA-446 anti-inflammatory fatty acid levels.25
Unraveling complex environmental and genetic disease etiologies demands that both environmental exposures and genetic variation are reliably measured. Characterizing the exposome represents a technological challenge at least as large as that for the human genome project, which began when DNA sequencing was in its infancy.22 The data generated by application of omics technologies, potentially in a longitudinal fashion, poses enormous bioinformatics and biostatistics challenges, requiring substantive interdisciplinary collaboration. However, exposomics methods and goals are not as standardized as they were for DNA sequencing, and, consequently, the exposome does not translate as easily to scale-up as did sequencing the human genome.18 Nevertheless, Systems Biology and Systems Toxicology, when linked to epidemiology, offer the prospect of examining gene–environment interactions and identifying biomarkers for causative pathways of diseases, monitoring their progression, and guiding treatments.
3. Large-Scale Molecular Measurement
A fundamental building block of Systems Toxicology is that exposures lead to changes at the molecular level, some of which may induce morphological or functional changes at the cellular and organism level that contribute to toxic outcomes. Over the past decade, there has been unprecedented development in technologies that enable the collection of very large-scale data sets composed of quantitative information regarding molecular responses on the basis of changes in all major classes of biomolecules in living organisms. Furthermore, advanced imaging methods enable the quantitative analysis of cell-level changes. Hereafter, we will highlight a few of the recent developments in transcriptomic, proteomic, metabolomic, and lipidomic measurement technologies as well as high-content screening as the main emerging molecular methods that are critical enablers of Systems Toxicology. A broader range of technologies, including epigenetics,26−28 and their applications to toxicology have been described recently.2,29
3.1. Transcriptomics
Transcriptomics or gene-expression profiling is perhaps the most widely used measurement technology in Systems Toxicology and is used to study the changes in expression of all mRNAs in a cell population, organ, or organism. Transcriptomic analysis is also the best established approach for identifying perturbed biological networks (see below) and thereby gaining mechanistic insight into the system’s response to an exposure.30,31 The current technological basis of transcriptomics measurements for Systems Toxicology studies has involved oligonucleotide microarrays and, more recently, next-generation sequencing (NGS).
Oligonucleotide microarrays represented the major technological advance in transcriptomics of the past decade and were introduced and generalized with the invention of high-density array printing by Affymetrix. Today, it is possible to design an array of oligomer probes that covers the whole transcriptome of any organism for which the genome sequence is known and the possible open reading frames and gene models have been identified using well-established bioinformatics analysis pipelines. These microarrays enable high-throughput parallel analysis of many samples derived from model systems as well as clinical or environmental samples. With the concomitant progress in full cDNA sequencing and RNA sequencing by next-generation sequencing technologies, current microarrays can cover each gene or its exons; such exon arrays are now available for human, mouse, and rat. The use of exon arrays has improved the analysis of alternatively spliced RNA transcripts as well as the accuracy of the overall gene-expression measurements.
NGS technologies30,31 are emerging as the measurement methods that may supersede microarray technologies in the near future on the basis of greater accuracy, providing exact transcript counts and results that closely approach quantitative PCR. Furthermore, NGS methods are more flexible as they enable gene-expression studies in organisms for which microarrays are not available, such as model systems used in environmental toxicology. Finally, they are likely to offer a higher throughput than microarrays as new developments will likely allow for the analysis of thousands of transcriptome samples in a single sequencing run.
3.2. Proteomics
The proteome represents the full complement of the proteins in a cell, organ, or organism, and proteomics is a systematic approach to characterizing all or an enriched subset of proteins therein. Measuring changes in levels and modifications of proteins is applied to diagnostics, drug discovery, and investigating toxic events. Proteomic data are of particular value in Systems Toxicology because the proteome is an important mediator of altered biological responses as a consequence of exposure to active substances. Increases or decreases in protein levels may be a direct consequence of corresponding mRNA-expression changes, but increases or decreases in protein function may also be influenced by post-translational modifications. For example, protein phosphorylation, which can be further addressed by high-throughput phosphoproteomics, enables the characterization of molecular events proximal to disease-related signaling mechanisms.32
Mass spectrometry (MS) is widely considered to be the central technology for modern proteomics, mainly because of its unsurpassed sensitivity and throughput. Thanks to the high accuracy of MS, peptides in the subfemtomolar range can be detected in biological samples with a mass accuracy of less than 10 ppm. This level of accuracy is necessary to compare proteins between samples derived from exposed and control systems. Isotope tagging for relative and absolute quantification (iTRAQ) is used in comparative proteomics for Systems Toxicology because it enables the relative quantification of protein species between samples in a nontargeted manner. This method can be further complemented with a targeted method of even higher accuracy: selected reaction monitoring (SRM). SRM enables the precise quantification of predefined proteins. It leverages a unique property of MS to quantify peptides, produced by a controlled enzymatic digestion of the proteome, as a way to quantify their corresponding proteins. Because it is a targeted approach, any proteomics analysis by SRM requires the a priori selection of the proteins to quantify. This is generally achieved using the results of previous experiments using nontargeted approaches such as iTRAQ combined with a review of the scientific literature to leverage prior knowledge. The list of selected proteins is then processed with bioinformatics tools to identify at least two proteolytic peptides that optimally represent the protein and distinguish it from all others. This step is followed by the selection of SRM transitions for each of those peptides and several optimization and validation steps to ensure unique identification and accurate quantification. This method enables a multiplexed approach by which hundreds of proteins can be quantified in a single MS run.
Targeted approaches for protein quantification have been developed to produce accurate and high-sensitivity data for defined subproteomes. An emerging example is the development of surface-capture probes to interrogate the surface-reactive subproteome, a critical response gateway for active substances, as recently demonstrated for hepatocytes.33 Antibody-based approaches are also important in targeted proteomics. For instance, reverse protein arrays (RPAs) have evolved from classical enzyme-linked immunosorbent assays (ELISA) and enable high-throughput analysis of up to 192 different lysates, including post-translationally modified proteins, on a ZeptoCHIP with over 200 validated antibodies. Other antibody-based targeted methods, such as Luminex, provide alternative measurement platforms for multiplexed, high-sensitivity and -specificity targeted proteomic data generation.32 These data can lead to the refinement of mechanistic details of toxicology pathways that were mainly based on transcriptomic data.
3.3. Metabolomics
Metabolomics involves a comprehensive and quantitative analysis of all metabolites or low molecular weight organic or inorganic chemicals that are products or substrates of enzyme-mediated processes.34−40 In the context of Systems Toxicology, metabolomics is unique because it can be used to define amounts of internalized xenobiotic chemicals and their biotransformation products23,41 as well as the perturbed endogenous metabolome, which represents the ultimate change in the levels of chemical species resulting from molecular perturbations at the genomic and proteomic levels.38−40 In the first case, an understanding of the kinetic behavior of xenobiotic toxicants and their metabolites (as well as related biomolecular adducts)42 is necessary to identify candidate biomarkers of exposure for human and environmental monitoring. The second is a more conventional Systems Biology perspective of metabolomics, which involves both identification of metabolites and quantification of changes in their abundance and rates of production caused by an exposure.
From a technical perspective, metabolomics most commonly involves NMR spectroscopy and/or MS analysis techniques in untargeted profiling or targeted analysis strategies.38,43 Profiling of a metabolome may entail global detection and relative quantification of a large number of metabolites without a priori knowledge. However, targeted experiments involve the absolute quantification of a small number of metabolites (around 20 or less) to test a defined hypothesis. In Systems Toxicology, metabolomics may be accompanied by concomitant transcriptomic and proteomic measurements to provide the full context of the exposure. Integrated analysis of these diverse data types is necessary to enable a full understanding of the mechanistic events driving metabolomic changes. Finally, emerging application areas include the characterization of effects of mixtures on chemical responses in microfluidic organ model systems.44,45
3.4. Lipidomics
The lipidome is the complete lipid profile of a biological system and is an example of a specialized subset of the metabolome. By extension, lipidomics is a systematic approach to characterize and quantify lipids in biological samples using, as with metabolomics, analytical methods based on NMR and MS. It has been recognized recently that alterations of lipid homeostasis contribute to several pathophysiological conditions. Lipids are not only an important energy store but are also essential constituents of all cellular membranes and exert a number of signaling functions. Indeed, profound changes in the cellular and tissue lipid composition occur in response to exposure to active substances and environmental factors. Lipidomics is thus a powerful method to monitor the overall lipid composition of biological matrixes and has been shown to have great potential to identify and detect candidate biomarker signatures indicative of toxicity. There have been rapid recent advances in MS-based techniques capable of identifying and quantifying hundreds of molecular lipid species in a high-throughput manner.46,47 Large sample collections can be analyzed by automated methods in a 96-well format.48 The lipid extracted from various biological matrices can then be analyzed by multiple MS platforms, either by detecting the lipids by shotgun lipidomics or after separation by liquid chromatography to detect and quantify lipids of lower abundance. Performing concomitant transcriptomics analysis in samples subjected to lipidomic profiling enables the integrated analysis of these complementary data sets to derive mechanistic information regarding the toxicological meaning of lipid alterations.49
3.5. High-Content Screening
Molecular changes measured in Systems Toxicology studies should, as often as possible, be correlated with cellular or tissue-level changes measured under the same conditions. Although histopathology is the main approach to gather such data, phenotypic assessment of cells in culture can be performed using high-content screening (HCS) methods.50 This technique is based on computer-aided visual detection of a panel of functional biomarker measurements providing precise temporal, spatial, and contextual information that define the biological status of the cells or organs and structure of small organisms (e.g., early life stages in zebrafish). The measurement of the different biomarkers can be achieved on either a fixed specimen labeled with fluorescent reagents or directly on a living specimen during the time of the exposure. A broad panel of biomarkers can be analyzed in a multiplexed manner to provide quantitative measurements of the level of abundance and localization of proteins (e.g., phosphorylated protein) and/or changes in the morphology of the cell. Each biomarker can be adapted to the cell type relevant to a particular exposure modality to quantify key cellular events such as (1) apoptosis and autophagy, (2) cell proliferation, (3) cell viability, (4) DNA damage, (5) mitochondrial health, (6) mitotic index, (7) cytotoxicity and oxidative stress, (8) nascent protein synthesis, and (9) phospholipidosis and steatosis. HCS is enabled by computer-aided automated digital microscopy (e.g., ArrayScan) or flow cytometry for data analysis and storage.
4. Data and Information Management
Advanced experimental technologies for Systems Toxicology produce unprecedented amounts of data, the management of which is a major challenge for scientists. It is expected that this trend will accelerate because of ongoing technology developments and concomitant cost decreases. Therefore, we are witnessing many efforts globally to develop methods and tools to manage, integrate, and process this data. As a consequence, a wide range of open-source software solutions (e.g., EMMA51 and MIMAS52) are continuously being developed and made available to store and manage data along with the information to describe accurately the experimental setup and the conditions that led to the data. This latter point is of crucial importance to data integration, experimental reproducibility, and proper data analysis and interpretation. The Institute of Medicine of the U.S. National Academy of Sciences has recently specified experimental transparency as one of the major requirements for translational omics.53 Minimum information about microarray experiment (MIAME )-based54 exchange format MAGE-TAB55 provides a solution for microarray data, which has also become the standard for data sets deposited in public databases such as ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) or Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). A similar standard exists for proteomics experiments, namely, the minimum information about a proteomics experiment (MIAPE).56
The analysis of complex data sets is greatly facilitated by computational pipelines or workflows that automate a sequence of computational tasks, often performed by independent software components. In this context, Computational Biology workflow management systems (e.g., Galaxy57 and Taverna58) have been developed to implement reproducible data analysis processes. caArray and caGrid59 from the National Cancer Institute Biomedical Informatics Grid (NCI caBIG) are prominent examples of a Systems Biology data-integration environment that includes knowledge management as well as workflow systems. For instance, this environment allows integration with commercial data analysis and biological pathway analysis systems.
5. Computational Platforms for Systems Toxicology
As outlined above, the core objective of Systems Toxicology is to elucidate the mechanisms that causally link a well-characterized exposure to active substances with quantifiable adverse events and disease. It requires collection of experimental data reflecting molecular changes in the context of quantifiable cellular, tissue-level, or physiological changes that are linked to disease phenotypes or adverse events at the organism level. Thus, Systems Toxicology heavily relies on computational approaches to manage, analyze, and interpret the data generated by the large-scale experimental approaches described earlier. The ultimate goal is to develop predictive in silico models that can be used in risk assessment. To reach this ambitious goal, computational Systems Toxicology has four major areas of focus:
-
•
Analyzing the massive amounts of systems-wide data generated by high-throughput methods and their integration with the structural description of the substances involved in the exposure.
-
•
Representing the relevant mechanisms leading to adverse outcome as biological network models that adequately describe both the normal state and the causal effect of their perturbations upon exposure to active substances (Figure 3).
-
•
Quantifying the dose-dependent and time-resolved perturbations of these biological networks upon exposure and assessing their overall biological impact.
-
•
Building and validating adequate computational models with predictive power that can be applied to risk assessment (Figure 3).
Figure 3.
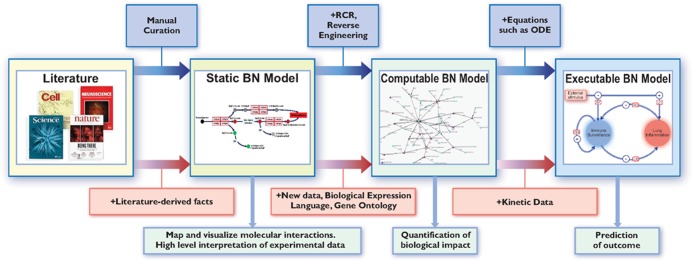
Biological network model-development process. Initial static BN models can be constructed using biological facts derived from the literature. This process mainly involves manual curation. These initial models can serve as the basis to guide the development of computable BN models. These models rely on biological facts (key events) derived from both the literature and new experimental data and are expressed in a computable format such as the biological expression language (BEL). Computational methods such as reverse causal reasoning and reverse engineering are used to support the model-building process. Such networks can then serve as the foundation to build executable BN models in which edges are expressed with equations such as ordinary differential equations. The main applications of these broad classes of BN models are shown in light blue boxes. Artwork by Samantha J. Elmhurst (www.livingart.org.uk).
Hereafter, we highlight just a few applications of computational Systems Toxicology covering Computational Toxicology, biological network model building, biological impact quantification, and predictive modeling to exemplify some recent developments in these four areas.
5.1. Computational Toxicology
Computational Toxicology, recently reviewed by Kavlock and colleagues,60 is a growing area of research that integrates molecular and cell biology with chemistry and computational approaches. The objective is to increase the predictive power of toxicology by more efficiently and effectively ranking chemicals based on risk. With the advent of high-throughput experimental setups, relevant cell-based assay systems can be exposed to a large number of substances in a dose-dependent manner, and many phenotypic end points can be measured quantitatively by high-content (or visual) screening methods. Furthermore, cell-free enzymatic and ligand-binding assays, including G-protein-coupled and nuclear receptors, kinases, phosphatases, cytochrome P450s, histone deacetylases, ion channels, and transporters, can be used in a high-throughput screening mode to profile the mechanism(s) of action of chemicals. These approaches enable the screening of numerous substances for their impact on toxicity pathways. This will eventually lead to the creation of a comprehensive landscape of adverse cellular effects induced by biologically active substances. The integration of the biological end points with the chemical structures of the tested substances enables computational mapping (interpolating) of untested chemical analogues into the toxicity data, thereby providing a prediction of their relative toxicological hazard potential.
ToxCast is a major initiative in Computational Toxicology supported by the U.S. EPA. ToxCast aims to understand the potential health risks of a large number of diverse chemicals including failed pharmaceuticals, commodity chemicals, and pesticides by employing a large array of high-throughput screening methodologies. The first phase, or proof of concept, involved the use of almost 300 chemicals that had been well-characterized for their toxicological potential in animal models that were studied in more than 600 high-throughput screening assays.61 The results of this phase included a large number of expected perturbed biological pathways and the development of several predictive models based on a multifactorial data analysis.62 The second phase of the ToxCast initiative (http://www.epa.gov/ncct/toxcast/) is currently screening over 1000 such compounds against many of the same targets employed in phase I, and the data for this component has been released (see ToxCast website). A key element of the ToxCast program involves the interpretation of the in vivo relevance of in vitro activity concentrations through a process called reverse toxicokinetics.63 Combining this information with high-throughput exposure assessments, as introduced by Wambaugh and colleagues,11 provides for a risk-based process of prioritization of chemicals for more intensive study of their potential to impact human health.
5.2. Building Computable Biological Networks
By necessity, all descriptions of complex systems need to be simpler than the system itself. Indeed, BN models are represented as diagrams of nodes (entities) and edges (relationships). Initial models can be constructed using existing knowledge drawn from both pathway databases and literature-derived information (Figure 3). Although publicly accessible databases of manually curated biological networks and pathways, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG)64 and WikiPathways,65 are available, they generally provide a high-level static representation of our understanding of biological processes and offer only a limited representations of the adverse event mechanisms in the context of a particular exposure. Therefore, they need to be further supplemented with new nodes and edges derived from experiments adequately designed to link observed molecular changes with phenotypic changes in the context of a given exposure. To extract the key molecular entities and their relationships from these data sets, sophisticated reverse-engineering algorithms, such as GNS Healthcare’s reverse-engineer and forward simulation predictive framework66,67 or those reviewed by Lecca and Priami68 can be applied. Second, these high-level pathway maps have limited computational value for the analysis of Systems Toxicology data sets. Even though recent network informatics tools allow us to “paint” pathway maps according to the differential abundance of molecular entities (such as gene-expression data), they generally fall short of providing a computational infrastructure for causal reasoning and hence a more complete interpretation of high-throughput data sets. The transition from static to computable BN models is thus a necessary step to analyze experimental data fully and to build our collective knowledge of the toxicological effects of exposure to biologically active substances. This transition requires a formal language to describe the causal nature of the interactions between nodes to complement the gene ontology69 that already provides a coherent framework for the description of the nodes themselves. The recent development of the biological expression language (BEL), for instance, enables this transition because it allows the semantic representation of life-science relationships in a computable format70 and thus the development of computable BN models composed of cause-and-effect relationships. BEL was designed to capture biological cause-and-effect relationships with associated experimental context from disparate sources. BEL thereby facilitates the encoding of directional relationships within computable biological network models. The BEL framework is an open-source technology for managing, publishing, and using structured life-science knowledge (http://www.openbel.org/).70 This is unlike the Biological Pathway Exchange (BioPAX), which focuses on the integration and exchange of biological pathway data across a large array of existing pathway resources.71
Building causal computable BN models is a process that has been previously described in several publications and is briefly summarized below.72−76 These models consist of qualitative causal relationships to represent biological processes and are coded in BEL. The model-building process starts with the definition of its contents and boundaries by including all necessary mechanistic building blocks relevant to the BN under consideration. This step is guided by literature investigations of the signaling pathways relevant to the BN model under construction. These network boundaries are then used to screen the literature and to derive a collection of cause-and-effect relationship statements describing the relevant and known biology linked to the BN model in a given tissue context. From this, a literature model is constructed. The model consists of nodes, representing biological entities, such as mRNA expression or protein abundance, and edges, representing causal relationships between the nodes. During this model-building process, reverse causal reasoning (RCR)77 analysis of relevant gene-expression data sets is used to guide the selection of nodes for model building. RCR interrogates the experimental data against a knowledgebase of possible causal relationships for all of the context-specific nodes and edges in the literature model to identify upstream controllers of the mRNA state changes (gene-expression changes) observed in the data set. The potential upstream controllers identified by RCR are statistically significant potential explanations for the observed mRNA state changes. These potential upstream controllers are then included as new nodes in the BN model if they have literature support for a mechanistic role in the biological process and context of interest. This allows uncovering relevant nodes and edges that were not identified during the construction of the literature-based network. RCR-based augmentation of the network model can be performed using proprietary or publicly available gene-expression data sets (e.g., GEO or ArrayExpress). As a final step in the construction of the network model, the nodes and edges are manually reviewed and refined to include relevant context and pathways (e.g., by additional literature review), producing the final network model.
The review process should be conducted with each model periodically to ensure that newly available data sets and new literature are taken into account and thereby ensure that each model is kept up to date. Furthermore, BN models need to be verified to prove that they capture context-specific nodes and edges that are supported by experimental observations. This verification can be achieved on three levels. First, existing data sets not considered during model building can be used to verify specific areas of the model. This approach is, however, constrained by the availability of relevant publicly accessible data sets that fulfill the context-specific boundary criteria. Second, the structure of the model can lead to the design and conduct of new experiments designed for verification purposes, as exemplified by recent work on the computable cellular proliferation network model for normal lung cells.78 Finally, research communities can be engaged to curate network models79 as implemented in WikiPathways80 or through collaborative competition leveraging the wisdom of the crowd.81
By systematically investigating and describing the major mechanisms involved in toxicology, the scientific community will gradually build a compendium of computable BN models covering the crucial mechanisms involved in toxicity. Three layers of information and knowledge need to be addressed to tackle this daunting task. First, the molecular entities (i.e., the nodes in the BN models) will need to be further characterized, and their often diverse biological functions need to be captured in curated knowledge bases such as UniProt.82 Second, BN model construction will need to focus on the identification of new nodes and connections in a context-sensitive manner, where cell type and tissue environment are taken into account. Finally, identified mutations and their contributions to adverse manifestations need be the catalogued and mapped into the BN models in a consistent manner. This is essential to Systems Biology in general and Systems Toxicology in particular and will require the coordination of efforts79 that can easily dwarf the Human Genome Project.
5.3. Network-Based Biological Impact Assessment
The ability to predict the potential health risk of long-term exposure to biologically active substances is an ultimate goal in Systems Toxicology. Such risks are typically assessed a posteriori with epidemiological studies. However, disease or population-level responses might take decades to manifest, at which point changes in therapeutic regime, life style, or exposure would not prevent disease onset. It is therefore necessary to develop methods that are predictive and quantitative for risk assessment. It is well-understood that exposure to biologically active substances leads to modifications in the abundance of molecular entities within a living system, and, in some cases, modifications in the very nature of these entities, such as splice variants and post-translational modifications of proteins, lead to BN perturbations, which in turn can later lead to tissue-level changes and adverse events or disease. As described earlier, causal computable BN models are designed to describe these causal links between biological entities and thus provide a possible quantitative framework for biological impact assessment. Indeed, these models can be used to derive a data-driven quantitative assessment of the perturbation of a given BN. Recent toxicogenomics approaches83 have proven the potential of mechanism-based assessment methods by using a simple scoring method to evaluate the overall gene set-level expression changes to profile biologically active substances. Although the use of gene sets provides a first assessment based on gene lists derived from, for example, KEGG and other pathway databases, BN models will provide a more accurate and detailed analysis of the mechanistic effects of the exposure as well as a quantitative evaluation of the perturbation of each model under consideration and an overall pan-model biological impact. By way of example, we have published a five-step approach to derive such a biological impact factor (BIF)84 for a given exposure or stimulus. This approach employs causal computable BN models as the substrate to analyze high-throughput data sets derived from in vitro and in vivo dose–response and time-course experiments. Briefly, quality-controlled measurements generated in vitro or in vivo constitute systems response profiles (SRPs) for each given exposure in a given experimental system. These SRPs express the degree to which each individual molecular entity is changed as a consequence of the exposure of the system. These SRPs are then analyzed in the context of a collection of BN models designed to cover the molecular mechanisms perturbed by the exposure. This analysis is designed to answer questions about the completeness of the models (i.e., whether additional nodes and edges need to be added to the model to cover all of the exposure effects and to identify any new mechanisms not yet reported in the literature). This analysis employs RCR, by which downstream measurements (e.g., gene expression) are causally mapped as effects of an activity of individual elements in the model. Once this step is completed, we apply specific algorithms to derive the specific network perturbation amplitude (NPA) scores.85 Specifically, the NPA algorithm translates gene fold-changes (or other downstream measurements) into the perturbation of individual elements in the model and finally aggregates these perturbations into a BN-specific score.85,86 By providing a measure of biological network perturbation, NPA scores allow correlation of molecular events with phenotypes that characterize the network at the cell, tissue, or organ level. Finally, the overall biological impact87 of a perturbing agent can be estimated by aggregating the NPA scores from several BN models into one holistic value, the BIF, that expresses the overall impact on the entire biological system. This BIF can be readily correlated with other measurable end points (e.g., collected at a higher level of biological organization through phenotypic changes) or used to generate testable hypotheses that could provide new insight into mechanisms of disease onset or progression.84,88
5.4. From Biological Network Models to the Virtual Organism and Beyond
The BN models described above provide either a static or a computable environment to contextualize experimental data in the framework of current knowledge. Although these approaches enable a detailed understanding of molecular processes and the identification of new nodes and edges to supplement existing BN in a context-sensitive manner (e.g., particular cell type in a specific tissue environment), they do not exhibit the simulation capabilities necessary to understand the dynamic response of a molecular system to an exposure or more importantly to predict the effect of new exposures a priori. Key to this transition from descriptive to executable biology is the development of mathematical models that represent the dynamic properties of biological processes and their corresponding computational models that can be executed in silico. Therefore, the ultimate objective of computational Systems Toxicology is to develop dynamic (executable) mathematical models of these BNs in such a way that their simulation correctly represents the observed behavior of the biological process (Figure 3). Such models will eventually permit true predictions, such as the emergence of new network behaviors following in silico perturbations. The development of such models is currently still in its infancy and represents a major frontier in Systems Biology and Systems Toxicology. One effort in this area has been initiated by the U.S. EPA and aims to build a virtual embryo model89in silico to predict the potential for environmental chemicals to affect embryonic development. The predictive power of the model will be tested with a selection of well-characterized chemicals with known health effects in animal studies. Another example of predictive modeling is provided by the PhysioLab platform developed by Entelos (Foster City, CA, USA). Several large-scale dynamic mathematical models were developed and include both human and mouse models of diseases: (1) virtual NOD mice (Type 1 Diabetes PhysioLab),90 (2) virtual ApoE–/– mice (ApoE–/– Mouse PhysioLab),91 (3) virtual humans in both Rheumatoid Arthritis92 and (4) the Skin Sensitization PhysioLab.93 All of these PhysioLab platforms are large multiscale biological models where molecular and cellular interactions are generally represented by ordinary differential equations, the numerical solution of which simulates the dynamic response of the biological system across several layers of organization. Although most of these computational systems are primarily designed to plan and understand, for example, clinical studies, two of them are sample applications for Systems Toxicology. For instance, the ApoE–/– PhysioLab was designed to understand the toxicological effects of cigarette smoke and cessation on atherosclerotic plaque growth.91 The objective was to build a framework to translating experimental ApoE–/– mouse results to the human setting despite the uncertainty around the mechanisms underlying disease etiology, the relative importance of these mechanisms as drivers of progression, and how these roles change in response to lifestyle changes. This model may be used to optimize in vivo experiments and to pave the way for a similar modeling approach for human disease. The second example is the DILIsym modeling software, a multiscale representation of drug-induced liver injury (DILI)94,95 that is associated with SimPops (see http://dilisym.com/Products-Services/simpops.html), a collection of simulated individuals that match a particular range and distribution of parameters and patient characteristics, allowing the exploration of the interindividual variability in response to potential DILI-causing drugs. The third example is driven by recent changes in European Union legislation, prohibiting the use of in vivo experimental animal models for testing cosmetics (EU Cosmetics Regulation).5 As a consequence, nonanimal approaches to provide the data for skin sensitization risk assessment need to be further developed. In this context, Maxwell and MacKay93 have developed an in silico model of the induction of skin sensitization to characterize and quantify the contribution of each pathway to the overall biological process. The resulting Skin Sensitization PhysioLab is thus an important attempt to integrate data derived from different forms of nonanimal hazard data to inform new skin sensitization risk-assessment decisions and demonstrates how computational Systems Toxicology can contribute to hazard evaluation in a new framework for consumer safety risk assessment.93 Predictive model development has also been started in nonhumans, namely, fish, and focuses on the reproductive effects resulting from disturbance of the endocrine axis by environmental chemicals in a research program led by the U.S. EPA.96
The development of dynamic (executable) mathematical models of AOPs linking exposure to individual and population-wide adverse outcomes is the grand challenge of computational Systems Toxicology. Although new computational methods and mathematical tools are certainly needed, the major challenge in building such models is our limited understanding of how BN perturbations are linked to the KEs in the AOPs at a higher level of biological organization. Although this obviously requires a deep understanding of the dynamic behavior of the causal link between BN perturbations and KEs, it also requires a far deeper understanding of the causal link between these KEs and the adverse outcome for the individual and how these translate to the population. Although the dynamic models described above are a step toward understanding the links between BN perturbations and individual adverse outcomes, the last step could be achieved by creating large sets of mechanistically distinct virtual humans that, upon simulation, statistically match the prevalence of phenotypic variability reported in human studies. The recently reported algorithm termed mechanistic axes population ensemble linkage (MAPEL),97 which utilizes a mechanistically based weighting method to match clinical trial statistics, may be an approach enabling such population-based risk assessment and expands on the concept of SimPops. Similar model can be developed for other organisms, such as fish or plants. Such dynamic models will eventually allow for simulating the behavior of biological systems at both the individual and population levels to generate new hypotheses and insights into the potential effect of an exposure a priori. Their validation will eventually provide a computational foundation for risk assessment.
6. Systems Toxicology and Risk Assessment
The needs of risk assessment are context-dependent and can vary from simple classification of a substance for hazard (e.g., is it genotoxic or not) to prioritization by the nature and severity of hazard for further investigation to quantitative estimates of risk to determine the urgency and nature of any risk-management action. Although existing approaches have served society well, there is increasing concern, some real and some hypothetical, that there may be important weaknesses in the assumptions used in current risk assessment. These include the shape of the dose–response relationship under human-relevant exposures, whether biological thresholds exist and for what end points, the extent of population variability in toxicological response, and the influence of factors such as life stage on response. Despite a substantial amount of research, some in large groups of animals, it has not been possible to resolve these concerns. This shortcoming is due to the intrinsic limitation of all experimental observations by the power of the study. Hence, robust resolution of these questions will require a mechanistic approach. As discussed above, Systems Toxicology has real potential to provide the data as well as the quantitative mechanistic models to address these issues. In this sense, Systems Toxicology can provide a deep mechanistic understanding of toxicological effects, permitting prediction of responses to chemicals. If adequately described, a systems description should enable prediction of responses for which experimental data were not available (i.e., the system will exhibit emergent properties entailing novel patterns and properties arising from the inherent structure of the system).
The development of such models is a major and complex undertaking. Hence, it will be some time before they can be used in risk assessment. Furthermore, risk assessors are not often well-trained in relevant mathematical procedures and hence there is inherent reluctance to adopt computationally intensive approaches with which they are poorly familiar. Thus, a phased transition will be necessary. One way in which this might be achieved is by adapting the more familiar mode of action/adverse outcome pathway paradigm. A MOA/AOP comprises a series of quantifiable key events that are necessary but individually usually not sufficient for a toxicological response to a chemical. It should be possible to use a modular approach to the development of Systems Toxicology, where one starts with a systems-based model of a single key event and incorporates that into a quantitative biological model with dose–response data for the other key events at an operational level. In this way, one could progressively add systems-based modules describing each key event until a full systems-based description of the response is achieved. This approach has the major advantage that each stage is readily verifiable experimentally. This will also allow incorporation of Systems Toxicologically in a progressive fashion into risk assessment, while providing risk assessors the opportunity to become familiar and comfortable with evaluating such data.
Major efforts are underway to develop nonanimal test methods, such as the high-throughput ToxCast program discussed earlier. However, in themselves, these platforms are unlikely to meet the needs of risk assessment beyond hazard identification. Taking a further step will require additional quantitative approaches, for which Systems Toxicology would be ideally suited. However, development of suitable models will be a complex and time-consuming undertaking. Mode of action can provide a translational bridge, enabling the incorporation of information from in vitro and other approaches into a systems-based description of key events, thereby enabling the stepwise development of a full Systems Toxicology characterization of the organ and eventually the organism
7. Outlook
We stand at the edge of an unprecedented transformation in the conduct of toxicological evaluations. A central tenant of the new toxicology involves applying modern molecular analysis techniques to elucidate mechanisms of toxicity and is being enabled by several factors. The first is the increasing power and availability of molecular measurement tools able to probe the functioning of biological networks inside organisms, organs, tissues, and cells. The second is the increasing affordability of high-throughput and high-content characterization approaches that can be applied to thousands of chemicals in short time periods rather than the chemical-by-chemical approach of the past 4 decades that involves thousands of animals and perpetual high costs and years of duration. The third enabler is the increasing computational power, data-storage capacity, and information-management tools now available to the scientific community that has facilitated the ability to employ complicated Systems Biology models. The fourth enabler is the acceleration in the development of adequate in vitro test systems to complement and gradually replace animal models.3 The fifth enabler is that of significant resource investment by governments throughout the world in funding efforts to develop the scientific foundation of Systems Toxicology. For example, DARPA (the Defense Advanced Research Projects Agency, USA) recently invested $70 million to develop up to 10 “organs on chips” within the next 5 years. Likewise, the EU has invested several hundred million Euros in FP7 (see http://www.axlr8.eu/) for various supporting projects such as SEURAT-1 (Safety Assessment Ultimately Replacing Animal Testing, see http://www.seurat-1.eu), with prospects to continue the investment in the European funding framework, Horizon 2020. Ultimately, these international efforts need to be combined to produce a real shift in the risk-assessment paradigm that not only assures the highest levels of protection of public health and the environment but also enables economic growth and global trade. Finally, a key requirement in addressing the challenge is that contributors and stakeholders, whether from government, academia, industry, NGOs, regulatory bodies, or the public at large, reach a consensus that new approaches based on Systems Toxicology reach the standards they demand.
Acknowledgments
We thank all of the other speakers at the Systems Toxicology 2013 symposium (www.systox2013.ch): Ila Cote (EPA and University of Colorado, USA), Kevin Crofton (National Center for Computational Toxicology, EPA, USA), Joost van Delft (University of Maastricht, The Netherlands), Thomas Hartung (Johns Hopkins Bloomberg School of Public Health, USA), Hennicke Kamp (BASF Metanomics Health, Germany), Marcel Leist (University of Konstanz, Germany), Thomas Paterson (Entelos, USA), François Pognan (Novartis Institutes for BioMedical Research, Switzerland), Stephen Rappaport (University of California, Berkeley, USA), Nigel Skinner (Agilent Technologies, UK), Gustavo Stolovitzky (IBM Research, USA), Robert Turesky (University of Minnesota, Twin Cities, USA), and Ioannis Xenarios (Swiss Institute of Bioinformatics, Switzerland) as well as the symposium participants for insightful discussions.
Glossary
Abbreviations
- AOP
adverse outcome pathway
- ApoE–/–
apolipoprotein E double-KO mouse
- BEL
biological expression language
- BIF
biological impact factor
- BioPAX
biological pathway exchange
- BN
biological network
- DILI
drug-induced liver injury
- DNEL
derived no effect level
- EDC
endocrine-disrupting chemical
- ELISA
enzyme-linked immunosorbent assay
- EWAS
environment-wide association study
- GEO
Gene Expression Omnibus
- HCS
high-content screening
- iTRAQ
isotope tagging for relative and absolute quantification
- KE
key event
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LOAEL
lowest observed adverse effect level
- MAPEL
mechanistic axes population ensemble linkage
- MIAME
minimum information about microarray experiment
- MIAPE
minimum information about a proteomics experiment
- MS
mass spectrometry
- NCI caBIG
National Cancer Institute Biomedical Informatics Grid
- NGS
next-generation sequencing
- NOAEL
no observed adverse effect level
- NPA
network perturbation amplitude
- OECD
Organisation for Economic Co-operation and Development
- PCR
polymerase chain reaction
- RCR
reverse causal reasoning
- REACH
registration, evaluation, authorization, and restriction of chemicals
- RPA
reverse protein array
- SRM
selected reaction monitoring
- SRP
systems response profile
- TSCA
U.S. Toxic Substances Control Act
The authors are funded by their respective institutions. S. Sturla acknowledges support by the Swiss National Science Foundation (grant no. 136247).
The views expressed in this article are those of the author(s) and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Any mention of trade names, products, or services does not imply an endorsement by the U.S. Government or the United States Environmental Protection Agency. The EPA does not endorse any commercial products, services, or enterprises.
The authors declare no competing financial interest.
References
- Waters M. D.; Fostel J. M. (2004) Toxicogenomics and systems toxicology: Aims and prospects. Nat. Rev. Genet. 5, 936–948. [DOI] [PubMed] [Google Scholar]
- Hartung T.; van Vliet E.; Jaworska J.; Bonilla L.; Skinner N.; Thomas R. (2012) Food for thought ... systems toxicology. ALTEX 29, 119–128. [DOI] [PubMed] [Google Scholar]
- Basketter D. A.; Clewell H.; Kimber I.; Rossi A.; Blaauboer B.; Burrier R.; Daneshian M.; Eskes C.; Goldberg A.; Hasiwa N.; Hoffmann S.; Jaworska J.; Knudsen T. B.; Landsiedel R.; Leist M.; Locke P.; Maxwell G.; McKim J.; McVey E. A.; Ouédraogo G.; Patlewicz G.; Pelkonen O.; Roggen E.; Rovida C.; Ruhdel I.; Schwarz M.; Schepky A.; Schoeters G.; Skinner N.; Trentz K.; Turner M.; Vanparys P.; Yager J.; Zurlo J.; Hartung T. (2012) A roadmap for the development of alternative (non-animal) methods for systemic toxicity testing. ALTEX 29, 3–91. [DOI] [PubMed] [Google Scholar]
- Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes, Official Journal of the European Union, L 276, pp 33–78.
- Regulation (EC) no 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products, Official Journal of the European Union, L 342, 22.12.2009, pp 59–209.
- Regulation (EC) no 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the registration, evaluation, authorisation and restriction of chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) no 793/93 and Commission Regulation (EC) no 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC, Official Journal of the European Union, L 136, pp 3–277.
- Regulation (EC) no 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC, Official Journal of the European Union, L 309, pp 1–50.
- Regulation (EC) no 528/2012 of the European Parliament and of the Council of 22 May 2012 concerning the making available on the market and use of biocidal products, Official Journal of the European Union, L 167, pp 1–123.
- Toxicity and assessment of chemical mixtures, Scientific Committee on Health and Environmental Risks (SCHER), Scientific Committee on Emerging and Newly Identified Health Risks (SCENHIR), and Scientific Committee on Consumer Safety (SCCS), joint opinion adopted on 14th December 2011, http://ec.europa.eu/health/scientific_committees/environmental_risks/opinions/index_en.htm.
- Bai J. P. F.; Abernethy D. R. (2013) Systems pharmacology to predict drug toxicity: Integration across levels of biological organization. Annu. Rev. Pharmacol. Toxicol. 53, 451–473. [DOI] [PubMed] [Google Scholar]
- Wambaugh J. F.; Setzer R. W.; Reif D. M.; Gangwal S.; Mitchell-Blackwood J.; Arnot J. A.; Joliet O.; Frame A.; Rabinowitz J.; Knudsen T. B.; Judson R. S.; Egeghy P.; Vallero D.; Cohen Hubal E. A. (2013) High-throughput models for exposure-based chemical prioritization in the ExpoCast project. Environ. Sci. Technol. 47, 8479–8488. [DOI] [PubMed] [Google Scholar]
- Ankley G. T.; Bennett R. S.; Erickson R. J.; Hoff D. J.; Hornung M. W.; Johnson R. D.; Mount D. R.; Nichols J. W.; Russom C. L.; Schmieder P. K.; Serrrano J. A.; Tietge J. E.; Villeneuve D. L. (2010) Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Guidance document on developing and assessing adverse outcome pathways, series on testing and assessment, Organisation for Economic Co-operation and Development (OECD), no. 184n ENV/JM/MONO(2013)6.
- Systems Toxicology 2013 – From Basic Research to Human Risk Assessment, Ascona, Switzerland, April 28–May 1, 2013. http://www.systox2013.ch/.
- Ezzati M., Hoorn S. V., Lopez A. D., Danaei G., Rodgers A., Mathers C. D., and Murray C. J. L. (2006) Comparative quantification of mortality and burden of disease attributable to selected risk factors, in Global Burden of Disease and Risk Factors (Lopez A. D., Mathers C. D., Ezzati M., Jamison D. T., and Murray C. J. L., Eds.) Chapter 4, World Bank, Washington, DC. [PubMed] [Google Scholar]
- Rappaport S. M. (2012) Discovering environmental causes of disease. J. Epidemiol. Community Health 66, 99–102. [DOI] [PubMed] [Google Scholar]
- Manolio T. A.; Collins F. S.; Cox N. J.; Goldstein D. B.; Hindorff L. A.; Hunter D. J.; McCarthy M. I.; Ramos E. M.; Cardon L. R.; Chakravarti A.; Cho J. H.; Guttmacher A. E.; Kong A.; Kruglyak L.; Mardis E.; Rotimi C. N.; Slatkin M.; Valle D.; Whittemore A. S.; Boehnke M.; Clark A. G.; Eichler E. E.; Gibson G.; Haines J. L.; Mackay T. F.; McCarroll S. A.; Visscher P. M. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild C. P. (2005) Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol., Biomarkers Prev. 14, 1847–1850. [DOI] [PubMed] [Google Scholar]
- Wild C. P. (2012) The exposome: From concept to utility. Int. J. Epidemiol. 41, 24–32. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Hermens J. L. M. (2004) Internal exposure: Linking bioavailability to effects. Environ. Sci. Technol. 38, 455A–462A. [DOI] [PubMed] [Google Scholar]
- Patel C. J.; Cullen M. R.; Ioannidis J. P.; Butte A. J. (2012) Systematic evaluation of environmental factors: Persistent pollutants and nutrients correlated with serum lipid levels. Int. J. Epidemiol. 41, 828–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind P. M.; Risérus U.; Salihovic S.; Bavel Bv.; Lind L. (2013) An environmental wide association study (EWAS) approach to the metabolic syndrome. Environ. Int. 55, 1–8. [DOI] [PubMed] [Google Scholar]
- Rappaport S. M.; Smith M. T. (2010) Epidemiology. Environment and disease risks. Science 330, 460–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport S. M. (2012) Biomarkers intersect with the exposome. Biomarkers 17, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1993) WHO International Programme on Chemical Safety, Environmental Health Criteria 155, Biomarkers and risk assessment: Concepts and principles, World Health Organization, Geneva, Switzerland.
- Ritchie S. A.; Tonita J.; Alvi R.; Lehotay D.; Elshoni H.; Myat S.; McHattie J.; Goodenowe D. B. (2013) Low-serum GTA-446 anti-inflammatory fatty acid levels as a new risk factor for colon cancer. Int. J. Cancer 132, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. M.; Johnson A.; Tarapore P.; Janakiram V.; Zhang X.; Leung Y. K. (2012) Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 53, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talikka M.; Sierro N.; Ivanov N. V.; Chaudhary N.; Peck M. J.; Hoeng J.; Coggins C. R.; Peitsch M. C. (2012) Genomic impact of cigarette smoke, with application to three smoking-related diseases. Crit. Rev. Toxicol. 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribny I. P.; Rusyn I. (2013) Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 754, 215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeng J., Talikka M., Martin F., Ansari S., Drubin D., Elamin A., Gebel S., Ivanov N. V., Kenny R. D., Koegel U., Schlage W. K., Sewer A., Sierro N., Thomson T., and Peitsch M. C. (2014) Toxicopanomics: applications of genomics, transcriptomics, proteomics and lipidomics in predictive mechanistic toxicology, in Principle and Methods on Toxicology (Hayes A. W., Ed.) CRC Press, Boca Raton, FL, in press. [Google Scholar]
- Dopazo J. (2013) Genomics and transcriptomics in drug discovery. Drug Discovery Today [Online early access], DOI: 10.1016/j.drudis.2013.06.003, Published Online: June 15. [DOI] [PubMed] [Google Scholar]
- Schirmer K.; Fischer B. B.; Madureira D. J.; Pillai S. (2010) Transcriptomics in ecotoxicology. Anal. Bioanal. Chem. 397, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M. K., Chi A., Melas I. N., and Alexopoulos L. G. (2013) Phosphoproteomics in drug discovery. Drug Discovery Today [Online early access], DOI: 10.1016/j.drudis.2013.10.010, Published Online: Oct 18. [DOI] [PubMed] [Google Scholar]
- Bausch-Fluck D.; Hofmann A.; Wollscheid B. (2012) Cell surface capturing technologies for the surfaceome discovery of hepatocytes. Methods Mol. Biol. 909, 1–16. [DOI] [PubMed] [Google Scholar]
- Fiehn O. (2001) Combining genomics, metabolome analysis, and biochemical modeling to understand metabolic networks. Comp. Funct. Genomics 2, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. P. (2013) Metabolomics: Integration of a new “omics” with clinical pharmacology. Clin. Pharmacol. Ther. 94, 547–551. [DOI] [PubMed] [Google Scholar]
- Gomez-Casati D. F.; Zanor M. I.; Busi M. V. (2013) Metabolomics in plants and humans: Applications in the prevention and diagnosis of diseases. Biomed. Res. Int. 2013, 792527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan L. (2013) Metabolomics in nutrition research: Current status and perspectives. Biomed. Soc. Trans. 41, 670–673. [DOI] [PubMed] [Google Scholar]
- Robertson D. G. (2005) Metabonomics in toxicology: A review. Toxicol. Sci. 85, 809–822. [DOI] [PubMed] [Google Scholar]
- Bouhifd M.; Hartung T.; Hogberg H. T.; Kleensang A.; Zhao L. (2013) Review: Toxicometabolomics. J. Appl. Toxicol. 33, 1365–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez T.; Daneshian M.; Kamp H.; Bois F. Y.; Clench M. R.; Coen M.; Donley B.; Fischer S. M.; Ekman D. R.; Fabian E.; Guillou C.; Heuer J.; Hogberg H. T.; Jungnickel H.; Keun H. C.; Krennrich G.; Krupp E.; Luch A.; Noor F.; Peter E.; Riefke B.; Seymour M.; Skinner N.; Smirnova L.; Verheij E.; Wagner S.; Hartung T.; van Ravenzwaay B.; Leist M. (2013) Metabolomics in toxicology and preclinical research. ALTEX 30, 209–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athersuch T. J. (2012) The role of metabolomics in characterizing the human exposome. Bioanalysis 4, 2207–2212. [DOI] [PubMed] [Google Scholar]
- Rappaport S. M.; Li H.; Grigoryan H.; Funk W. E.; Williams E. R. (2012) Adductomics: Characterizing exposures to reactive electrophiles. Toxicol. Lett. 213, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D. I.; Atherton H. J.; Goodacre R.; Griffin J. L. (2011) Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 40, 387–426. [DOI] [PubMed] [Google Scholar]
- Jones O. A.; Swain S. C.; Svendsen C.; Griffin J. L.; Sturzenbaum S. R.; Spurgeon D. J. (2012) Potential new method of mixture effects testing using metabolomics and Caenorhabditis elegans. J. Proteome Res. 11, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Shintu L.; Baudoin R.; Navratil V.; Prot J. M.; Pontoizeau C.; Defernez M.; Blaise B. J.; Domange C.; Péry A. R.; Toulhoat P.; Legallais C.; Brochot C.; Leclerc E.; Dumas M. E. (2012) Metabolomics-on-a-chip and predictive systems toxicology in microfluidic bioartificial organs. Anal. Chem. 84, 1840–1848. [DOI] [PubMed] [Google Scholar]
- Wenk M. R. (2010) Lipidomics: New tools and applications. Cell 143, 888–895. [DOI] [PubMed] [Google Scholar]
- Vihervaara T., Suoniemi M., and Laaksonen R. (2013) Lipidomics in drug discovery. Drug Discovery Today[Online early access], DOI: 10.1016/j.drudis.2013.09.008, Published Online: Sept 17. [DOI] [PubMed] [Google Scholar]
- Jung H. R.; Sylvanne T.; Koistinen K. M.; Tarasov K.; Kauhanen D.; Ekroos K. (2011) High throughput quantitative molecular lipidomics. Biochim. Biophys. Acta 1811, 925–934. [DOI] [PubMed] [Google Scholar]
- De Léon H.; Boué S.; Peitsch M. C.; Hoeng J. (2013) Modulation of the hepatic lipidome and transcriptome of Apoe–/– mice in response to smoking cessation. J. Liver 2, 1000132-1–1000132-11. [Google Scholar]
- Lu S.; Jessen B.; Strock C.; Will Y. (2012) The contribution of physicochemical properties to multiple in vitro cytotoxicity endpoints. Toxicol. In Vitro 26, 613–620. [DOI] [PubMed] [Google Scholar]
- Dondrup M.; Albaum S. P.; Griebel T.; Henckel K.; Jünemann S.; Kahlke T.; Kleindt C. K.; Küster H.; Linke B.; Mertens D.; Mittard-Runte V.; Neuweger H.; Runte K. J.; Tauch A.; Tille F.; Pühler A.; Goesmann A. (2009) EMMA 2 – a MAGE-compliant system for the collaborative analysis and integration of microarray data. BMC Bioinf. 10, 50-1–50-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattiker A.; Hermida L.; Liechti R.; Xenarios I.; Collin O.; Rougemont J.; Primig M. (2009) MIMAS 3.0 is a multiomics information management and annotation system. BMC Bioinf. 10, 151-1–151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheel C., Nass S., and Omenn G. (2012) Evolution of translational omics: Lessons learned and the path forward, Institute of Medicine Consensus Report, National Academies Press, Washington, DC. [PubMed]
- Brazma A.; Hingamp P.; Quackenbush J.; Sherlock G.; Spellman P.; Stoeckert C.; Aach J.; Ansorge W.; Ball C. A.; Causton H. C.; Gaasterland T.; Glenisson P.; Holstege F. C.; Kim I. F.; Markowitz V.; Matese J. C.; Parkinson H.; Robinson A.; Sarkans U.; Schulze-Kremer S.; Stewart J.; Taylor R.; Vilo J.; Vingron M. (2001) Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 29, 365–371. [DOI] [PubMed] [Google Scholar]
- Rayner T. F.; Rocca-Serra P.; Spellman P. T.; Causton H. C.; Farne A.; Holloway E.; Irizarry R. A.; Liu J.; Maier D. S.; Miller M.; Petersen K.; Quackenbush J.; Sherlock G.; Stoeckert C. J. Jr.; White J.; Whetzel P. L.; Wymore F.; Parkinson H.; Sarkans U.; Ball C. A.; Brazma A. (2006) A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB. BMC Bioinf. 7, 489-1–489-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. F.; Paton N. W.; Lilley K. S.; Binz P. A.; Julian R. K. Jr.; Jones A. R.; Zhu W.; Apweiler R.; Aebersold R.; Deutsch E. W.; Dunn M. J.; Heck A. J.; Leitner A.; Macht M.; Mann M.; Martens L.; Neubert T. A.; Patterson S. D.; Ping P.; Seymour S. L.; Souda P.; Tsugita A.; Vandekerckhove J.; Vondriska T. M.; Whitelegge J. P.; Wilkins M. R.; Xenarios I.; Yates J. R. 3rd; Hermjakob H. (2007) The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 25, 887–893. [DOI] [PubMed] [Google Scholar]
- Goecks J.; Nekrutenko A.; Taylor J. (2010) Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86-1–R86-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull D.; Wolstencroft K.; Stevens R.; Goble C.; Pocock M. R.; Li P.; Oinn T. (2006) Taverna: A tool for building and running workflows of services. Nucleic Acids Res. 34, W729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz J.; Oster S.; Hastings S.; Langella S.; Kurc T.; Sanchez W.; Kher M.; Manisundaram A.; Shanbhag K.; Covitz P. (2006) caGrid: Design and implementation of the core architecture of the cancer biomedical informatics grid. Bioinformatics 22, 1910–1916. [DOI] [PubMed] [Google Scholar]
- Kavlock R. J.; Ankley G.; Blancato J.; Breen M.; Conolly R.; Dix D.; Houck K.; Hubal E.; Judson R.; Rabinowitz J.; Richard A.; Setzer R. W.; Shah I.; Villeneuve D.; Weber E. (2007) Computational toxicology–a state of the science mini review. Toxicol. Sci. 103, 14–27. [DOI] [PubMed] [Google Scholar]
- Dix D. J.; Houck K. A.; Martin M. T.; Richard A. M.; Setzer R. W.; Kavlock R. J. (2007) The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 95, 5–12. [DOI] [PubMed] [Google Scholar]
- Kavlock R.; Chandler K.; Houck K.; Hunter S.; Judson R.; Kleinstreuer N.; Knudsen T.; Martin M.; Padilla S.; Reif D.; Richard A.; Rotroff D.; Sipes N.; Dix D. (2012) Update on EPA’s ToxCast program: Providing high throughput decision support tools for chemical risk management. Chem. Res. Toxicol. 25, 1287–1302. [DOI] [PubMed] [Google Scholar]
- Wetmore B. A.; Wambaugh J. F.; Ferguson S. S.; Sochaski M. A.; Rotroff D. M.; Freeman K.; Clewell H. J. 3rd.; Dix D. J.; Andersen M. E.; Houck K. A.; Allen B.; Judson R. S.; Singh R.; Kavlock R. J.; Richard A. M.; Thomas R. S. (2012) Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci. 125, 157–174. [DOI] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S.; Sato Y.; Furumichi M.; Tanabe M. (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico A. R.; Kelder T.; van Iersel M. P.; Hanspers K.; Conklin B. R.; Evelo C. (2008) WikiPathways: Pathway editing for the people. PLoS Biol. 6, e184-1–e184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H.; McDonagh P. D.; Bienkowska J.; Cashorali T.; Runge K.; Miller R. E.; Decaprio D.; Church B.; Roubenoff R.; Khalil I. G.; Carulli J. (2011) Causal modeling using network ensemble simulations of genetic and gene expression data predicts genes involved in rheumatoid arthritis. PLoS Comput. Biol. 7, e1001105-1–e1001105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Kogel U., Gebel S., Peck M. J., Peitsch M. C., Akmaev V. R., Hayete B., Parikh J., Young L., Caprice J., Hoeng J., and Khalil I. (2012) Discovery of emphysema/COPD-relevant molecular networks from an A/J mouse COPD inhalation study by reverse engineering and forward simulation (REFSTM). German Conference on Bioinformatics, Jena, Germany, September 19–22.
- Lecca P.; Priami C. (2013) Biological network inference for drug discovery. Drug Discovery Today 18, 256–264. [DOI] [PubMed] [Google Scholar]
- (2013) Gene ontology annotations and resources. Nucleic Acids Res. 41, D530–D535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater T.; Song D. H. (2012) Saved by the BEL: Ringing in a common language for the life sciences. Drug Discovery World 75–80. [Google Scholar]
- Demir E.; Cary M. P.; Paley S.; Fukuda K.; Lemer C.; Vastrik I.; Wu G.; D’Eustachio P.; Schaefer C.; Luciano J.; Schacherer F.; Martinez-Flores I.; Hu Z.; Jimenez-Jacinto V.; Joshi-Tope G.; Kandasamy K.; Lopez-Fuentes A. C.; Mi H.; Pichler E.; Rodchenkov I.; Splendiani A.; Tkachev S.; Zucker J.; Gopinath G.; Rajasimha H.; Ramakrishnan R.; Shah I.; Syed M.; Anwar N.; Babur O.; Blinov M.; Brauner E.; Corwin D.; Donaldson S.; Gibbons F.; Goldberg R.; Hornbeck P.; Luna A.; Murray-Rust P.; Neumann E.; Ruebenacker O.; Samwald M.; van Iersel M.; Wimalaratne S.; Allen K.; Braun B.; Whirl-Carrillo M.; Cheung K. H.; Dahlquist K.; Finney A.; Gillespie M.; Glass E.; Gong L.; Haw R.; Honig M.; Hubaut O.; Kane D.; Krupa S.; Kutmon M.; Leonard J.; Marks D.; Merberg D.; Petri V.; Pico A.; Ravenscroft D.; Ren L.; Shah N.; Sunshine M.; Tang R.; Whaley R.; Letovksy S.; Buetow K. H.; Rzhetsky A.; Schachter V.; Sobral B. S.; Dogrusoz U.; McWeeney S.; Aladjem M.; Birney E.; Collado-Vides J.; Goto S.; Hucka M.; Le Novère N.; Maltsev N.; Pandey A.; Thomas P.; Wingender E.; Karp P. D.; Sander C.; Bader G. D. (2010) The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 28, 935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra J. W.; Schlage W. K.; Frushour B. P.; Gebel S.; Catlett N. L.; Han W.; Eddy S. F.; Hengstermann A.; Matthews A. L.; Mathis C.; Lichtner R. B.; Poussin C.; Talikka M.; Veljkovic E.; Van Hoose A. A.; Wong B.; Maria M. J.; Peitsch M. C.; Keeney R. D.; Hoeng J. (2011) Construction of a computable cell proliferation network focused on non-diseased lung cells. BMC Syst. Biol. 5, 105-1–105-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlage W. K.; Westra J. W.; Gebel S.; Catlett N. L.; Mathis C.; Frushour B. P.; Hengstermann A.; Van Hooser A.; Poussin C.; Wong B.; Lietz M.; Park J.; Drubin D.; Veljkovic E.; Peitsch M. C.; Hoeng J.; Deehan R. (2011) A computable cellular stress network model for non-diseased pulmonary and cardiovascular tissue. BMC Syst. Biol. 5, 168-1–168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebel S.; Lichtner R.; Frushour B.; Schlage W.; Hoang V.; Talikka M.; Hengstermann A.; Mathis C.; Veljkovic E.; Müller T.; Peck M.; Peitsch M. C.; Deehan R.; Hoeng J.; Westra J. (2013) Construction of a Computable Network Model for DNA Damage, Autophagy, Cell Death, and Senescence. Bioinf. Biol. Insights 7, 97–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeng J.; Park J. S.; Schlage W. K.; Frushour B. P.; Talikka M.; Toedter G.; Gebel S.; Deehan R.; Veljkovic E.; Westra J. W.; Peck M. J.; Boué S.; Kogel U.; Gonzalez-Suarez I.; Hengstermann A.; Peitsch M. C. (2013) Construction of a computable network model of tissue repair and angiogenesis in the lung. J. Clin. Toxicol. S12-002-1–S12-002-14. [Google Scholar]
- Westra J. W.; Schlage W. K.; Hengstermann A.; Gebel S.; Mathis C.; Thomson T.; Wong B.; Hoang V.; Veljkovic E.; Peck M.; Lichtner R. B.; Weisensee D.; Talikka M.; Deehan R.; Hoeng J.; Peitsch M. C. (2013) A modular cell-type focused inflammatory process network model for non-diseased pulmonary tissue. Bioinf. Biol. Insights 7, 167–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett N. L.; Bargnesi A. J.; Ungerer S.; Seagaran T.; Ladd W.; Elliston K. O.; Pratt D. (2013) Reverse causal reasoning: Applying qualitative causal knowledge to the interpretation of high-throughput data. BMC Bioinf. 14, 340-1–340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcastro V.; Poussin C.; Gebel S.; Mathis C.; Schlage W.; Lichtner R.; Quadt-Humme S.; Wagner S.; Hoeng J.; Peitsch M. C. (2013) Systematic verification of upstream regulators of a computable cellular proliferation network model on non-diseased lung cells using a dedicated dataset. Bioinf. Biol. Insights 7, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H.; Ghosh S.; Matsuoka Y. (2011) Social engineering for virtual ‘big science’ in systems biology. Nat. Chem. Biol. 7, 323–326. [DOI] [PubMed] [Google Scholar]
- Kelder T.; van Iersel M. P.; Hanspers K.; Kutmon M.; Conklin B. R.; Evelo C. T.; Pico A. R. (2012) WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 40, D1301–D1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The sbv IMPROVER project team (in alphabetical order)Ansari S.; Di Fabio A.; Binder J.; Boué S.; Hayes W.; Hoeng J.; Iskandar A.; Kleiman R.; Norel R.; O’Neel B.; Peitsch M. C.; Poussin C.; Pratt D.; Rhrissorrakai K.; Schlage W. K.; Stolovitzky G.; Talikka M. (2013) On crowd-verification of biological networks. Bioinform. Biol. Insight 7, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41, D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyosawa N.; Manabe S.; Yamoto T.; Sanbuissho A. (2010) Practical application of toxicogenomics for profiling toxicant-induced biological perturbations. Int. J. Mol. Sci. 11, 3397–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeng J.; Kenney R. D.; Pratt D.; Martin F.; Sewer A.; Thomson T. M.; Drubin D. A.; Waters C. A.; de Graaf D.; Peitsch M. C. (2012) A network-based approach to quantify the impact of biologically active substances. Drug Discovery Today 17, 413–418. [DOI] [PubMed] [Google Scholar]
- Martin F.; Thomson T. M.; Sewer A.; Drubin D. A.; Mathis C.; Weisensee D.; Pratt D.; Hoeng J.; Peitsch M. C. (2012) Assessment of network perturbation amplitude by applying high-throughput data to causal biological networks. BMC Syst. Biol. 6, 54-1–54-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskandar A. R.; Martin F.; Talikka M.; Schlage W. K.; Kostadinova R.; Mathis C.; Hoeng J.; Peitsch M. C. (2013) Systems approaches evaluating the perturbation of xenobiotic metabolism in response to cigarette smoke exposure in nasal and bronchial tissues. BioMed. Res. Int. 512086-1–512086-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson T. M.; Sewer A.; Martin F.; Belcastro V.; Frushour B.; Gebel S.; Park J.; Schlage W. K.; Talikka M.; Vasilyev D.; Westra J. W.; Deehan R.; Hoeng J.; Peitsch M. C. (2013) Quantitative assessment of biological impact using transcriptomic data and mechanistic network models. Toxicol. Appl. Pharmacol. 272, 863–878. [DOI] [PubMed] [Google Scholar]
- Hoeng J., Talikka M., Martin F., Sewer A., Yang X., Iskandar A., Schlage W., and Peitsch M. C. (2013) Case study: The role of mechanistic network models in systems toxicology. Drug Discovery Today [Online early access], DOI: 10.1016/j.drudis.2013.07.023, Published Online: Aug 9. [DOI] [PubMed] [Google Scholar]
- Knudsen T. B.; Kleinstreuerm N. C. (2011) Disruption of embryonic vascular development in predictive toxicology. Birth Defects Res., Part C 93, 312–323. [DOI] [PubMed] [Google Scholar]
- Shoda L.; Kreuwel H.; Gadkar K.; Zheng Y.; Whiting C.; Atkinson M.; Bluestone J.; Mathis D.; Young D.; Ramanujan S. (2010) The type 1 diabetes PhysioLab platform: A validated physiologically based mathematical model of pathogenesis in the non-obese diabetic mouse. Clin. Exp. Immunol. 161, 250–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. R.; Vuillaume G.; Bever C.; Lebrun S.; Lietz M.; Steffen Y.; Stolle K.; Wahba K.; Wang X.; Moodie S.; Hoeng J.; Peitsch M. C.; Powell L. M. (2012) The Apoe–/– mouse PhysioLab® platform: A validated physiologically-based mathematical model of atherosclerotic plaque progression in the Apoe–/– mouse. BioDiscovery 3, 2-1–2-13. [Google Scholar]
- Rullmann J. A.; Struemper H.; Defranoux N. A.; Ramanujan S.; Meeuwisse C. M.; van Elsas A. (2005) Systems biology for battling rheumatoid arthritis: application of the Entelos PhysioLab platform. Syst. Biol. 152, 256–262. [DOI] [PubMed] [Google Scholar]
- Maxwell G.; Mackay C. (2008) Application of a systems biology approach to skin allergy risk assessment. Altern. Lab. Anim. 36, 521–556. [DOI] [PubMed] [Google Scholar]
- Howell B. A.; Yang Y.; Kumar R.; Woodhead J. L.; Harrill A. H.; Clewell H. J. 3rd.; Andersen M. E.; Siler S. Q.; Watkins P. B. (2012) In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym: a mechanistic, mathematical model of DILI. J. Pharmacokinet. Pharmacodyn. 39, 527–541. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Shoda L. K.; Zhang Q.; Woods C. G.; Howell B. A.; Siler S. Q.; Woodhead J. L.; Yang Y.; McMullen P.; Watkins P. B.; Andersen M. E. (2012) Modeling drug- and chemical-induced hepatotoxicity with systems biology approaches. Front. Physiol. 3, 462-1–462-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen M.; Villeneuve D. L.; Ankley G. T.; Bencic D. C.; Breen M. S.; Watanabe K. H.; Lloyd A. L.; Conolly R. B. (2013) Developing predictive approaches to characterize adaptive responses of the reproductive endocrine axis to aromatase inhibition: II. Computational modeling. Toxicol. Sci. 133, 234–247. [DOI] [PubMed] [Google Scholar]
- Schmidt B. J.; Casey F. P.; Paterson T.; Chan J. R. (2013) Alternate virtual populations elucidate the type I interferon signature predictive of the response to rituximab in rheumatoid arthritis. BMC Bioinf. 14, 221-1–221-16. [DOI] [PMC free article] [PubMed] [Google Scholar]