

Abstract
Regulation of cell growth is a fundamental process in development and disease that integrates a vast array of extra- and intracellular information. A central player in this process is RNA polymerase I (Pol I), which transcribes ribosomal RNA (rRNA) genes in the nucleolus. Rapidly growing cancer cells are characterized by increased Pol I–mediated transcription and, consequently, nucleolar hypertrophy. To map the genetic network underlying the regulation of nucleolar size and of Pol I–mediated transcription, we performed comparative, genome-wide loss-of-function analyses of nucleolar size in Saccharomyces cerevisiae and Drosophila melanogaster coupled with mass spectrometry–based analyses of the ribosomal DNA (rDNA) promoter. With this approach, we identified a set of conserved and nonconserved molecular complexes that control nucleolar size. Furthermore, we characterized a direct role of the histone information regulator (HIR) complex in repressing rRNA transcription in yeast. Our study provides a full-genome, cross-species analysis of a nuclear subcompartment and shows that this approach can identify conserved molecular modules.
INTRODUCTION
An aberrant increase in nucleolar size in cancer cells was documented more than a century ago (1). Subsequently, this observation, combined with further research, led to the conclusion that nucleolar hypertrophy is a common feature in cancer and that nucleolar size can be used as a histopathological marker to grade the malignancy of tumors (2, 3). Despite this early association of enlarged nucleoli and cancer, the genetic network underlying the regulation of nucleolar size is still poorly understood.
The nucleolus assembles around nucleolar organizer regions (NORs) (4), which are chromosomal regions that contain ribosomal RNA (rRNA)–encoding gene clusters [ribosomal DNA (rDNA)]. Transcription of rDNA by RNA polymerase I (Pol I) is a major determinant of the cellular ribosome concentration and positively correlates with both the magnitude of protein biosynthesis and the rate of growth (2, 5, 6). Several studies have demonstrated that the size of the nucleolus reflects the rate of rDNA transcription (6). Rapidly growing yeast cells contain big nucleoli and show increased rDNA transcription, and up to 80% of their total nucleic acid content is composed of rRNA molecules (7). The high rate of energy consumption during rDNA transcription requires cells to tightly control their rRNA production rate, especially under nonfavorable growth conditions. Consequently, nutrient starvation or inhibition of signals promoting cell growth [for example, the target of rapamycin (TOR) pathway] results in a marked decrease in nucleolar size and a concurrent reduction in Pol I–mediated transcription and rRNA abundance (8).
Several studies have linked the growth of tumors with nucleolar hypertrophy. Oncogenes like c-Myc stimulate rDNA transcription and induce nucleolar hypertrophy upon overexpression (9–11). Moreover, studies in Drosophila have demonstrated that nucleolar hypertrophy is a conserved feature of tumor formation (12–14) and that tumor stem cells in different tissues are highly dependent on growth-promoting molecular feedback loops (15). Currently, it is less clear whether mutations that cause an increase in rDNA transcription are a general class of cell growth suppressors. Similarly, a systematic understanding of the genes that are required to sustain rDNA transcription is still lacking because comprehensive studies focused on nucleolar function have not yet been performed. Because interfering with Pol I–mediated transcription has been proposed as a beneficial strategy in the treatment of cancer (2, 16, 17), understanding how cells regulate rDNA transcription has medical implications and might suggest new strategies of how to interfere with unrestricted growth.
To identify both evolutionarily conserved and species-specific genetic networks that influence nucleolar size regulation and rDNA transcription, we performed a comparative, genome-wide phenotypic analysis of nucleolar size in Saccharomyces cerevisiae and Drosophila melanogaster. This analysis allowed us to directly assess the conservation of gene function at the level of nucleolar phenotypes. We combined our genetic results with a mass spectrometry (MS)–based analysis of the 35S rDNA promoter–associated proteome in S. cerevisiae to systematically identify direct regulators of rDNA transcription. Together, these approaches enabled us to generate a comprehensive map of the conserved core set of factors that control nucleolar size. Our analysis reveals a complex cellular network underlying nucleolar size regulation and serves as a useful resource for further studies of cell growth and Pol I–mediated transcription. In addition, we demonstrate a direct role for the histone information regulator (HIR) complex in repressing rDNA transcription in yeast. Overall, our analysis illustrates the power of performing parallel loss-of-function analyses in different species to establish a comprehensive network of evolutionarily conserved regulators for a given process.
RESULTS
Genome-wide analysis of nucleolar size in yeast
To analyze nucleolar phenotypes in yeast on a genome-wide scale, we first established a “nucleolar query” strain compatible with the synthetic genetic array (SGA) platform (18). We constructed a yeast strain with fully functional fluorescent reporter proteins that differentially label three cellular compartments: nucleolus [Nop10–GFP (green fluorescent protein)], nucleus (Hta2-mCherry), and cytoplasm (tdTomato) (fig. S1, A and B). Epitope tagging of the essential Nop10 protein and the histone H2A protein, as well as the expression of a cytoplasmically localized tdTomato, did not interfere with the growth rate of haploid cells (fig. S1C). The nucleolar size of our nucleolar query strain during different growth phases was similar to that of wild-type strains (8). First, we observed a significant decrease in nucleolar size in stationary phase as compared to cells in mid-log phase (fig. S1D). In addition, cells treated with the TOR inhibitor rapamycin had smaller nucleoli than untreated cells, in agreement with previous reports (16, 19–23).
We next measured nucleolar morphology and size for a genome-wide set of haploid yeast strains (18), each containing the reporter genes and a deletion allele for each of the nonessential genes (24) or a temperature-sensitive allele for a large set of essential genes (25). We used high-throughput automated confocal microscopy to image the reporter proteins in mid-log phase yeast cells and subsequently quantified nucleolar size and nucleolar fragmentation, as hallmarks for altered Pol I–mediated transcription (26), using automated image analysis (Fig. 1A and figs. S1, A, E, and F, and S2, A and B). For the temperature-sensitive collection, we analyzed nucleolar fragmentation at both permissive (25°C) and nonpermissive (37°C) temperatures and observed nucleolar fragmentation for many temperature-sensitive mutants of genes that are involved Pol I–mediated transcription (fig. S1E).
Fig. 1. SGA screen for nucleolar defects in S. cerevisiae.

(A) Computer-based detection of nuclear (green outlines) and nucleolar (red outlines) size and morphology changes in the RNA Pol I transcription initiation mutant rrn3-S213P or wild-type (WT) cells. Images are representative of four independent biological replicates. (B) Scatter plot illustrating the dependence of nucleolar area [x axis: (per plate) normalized nucleolar area] on cell number (number of nucleoli per well). Black dots, WT control wells; blue dots, loss-of-function alleles that are similar to WT; red dots, wells that were classified as phenotypes based on their localization outside the WT region. Phenotypic strength was calculated as the distance to the regression line (gray). (C) Representative examples of different nucleolar phenotypes in the essential and nonessential gene set collections (close-ups show individual representative nucleoli of the respective genotype). Images are representative of four independent biological replicates.
We quantified nucleolar area on a genome-wide scale by fitting a regression curve through the nucleolar data obtained from wild-type wells. The regression analysis yielded an estimated range of nucleolar areas specific to particular cell concentrations that allowed us to score phenotypic changes in nucleolar area that significantly deviated from wild type (Fig. 1B and fig. S2, A and B). The severity of a phenotype was calculated as the distance (Δ) of a data point to the fitted model. In addition, we quantified nucleolar fragmentation and observed nucleolar phenotypic changes in mutants of genes that alter Pol I–mediated transcription (fig. S1, E and F), which is consistent with observation described in previous reports. For example, yeast strains with temperature-sensitive mutations in genes encoding the Pol I subunits Rpa190 or Rpc40 (27, 28), the Pol I–specific transcription initiation factor Rrn3 (29), the TATA box–binding protein (TBP)–associated protein Mot1 (30), the nucleolar proteins Nop1 or Nop2, or several previously identified components of the secretory pathway (31) showed a decrease in nucleolar size and a concurrent increase in nucleolar fragmentation (Fig. 1, A and C, and fig. S1E).
The cellular network underlying nucleolar size regulation in yeast
The SGA analysis uncovered 388 genes (representing ~6% of the yeast genome) for which mutations caused either a significant change in nucleolar size and/or nucleolar fragmentation (Fig. 1B, fig. S1, E and F, and table S1). From those, we identified 113 genes as high-confidence candidates because multiple mutant alleles of a specific gene showed similar phenotypes (table S1). The phenotypic rate varied between the essential (~23%) and the nonessential (~2%) gene data sets. For the temperature-sensitive candidates, ~88% (72 of 82) of the mutants with an apparent nucleolar fragmentation phenotype had an observable phenotype only at high temperature (fig. S1E). The high frequency of candidates in the temperature-sensitive class and the requirement for a shift to nonpermissive temperature are consistent with the idea that strong perturbations in rDNA transcription cause inviability.
Next, we used publicly available data sets from genome-wide subcellular localization studies in yeast (32) to analyze the subcellular distribution of the gene products identified in our screen. This analysis revealed a strong enrichment for genes encoding proteins that localize to the nucleolus, nucleus, and endoplasmic reticulum (ER)–Golgi network, indicating an important role of these compartments in Pol I–mediated transcription (Fig. 2A). Gene products localizing to other compartments, such as peroxisomes, endosomes, lipid particles, or the vacuole, were conversely underrepresented in our data set, suggesting a negligible role for these compartments in nucleolar size regulation.
Fig. 2. Identification of direct regulators of rDNA transcription through promoter proteomics in S. cerevisiae.

(A) Subcellular localization of proteins whose corresponding genes resulted in a decrease (blue nodes) or an increase (red nodes) of nucleolar size upon loss of function. (B) Examples of molecular complexes implicated in transcriptional processes identified in the SGA screen [red: increase in nucleolar size, blue: decrease in nucleolar size (color intensity correlates with nucleolar size phenotypic strength), gray: not screened; asterisks denote proteins identified in the TALO8 purification]. (C) Representative silver-stained polyacrylamide gel of the purified proteins associated with TALO8 and TALO8–35S rDNA (M, marker; L, lysate; S, supernatant; E, eluate; B, beads). Images are representative of two independent biological replicates. (D) Venn diagram representing the overlap between SGA screen and TALO8 purification. P value (P = 6.82 × 10−4) was calculated using a hypergeometric test. (E) 35S pre-rRNA abundance is significantly increased in eap1 and rsc2 mutants compared to WT as measured by qRT-PCR. Bars represent the means ± SD of five independent biological replicates. ***P < 0.001.
To identify the functional categories that are associated with mutations causing a change in nucleolar size, we performed GO (Gene Ontology) term enrichment analyses. Consistent with the subcellular distribution results, genes regulating ER–to–Golgi vesicle–mediated transport, rRNA biosynthesis, nucleosome assembly, general regulation of transcription, or histone acetylation were enriched in the “decreased nucleolar size” category (table S2). Conversely, mutations in various genes involved in cell division conferred an increase in nucleolar size, suggesting that this phenotype might be common with defects in the cell cycle machinery. This is consistent with the observation that cell cycle regulators, such as Cdc14 (33), can directly modulate rDNA transcription. Together, the overall coverage of identified genes that influence nucleolar size control in our data set, the enrichment of genes encoding factors that function in the nucleus or nucleoli, and the results of the GO term enrichment analyses suggest that our data set is of high quality and should be useful for identifying potential new regulators of nucleolar size.
To further reduce residual experimental noise and to identify regulatory modules in our data set, we identified the molecular complexes, as defined by protein-protein interaction (PPI) evidence, which underlie nucleolar size regulation. To do this, we used COMPLEAT [a database containing all publicly available high-quality PPIs (34)] to perform an enrichment analysis to identify defined molecular complexes in the SGA data set. This approach provides an unbiased strategy to uncover regulatory modules controlling nucleolar size because it integrates PPI data and phenotypic consistency of individual complex members. This analysis allowed us to identify 339 molecular complexes that influence nucleolar size upon loss of function (Fig. 2B, fig. S4A, and table S3). Moreover, this complex-based analysis revealed insights that would have not been possible by solely using GO term or pathway enrichment analyses. For example, the HIR complex was identified in this analysis as a high-confidence regulator of nucleolar size based on the phenotypic consistency of the complex members. In addition, this analysis revealed roles in nucleolus size regulation for the FACT (facilitates chromatin transcription) complex, several molecular complexes regulating the chromatin state, as well as molecular modules regulating RNA processing, intracellular trafficking, or splicing.
Identification of the rDNA promoter–associated proteome
Because the SGA screen does not allow us to distinguish between direct and indirect regulators of rDNA transcription, we next sought to comprehensively characterize the proteomic composition of the 35S rDNA promoter and complement the phenotypic SGA data set. We purified the 35S rDNA promoter region by isolating minichromosomes from wild-type yeast cells that contained either the TALO8 (control) or the TALO8–35S rDNA minichromosome and determined the associated proteins by MS analysis (fig. S3A) (35, 36). As expected, the control and 35S rDNA minichromosomes exhibited different protein banding patterns but similar amounts of both histones and the 3×FLAG-LacI repressor protein (Fig. 2C), suggesting the control and the rDNA promoter had different protein compositions. Similar to previous reports, most proteins identified in both purifications nonspecifically associated with 3×FLAG-LacI or interacted with DNA (for example, histones or proteins that are involved in DNA replication, such as the minichromosome maintenance protein complex).
From the MS analysis, we identified a total of 308 proteins that were specific for the 35S rDNA promoter plasmid (table S4). We identified many factors that have been previously shown to be involved in 35S rDNA transcription, including all the components of the Pol I upstream activating factor complex (UAF) (Rrn5, Rrn9, Rrn10, and Uaf30), the TBP, the Pol I enhancer binding protein Reb1, as well as the components of the RENT complex (Cdc14, Net1) (fig. S3B). Whereas Net1 stimulates, Cdc14 represses Pol I–mediated transcription (33, 37–39), suggesting that the purification can identify regulators that can stimulate or inhibit. To analyze the functional categories that are associated with the 35S rDNA promoter proteome, we performed functional and localization GO term enrichment analyses (fig. S3, C and D). As expected, most of the identified proteins localize to the nucleolus or nucleus, and certain GO categories such as “transcription” or “regulation of transcription” and identified proteins with chromatin remodeling and modifying activities were enriched. To identify potential direct mediators of rDNA transcription, we compared the yeast SGA screen to the 35S rDNA promoter proteome and found an overlap of 44 proteins (Fig. 2D). Because many of the Pol I–associated core transcription factors are not covered by temperature-sensitive alleles in the SGA screen, we could not identify these proteins in the overlap, although many were found in the purification (fig. S3B). These overlapping proteins [including Nop1, Nop2, Mot1, Rpa190, Rpc40, Taf5, Taf6, Mtr4, Abf1, Eap1, Sub2, Rpl16b, Hir2, Spt5, the FACT complex (Spt16, Pob3), and members of the RSC complex (Rsc2, Rsc8, Sth1, Arp7)] are mainly implicated in transcriptional regulation, suggesting a direct role for these factors in Pol I–mediated transcription (Fig. 2B and fig. S4B).
We next sought to validate candidates found in both assays by analyzing 35S pre-rRNA abundance using quantitative reverse transcription polymerase chain reaction (qRT-PCR). As a proof of principle, qRT-PCR analysis indicated decreased 35S pre-rRNA abundance in the rpa190 and rrn3 temperature-sensitive mutants, consistent with the SGA nucleolus phenotypes (fig. S4A). For all positive controls tested, we found that a decrease in nucleolar size correlated with decreased amounts of 35S pre-rRNA (fig. S4A). In contrast, mutations resulting in an enlarged nucleolus, such as mutations in EAP1, a gene that encodes a protein implicated in TOR signaling (40), or in RSC2, a component of the RSC chromatin remodeling complex (41), showed increased abundance of 35S pre-rRNA (Fig. 2E). Among the tested candidates, we identified several members of the RSC complex that have not been previously implicated in rDNA transcription. Moreover, Hir2, a component of the HIR histone chaperone complex, and the FACT complex members, Spt16 and Pob3, were identified by both approaches. These data suggest that RSC, HIR, and FACT are direct regulators of rDNA transcription in yeast (Fig. 2B). Together, our integrative approach allowed us to identify putative direct regulators of Pol I–mediated transcription (of which we validate the HIR complex in sections below) in our phenotypic-based SGA data set.
A comparative RNA interference screen in Drosophila to identify conserved regulators of nucleolar function
The results of our phenotypic screen in yeast indicate that we can identify molecular complexes altering nucleolar size upon loss of function with high confidence using a combined genetic and proteomic approach. Nevertheless, S. cerevisiae deviates in several cell biological aspects from higher eukaryotes, obfuscating the extent of evolutionary conservation. To identify proteins and protein complexes with an evolutionarily conserved role in nucleolar size regulation, we designed a genome-wide RNA interference (RNAi) screen for nucleolar-size phenotypes in D. melanogaster cultured cells. Similar to the yeast SGA screen, we used three reporters that differentially labeled the nucleolus (fibrillarin), the nucleus [4′,6-diamidino-2-phenylindole (DAPI)], and the cell border (phalloidin) (fig. S5). We performed an immunofluorescence-based genome-wide RNAi screen (42) at 1.7 double-stranded RNAs (dsRNAs) per gene coverage using high-throughput confocal microscopy combined with automated image analysis. This approach identified 757 genes (~5% of all protein-coding genes) that scored greater than or less than a z score cutoff of ±2 (Fig. 3, A and B, and table S5). As expected, we found numerous genes previously implicated in nucleolar size regulation in Drosophila or other species, including the tumor suppressor brat (12), the transcriptional repressor sin3a (8), nucleolar proteins such as Nop5, Nop56, and Nopp140 (43), as well as transcription factors such as diminutive (dm), the Drosophila myc homolog (11), and tif1a (44), which have been implicated in Pol I–mediated transcription (Fig. 3, A to C). To determine whether changes in nucleolar size indeed reflect differences in rDNA transcription, we quantified pre-rRNA abundance in selected positive controls. We consistently observed a decrease in pre-rRNA abundance upon knockdown of tif1a, nopp140, or reptin (Fig. 3C), suggesting that the quantification of nucleolar size in Drosophila cell culture can be used to infer the Pol I transcriptional state.
Fig. 3. A genome-wide RNAi screen for nucleolar size defects in Drosophila cell culture.

(A) Representative images of different nucleolar phenotypes upon RNAi-mediated loss of function. Images are representative of three or more independent biological replicates. (B) Z score–ordered rank plot of all screened Drosophila RNAi Screening Center (DRSC) amplicons. Red circles indicate the z score of the respective genes (red lines represent a z score of ±2 used for hit selection). (C) 35S pre-rRNA abundance is decreased upon RNAi targeting tif1a, nopp140, and pontin as measured by qRT-PCR. Bars represent the means ± SD of four independent biological replicates. *P < 0.05. (D) Examples of enriched molecular complexes regulating nucleolar size (red: increase in nucleolar size, blue: decrease in nucleolar size).
Analogous to the yeast SGA screen, we determined general trends in the data set by performing a functional GO term enrichment analysis, which, as expected, yielded categories such as “Pol I transcription factor activity,” “histone acetyltransferase complex activity,” and “chromatin remodeling activity” in the reduced nucleolar size category. Similar to the yeast SGA screen, GO terms associated with the cell cycle were enriched in the increased nucleolar size category. Additionally, knockdown of genes with roles in RNA splicing, ribosomal function, and general translation is associated with increased nucleolar size (Fig. 4A and table S2). Because large-scale RNAi experiments are inherently noisy (45, 46), we next identified the molecular complexes that were enriched in the Drosophila data set. This analysis revealed 192 molecular complexes that influence nucleolar size upon loss of function and included many complexes that contain regulators of Pol I–mediated transcription (Fig. 3D and table S6). We identified several molecular complexes for which mutations were not analyzed in the orthologous yeast genes because they are essential for viability and were not present in the temperature-sensitive collection. For example, four members of the box C/D small nucleolar (sno) RNP complex—nop56, fib, hoip, and nop5—were identified in the screen (Fig. 3D). The complex is required for ribose-2′-O-methylation of rRNA, which is essential for ribosome function and conserved from yeast to humans (47, 48). Together, these data demonstrate that the Drosophila RNAi screen enriched the combined data set for genes that are required for nucleolar size regulation and identified previously unknown factors that might have a direct function in regulating rDNA transcription.
Fig. 4. Comparative analysis between the S. cerevisiae SGA and the D. melanogaster RNAi screens.

(A) Heatmap displaying the natural log–transformed P values for a selected set of GO terms [D. melanogaster, S. cerevisiae, decrease (−) or increase (+) in nucleolar size; a comprehensive heatmap displaying the results of the GO term analysis is given in fig. S6]. (B) Examples of evolutionarily conserved molecular complexes regulating nucleolar size in D. melanogaster and S. cerevisiae (red: increase in nucleolar size, blue: decrease in nucleolar size, gray: genes that have not been screened or for which no homolog exists in the respective species, white: no nucleolar size phenotype detected; blue lines denote PPIs, and gray lines connect homologous genes).
Identification of evolutionarily conserved modules by cross-species analysis
To probe the extent of evolutionary conservation in the regulation of nucleolar size between S. cerevisiae and D. melanogaster, we performed a comparative GO enrichment analysis (Fig. 4A, fig. S6, and table S2). This analysis revealed extensive similarities in the overall trends of nucleolar size regulation between the two organisms. A decrease in nucleolar size is observed in both species upon loss of genes whose products are annotated to participate in processes such as chromatin organization, histone acetylation, nucleolar processes, or intracellular transport. Conversely, an increase in nucleolar size is observed upon loss of genes whose products function in cell division, mRNA processing, or DNA replication. The similarities at the level of GO terms were also reflected in a comparative analysis of molecular complexes. We identified 74 molecular complexes that are comparably enriched in both species (Fig. 4B and table S7). We found evidence for an evolutionarily conserved role for molecular complexes mediating histone acetylation or the TRAMP complex, which has been implicated in pre-rRNA surveillance, in the regulation of nucleolar size. Molecular complexes that participate in various different cellular functions appear to share a conserved role in regulating nucleolar size. Depletion or deletion of components of molecular complexes that mediate “mRNA export from the nucleus,” “general transcriptional regulation,” or “ER–to–Golgi vesicle–mediated transport” caused nucleolar phenotypes in both yeast and Drosophila. These data unravel a complex, evolutionarily conserved genetic network underlying nucleolar size control.
In addition to identifying similarly enriched GO terms, we also identified terms that were only associated with one species because incomplete coverage of the respective genes in the screening pipeline of the other species precluded their systematic analysis. For example, we identified several ribosomal protein genes in D. melanogaster that resulted in a pronounced increase in nucleolar size upon knockdown (Fig. 4A and fig. S7A). Consistent with this finding, pre-rRNA abundance was significantly increased upon knockdown of either rpl27a or rpl23a (fig. S7B). Because the yeast mutant collections that we screened do not contain all ribosome protein gene mutants, we did not identify the ribosome as an enriched GO term in the SGA screen. However, the results of the complex-based analysis and the phenotypes of individual yeast ribosomal protein mutants revealed an increase in nucleolar size, suggesting that the nucleolar size phenotype upon loss of ribosomal proteins is evolutionarily conserved (fig. S7A). To test this idea, we measured 35S pre-rRNA abundance in mutants for ribosomal protein genes that showed an increase in nucleolar size in the yeast SGA screen. Consistent with the observed phenotype, rpl34aΔ and rpl22aΔ mutant yeast strains showed an about fivefold increase in 35S pre-rRNA abundance as compared to wild-type (fig. S7B), demonstrating that ribosomal integrity is a critical evolutionarily conserved parameter in maintaining proper nucleolar size. Overall, our comparative, complex-based analysis suggests that the main architecture of the cellular network that controls Pol I–mediated transcription is similar between S. cerevisiae and D. melanogaster, and reveals considerable evolutionary conservation in the regulation of nucleolar size.
Use of complementary phenotypic analysis to complement high-throughput data sets
The yeast SGA screen as well as the TALO8 purification identified molecular complexes for which we did not obtain evidence in the Drosophila RNAi screen, potentially due to nonfunctional RNAi constructs. One of these complexes is the chromatin remodeling complex FACT, which is required for rDNA transcription in mammalian cells (49). Mutations in either one of the two yeast genes encoding FACT members, SPT16 or POB3, resulted in significantly smaller and condensed, round nucleoli as compared to wild type (Figs. 2B and 5A). These results, along with a previous study (49), prompted us to test for a role for the FACT complex in controlling nucleolar size and cell growth in Drosophila. To do this, we analyzed nucleolar size in third instar salivary glands upon knockdown of spt16 or ssrp (the Drosophila homolog of POB3). Consistent with the data obtained in yeast, depletion of either Spt16 or Ssrp resulted in a significant reduction of nucleolar size, suggesting that the FACT complex has an evolutionarily conserved role in controlling nucleolar size (Fig. 5, B and C, and fig. S8A).
Fig. 5. The FACT complex is an evolutionarily conserved regulator of nucleolar size.

(A) Small nucleolus phenotype in S. cerevisiae FACT complex mutants. Images are representative of five independent biological replicates. (B) Examples of nucleolar size phenotypes in D. melanogaster third instar larval salivary glands upon knockdown of the indicated genes using transgenic RNAi. Images are representative of five independent biological replicates. (C) Quantification of the indicated nucleolar size phenotypes is shown in (B). Bars represent means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05. (D) Loss of germline cells upon knockdown of spt16 using two independent, non-overlapping RNAi constructs. Images are representative of four independent biological replicates. (E) Suppression of neural stem cell tumor growth in D. melanogaster (assayed by tumor-specific luciferase expression) upon targeting genes that result in smaller nucleoli upon transgenic RNAi. Bars represent the means ± SEM of four independent biological replicates. ***P < 0.001.
To determine whether the FACT complex is also required for cell growth regulation in Drosophila, we analyzed germline stem cell maintenance and tumor growth upon loss of FACT complex members. Germline stem cells in Drosophila asymmetrically retain the cell growth–promoting nucleolar protein Wicked, and reduction of a high cell growth rate results in stem cell maintenance defects (13, 50). Consistent with a role for the FACT complex in cell growth regulation, we observed germ-line stem cell maintenance defects upon knockdown of spt16 using two independent, non-overlapping short hairpin RNA constructs (Fig. 5D). To determine whether the FACT complex is also required for tumor growth, we generated a brain tumor model in which we could monitor the growth rate over prolonged periods using luciferase activity as a readout (51). Because type II neuroblast lineages are not required for survival, we expressed a brat-RNAi construct from worniu-Gal4, asense-Gal80 to obtain viable flies with neural stem cell–derived tumors (52). To test the approach, we expressed RNAi constructs targeting nopp140, which completely disrupts nucleolar architecture, and dm (the Drosophila myc homolog), which results in a ~90% reduction of nucleolar area in salivary gland nuclei (Fig. 5B). Luciferase activity was reduced ~95 and ~50% with nopp140 and dm RNAi, respectively (Fig. 5E and fig. S8B). Consistent with a reduction in nucleolar size, depletion of FACT complex members with multiple non-overlapping RNAi constructs against spt16 or ssrp resulted in decreased luciferase activity in adult flies (Fig. 5E and fig. S8B), suggesting that the FACT complex is an evolutionarily conserved regulator of rDNA transcription required for sustaining proper cell growth rates. Together, these data demonstrate that cross-species comparison of loss-of-function assays can be used to complement an incomplete genetic screen from one species and thus obtain a more comprehensive view of a given genetic process.
Regulation of rDNA transcription by the HIR complex in S. cerevisiae
In addition to using our approach to identify evolutionarily conserved regulators of nucleolar size, we also determined if our cross-species analysis would allow us to identify molecular complexes that result in species-specific nucleolar phenotypes upon loss of function. One candidate complex for a nonconserved role in controlling nucleolar size was the HIR complex, for which an involvement in rDNA transcription has not previously been documented. In S. cerevisiae, the genes HIR1, HIR2, HIR3, and HPC2 encode components of the HIR complex, which is evolutionarily conserved from yeast to humans (53). The HIR complex acts as a histone H3–H4 chaperone in nucleosome assembly and has important functions in transcriptional regulation, elongation, gene silencing, cellular senescence, and aging (54–57). Moreover, the HIR complex regulates three of the four histone gene loci during the cell cycle in yeast (58, 59). In Drosophila, the HIRA complex consists of the proteins Hira and yemanuclein-α. Their function in nucleosome assembly is required during sperm decondensation by deposition of the histone H3.3 in the male pronucleus (60–64).
In the yeast SGA screen, we found that deletion of either HIR1 or HIR2 resulted in increased nucleolar size, and our complex-based analysis identified a statistical enrichment of the whole HIR complex, indicating that rDNA transcription might be increased in HIR complex mutants. We also identified Hir2 in the rDNA promoter proteome analysis, suggesting that the HIR complex might be a direct regulator of rDNA transcription in S. cerevisiae. To test this hypothesis, we analyzed binding of the HIR complex to the rDNA using chromatin immunoprecipitation (ChIP). Consistent with the SGA screen and the rDNA promoter proteome, we found a specific association of both myc-tagged Hir1 and Hir2 at the 35S rDNA region, whereas these proteins were absent from a nontranscribed region (Fig. 6A). Because deletion of the genes encoding individual HIR complex members results in an increase in nucleolar size, we hypothesized that the phenotype we observed might be a result of increased rRNA abundance. qRT-PCR analysis of 35S pre-rRNA abundance in hir1Δ, hir2Δ, hir3Δ, and hpc2Δ mutants consistently revealed increased abundance of the rRNA precursor (Fig. 6B). In agreement with these results, we found an increased association of Pol I at the rDNA locus in HIR mutants (fig. S9, A and B), demonstrating that rDNA transcription is increased upon loss of the HIR complex.
Fig. 6. The histone chaperone HIR complex regulates 35S rDNA transcription specifically in S. cerevisiae.

(A) The HIR complex is associated with the 35S rDNA region. ChIP qPCR analysis of myc-tagged Hir1 and Hir2 at the 35S rDNA region and the PMA1 control region, and a nontranscribed region (NTR) was performed with the indicated antibodies. Bars represent the means ± SD of five independent biological replicates. ***P < 0.001; **P < 0.01; *P < 0.05; NS, not significant. (B) Deletion of HIR complex members in S. cerevisiae results in increased 35S pre-rRNA abundance, as determined by qRT-PCR with specific oligonucleotides recognizing the 35S pre-rRNA. Values are normalized relative to the PMA1 control gene, and 35S pre-rRNA abundance for WT is set to 1. Bars represent the means ± SD of five independent biological replicates. ***P < 0.001. (C) Mutants of the HIR complex in S. cerevisiae show 35S rDNA silencing defects. Images are representative of three independent biological replicates. (D) HIR complex mutants show reduced histone H3 binding at the 35S rDNA region. ChIP using H3-specific antibodies followed by qPCR to determine the abundance of H3 at different loci in WT, hir1Δ, hir2Δ, or hir3Δ strains. Immunoprecipitation values were normalized to input values. Bars represent the means ± SD of four independent biological replicates. ***P < 0.001; *P < 0.05. (E) Representative images of D. melanogaster third instar larval salivary gland nucleoli of control and hira null (HR1) animals. Images are representative of three independent biological replicates. (F) Quantification of nucleolar size in control (hiraHR1/+) and hira null third instar larval salivary nuclei. Bars represent the means ± SD of three independent biological replicates. (G) qRT-PCR experiment assaying the 35S pre-rRNA abundance in control and hira null third instar larvae. Bars represent the means ± SD of three independent biological replicates. (H) qRT-PCR experiment in control (w1118) and hira null adult male flies assaying the 35S pre-rRNA abundance. Bars represent the means ± SD of three independent biological replicates.
Efficient regulation of rDNA transcription involves the epigenetic silencing of a subset of rDNA genes. To determine whether the increased rRNA abundance observed in HIR complex mutants might stem from defects in this process, we performed a silencing assay based on growth of yeast strains bearing a reporter gene in the rDNA repeats on selective or counterselective medium (65, 66). We constructed yeast strains containing a deletion of each gene encoding a HIR complex component as well as a modified URA3 reporter (mURA3) inserted into the 35S rDNA region. As reported previously, the mURA3 reporter gene was efficiently silenced in wild-type cells, and thus, the reporter gene strain grew poorly on SC (synthetic complete) medium lacking the amino acid uracil (−URA), but grew well on medium containing 5-fluoroorotic acid (FOA), which selects against URA3 expression (Fig. 6C). Deletion of HIR complex genes resulted in poor growth on FOA plates and enhanced growth on −URA plates, suggesting increased transcription of the reporter. These silencing defects, along with the histone H3–H4 chaperone activity of the HIR complex, suggested that nucleosomal occupancy might be reduced at rDNA genes in HIR complex mutants. ChIP experiments revealed a ~30 to 50% decrease in H3 occupancy at the 35S rDNA in HIR complex member deletions in comparison to wild type (Fig. 6D). In contrast, the abundance of H3 at a nontranscribed region or at the highly transcribed PMA1 gene did not appreciably change. This suggests that loss of HIR complex function results in increased rDNA transcription caused by diminished incorporation of histone H3–H4 dimers into nucleosomes at rDNA genes. These data show that the HIR complex is required for rDNA transcription in S. cerevisiae.
Further, we did not obtain evidence for a conserved role of the HIR complex in rDNA transcriptional suppression in the Drosophila RNAi screen, although Drosophila HIRA is associated with rDNA and is required for histone H3.3 deposition, presumably at rDNA genes (67). To clarify if the HIR complex indeed functionally diverges in Drosophila with respect to rDNA transcription, we took advantage of previously published hira null mutant flies (62). We did not detect a significant change in nucleolar size in hiraHR1 null mutant third instar salivary gland nuclei as compared to wild type (Fig. 6, E and F). Consistent with this result, we did not find changes in the pre-rRNA abundance in third instar hiraHR1 mutant larvae or adult hiraHR1 mutant flies when measured by qRT-PCR (Fig. 6, G and H), demonstrating that hira loss of function does not affect rDNA transcription or the regulation of nucleolar size in Drosophila.
Finally, we wanted to determine whether increased nucleolar size or increased Pol I transcriptional activity, as observed in HIR complex mutants, translates into an increased cell growth rate. We therefore examined several key parameters of cell growth and division across our hits and a variety of control strains (Fig. 7, A and B, and fig. S10, A to C). From our primary screen, we selected 50 strains carrying loss-of-function mutations in nonessential genes that conferred an increased nucleolar size and 10 negative control strains that did not show significant alteration of nucleolar size. We also included a panel of replicate wild-type strains (12) in parallel to determine the relative phenotypic variation between and within these groups. We did not observe differences in the growth kinetics between wild type and mutants with increased nucleolar size (including the HIR complex, RSC2, EAP1, RPL22A, and RPL34A). Similarly, we did not detect enhanced growth in mutants with increased Pol I transcription (SIN3 or HIR mutants), nor did we detect an increased resistance to rapamycin in HIR complex mutants. We therefore conclude that the enhanced rDNA transcriptional activity is frequently decoupled from cell growth regulation.
Fig. 7. Increased rDNA transcription does not translate into an increased growth rate in HIR mutants.
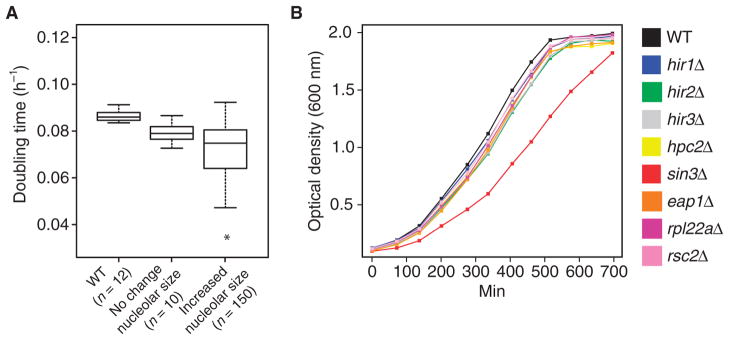
(A) Analysis of the cell growth rate by online optical density measurements of WT, mutant yeast strains with no phenotype in nucleolar size regulation (No change nucleolar size), and mutants with increased nucleolar size (Increased nucleolar size). n = 2 independent biological replicates. (B) Growth of WT and hir mutants and a selected set of mutants affecting nucleolar size and/or rRNA abundance. The HIR complex mutants show no difference in growth behavior compared to WT. Data are from one representative experiment of n = 3 independent biological replicates.
DISCUSSION
Comparative phenotypic analysis
Here, we describe a comparative, genome-wide phenotypic analysis in S. cerevisiae and D. melanogaster to map the cellular network regulating nucleolar size. This study revealed important insights into the use of performing comparative phenotypic analyses in two species. We demonstrate that this approach can overcome species-specific limitations such as incomplete genome coverage or experimental noise. As a result, the combined data set is improved in terms of both confidence and comprehensive coverage. Although the yeast SGA technology enables large-scale phenotypic analyses, not all genes can be analyzed because of experimental limitations. For example, we were able to assay 71% (4202 of 5882 protein-coding genes) of the genome in our analysis because of slow growth rate of some mutant temperature-sensitive strains (25). Likewise, RNAi-based methods in Drosophila have limitations, including experimental noise primarily due to off-target effects of the dsRNA constructs. To generate a high-confidence data set, we used phenotypic consistency at the protein complex level and cross-species comparison to filter and complement the single species-derived data sets. Overall, we demonstrate that comparative systems biology holds the promise to identify evolutionarily conserved core components as well as species-specific regulators of a biological process in a high-throughput manner.
Organelle-specific comparative phenotypic analysis
Organelles spatially segregate cellular processes into defined subcompartments. As a subnuclear organelle, the nucleolus assembles and adjusts in its size primarily in response to the rate of rDNA transcription. Consistent with the idea that Pol I is a convergence point for a vast array of cellular inputs, we found a complex cellular network underlying the regulation of nucleolar size. Our comparative approach provides evidence that at the level of nucleolar size regulation, the phenotypic manifestations upon loss of specific gene groups are conserved between S. cerevisiae and D. melanogaster. This is consistent with reports that suggest that core features of ribosome biogenesis are evolutionarily conserved (68, 69).
Nucleolar size has been used in several organisms to probe the cell growth state because the Pol I transcriptional rate is correlated with the protein biosynthesis rate (6). Our data suggest that this correlation holds for genes that are required for sustaining and promoting growth and whose loss-of-function phenotype is consequently associated with a decrease in nucleolar size. The identification of suppressors of cell growth solely through quantification of nucleolar size is, however, hindered by the fact that an increase in the Pol I transcriptional rate does not necessarily lead to an increased cell growth rate. Our data on mutations in ribosomal protein genes, SIN3, RSC2, EAP1, or HIR complex members demonstrate that Pol I–mediated transcription can be decoupled from cell growth regulation. Although increased Pol I–mediated transcription has been reported to be sufficient to increase the transcription in a subset of Pol II–regulated growth genes in yeast (70) and flies (44), our data suggest that enhanced rDNA transcription in many cases does not translate into an increased cell growth rate, suggesting that only a small subset of genes that induce nucleolar hypertrophy upon loss of function are bona fide growth suppressors. Consistent with this idea, the increase in rDNA transcription that occurs after overexpressing Tif-Ia in Drosophila is insufficient to increase the cellular growth rate (44). Increased cell growth as observed in cancer might rather be induced by a global amplification of growth-promoting transcriptional programs as has been suggested for the oncogene c-Myc (71, 72).
Our data allow us to identify positive regulators of cell growth with high confidence. These genes might be valuable targets for cancer therapy. We exemplify the use of this strategy by interfering with rDNA transcription in a Drosophila model of tumor formation. Collectively, these data show that interference with regulators of Pol I–mediated transcription might be a general and promising way to limit tumor growth as previously proposed (16). The Pol I inhibitor CX-3543, which shows antitumor properties in xenograft assays, has entered clinical trials (17). In this context, our study defines an evolutionarily conserved network of genes that is required to sustain cell growth and might provide new approaches to limiting cell growth in tumors. In contrast to vertebrates, in which the nucleolus can be divided into three subcompartments, in S. cerevisiae and D. melanogaster, the nucleolus follows a bipartite architecture (73). It will thus be interesting to extend phenotypic analysis to vertebrate nucleoli to determine whether core components of nucleolar size regulation are conserved despite these architectural differences.
Regulation of Pol I transcription by the HIR complex
The HIR complex has been identified as a transcriptional repressor of his-tone genes in yeast, and our data demonstrate that it similarly functions as a transcriptional repressor at the rDNA cluster. We provide several lines of evidence that the HIR complex is directly required for silencing of rDNA genes in S. cerevisiae. The HIR complex is associated with the rDNA cluster, and loss of the HIR complex subunits results in an increased nucleolar size and rDNA silencing defects. Consistently, Pol I occupancy and the amount of 35S pre-rRNA are increased in hir1, hir2, hir3, and hpc2 mutants.
Pol I–mediated transcription is regulated through two major mechanisms. First, short-term adjustments are established by posttranslational modifications of Pol I and associated transcription factors, which collectively modulate Pol I transcriptional activity (74, 75). Second, the overall fraction of actively transcribed rDNA genes is set epigenetically by modulating the chromatin state at individual units of the rDNA gene cluster (76). Silent rDNA genes are organized into canonical nucleosomal structures reminiscent of heterochromatin. Our data suggest that the HIR complex is required for nucleosome assembly and heterochromatin formation at the rDNA locus. Consistent with these functions, the HIR complex has been implicated in gene silencing at other genomic loci, such as at the telomeres and the mating type loci (53). Moreover, hira null mutant flies do not show an increase in nucleolar size nor is the amount of pre-rRNA increased, although Hira acts in nucleosome replacement at active rDNA genes (67). These data demonstrate that the nucleolar phenotypes that occur after HIR complex function is impaired are not conserved and suggest that the HIR complex might function in a different or redundant genetic network in flies, underlining that the molecular aspects of rDNA silencing have changed over evolutionary time.
MATERIALS AND METHODS
Strains and plasmids
The yeast strains and primers used in this study are listed in tables S8 and S9. All yeast strains are in the S288C genetic background. To analyze multiple proteins in the same S. cerevisiae background, we used wild-type cells with genomically tagged genes. Standard media were used, and strains were constructed by standard methods (crossing or transformation). All strains for follow-up experiments were confirmed by PCR, Western blot, or fluorescence microscopy. Double-mutant strains used to assess the potential for 35S rDNA readthrough were generated by crossing the appropriate parental strains of the opposite mating types. Diploid strains were then sporulated and subjected to tetrad dissection. Double mutants were identified by analysis of genetic markers and verified with PCR. For all growth tests, the appropriate yeast strains were grown to saturation overnight in YPD (yeast extract, peptone, and dextrose) medium at 30°C. Tenfold serial dilutions of each culture starting with about 105 cells per drop are shown. Plates were scanned after 2 to 4 days of incubation at 30°C. A detailed description of plasmid construction is available upon request.
In Drosophila, the salivary gland and neuroblast-specific driver line UAS-Dicer2; insc-Gal4, UAS-cd8::GFP (52) was used for quantification of nucleolar size. Luciferase-based quantification of tumor growth was done by crossing RNAi lines to the type II neuroblast–specific driver line (52) wor-Gal4, ase-Gal80/CyO; UAS-brat RNAi, UAS-luciferase/TM3, tubP-gal80ts. Subsequently, adult flies of the correct genotype were chosen and assayed with a standard luciferase luminescence kit (Promega). To control for a similar number of UAS (upstream activation sequences), a UAS-GFP fly strain served as the control. For quantification of nucleolar size in hira mutant animals, the hiraHR1 (62) null mutant knockout flies (in the w1118 background) were crossed to w1118 flies for two generations. Homozygous mutant animals were then compared to either w1118 or heterozygous hiraHR1/+ flies. For transgenic RNAi experiments, the following strains were used: dm-RNAi [JF01761, from the Transgenic RNAi Project at Harvard Medical School (HMS)], brat-RNAi [HMS01121 (HMS)], spt16-RNAi [GL00017 (germ line–specific) (HMS), HMS01332 (HMS), 1828R (National Institute of Genetics of Japan (NIG)], ssrp-RNAi [JF02120 (HMS), 44343 (Vienna Drosophila RNAi Center), 4817R-2 (NIG)], and nopp140- RNAi [HMS00564 (HMS)].
SGA screen
To perform the SGA screen, we first created a query strain endogenously expressing NOP10-GFP, HTA2-mCherry, and tdTomato (Y7039 MATα can1prΔ::RPL39pr-tdTomato-CaURA3can1Δ::STE2pr-LEU2 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 NOP10::GFP-HIS3 HTA2::mCherry-natMX). Epitope tagging was performed by homologous recombination of PCR-generated DNA sequences into the genome. Subsequently, the cassettes encoding the tag and a selection marker were integrated at the 3′ end of the gene, removing the endogenous stop codon. For epitope tagging of Nop10 at the C terminus, a GFP-HIS3 cassette was PCR-amplified (RN154, RN155), followed by transformation and selection on SD-HIS plates. Similarly, HTA2-mCherry was created by transformation of an mCherry-natMX PCR fragment (RN169, RN170) amplified from pKT-mCherry-natMX. Finally, a yeast strain constitutively expressing RPL39 promoter driving tdTomato (77) inserted in the CAN1 promoter region was crossed to the Nop10-GFP and HTA2-mCherry strain. After sporulation and subsequent tetrad dissection, the final haploid query strain was selected. The establishment of haploid cells containing the three fluorescent markers and individual knockout mutations or temperature-sensitive alleles using the SGA pipeline was performed as described in (18).
Haploid yeast strains derived from the SGA pipeline were grown in liquid synthetic medium(SD+methionine+NAT+G418)overnightina 96-well plate, diluted, and grown to mid-log phase (overnight) in low-fluorescence medium (LFD + methionine + NAT + G418). After a measurement of the optical density of every culture, cells were transferred into PerkinElmer Opera compatible plates and subsequently imaged with a PerkinElmer Opera microscope using a 60× objective. For nonessential genes, the plates were imaged at room temperature. For essential genes, plates containing strains expressing temperature-sensitive alleles of essential genes were imaged at room temperature, incubated at 37°C for 3 hours, and then re-imaged in a 37°C chamber. Single plain confocal images were recorded from 16 positions per well (images are available upon request). To avoid problems with movement of nonfixed yeast cells during the imaging procedure, red fluorescent protein and GFP were imaged simultaneously. Measurement of nucleolar size and determination of fragmentation phenotypes were performed with a dedicated Definiens IDE (integrated development environment) (v.7**) image processing pipeline.
Image acquisition and bioinformatics analyses
Data for the SGA screen were preprocessed as follows: Images containing fewer than 10 cells were discarded. To score a fragmentation phenotype, a fragmentation index (Φ) was introduced. For each imaged location, Φ was calculated in the following form: as the weighted average of a number of nucleoli per nucleus in a given image. In the above formula, nk represents the number of nucleoli identified in a nucleus, wk is a weight constant, and N is the total number of nucleoli detected in the image. The weights for each nucleolus were computed as a ratio of the number of fragmented nucleoli in the image versus the total number of nucleoli found in the image. The data generated with the image processing pipeline, as well as the calculated values for Φ, were averaged in every well to create a cumulative value for each feature.
Mutants with fragmented nucleoli were identified as follows: To identify fragmented phenotypes, we compared the probability distributions of the mean number of nucleoli per nucleus versus the mean in a well. The yeast nonessential gene collection did not exhibit any overfragmented wells; hence, the subsequent analysis was run for the essential gene collection only.
The quantile-quantile dependence shown in fig. S1F demonstrates that the arguments of the distribution functions are coupled by a piecewise linear transformation. Moreover, the distribution laws become identical after a salient point of the regression plot. We applied linear piecewise regression to model the relationship between averaged Φ and the number of nucleoli per nucleus and used residual values to compute z scores to identify the fragmentation mutants (fig. S1F). To eliminate images with a small number of cells and few fragmented nucleoli, we set the z score tolerance interval to [−2,2]. Our next step was to exclude non–wild-type wells clustered in the vicinity of the salient point of the regression plot and, therefore, considered to be similar to controls. To do this, we selected a threshold Φc, which is computed as Φc = Φ̄+σ(Φ), using wild-type fragmentation index measurements only. Here, σ denotes SD, and Φ̄ is mean value of the fragmentation index. The threshold Φc is equal to 1.097. All wells with z scores lying within the tolerance range, and for which the average fragmentation index exceeds Φc, are considered as hits.
Using this approach, we identified 82 genes whose loss of function was associated with apparent nucleolar fragmentation, and in ~88% of cases (72 of 82 genes), the phenotype was only observable at high temperature, validating the high specificity of the screening approach (fig. S1D). We observed nucleolar fragmentation for many temperature-sensitive mutations in genes previously implicated in Pol I–mediated transcription. The phenotypic analysis of all temperature-sensitive strains was carried out after 3 hours of incubation at nonpermissive temperature. Because nucleolar fragmentation is indicative of altered Pol I–mediated transcription, this phenotypic analysis was included in the data analysis along with the analysis in nucleolar size changes. The nucleolar fragmentation phenotype could be intermediate and might frequently translate into a complete loss of the nucleolus at later time points.
Mutants with a decrease or increase in nucleolar size were identified as follows: To detect genes regulating nucleolar size in yeast, we exploited the fact that the average nucleolar size decreases with an increased density of the culture. For simplicity, the analysis was carried out on log-transformed and per plate–normalized values (Fig. 1B). The relationship between nucleolar size and cell culture density was estimated using regression analysis. To detect mutant phenotypes, we introduced a distance measure (Δ), which is computed as a standard score of residual values to the regression line. The regression was performed on the wild-type wells only, providing a model of a maximum span of Δ. Distance thresholds estimated from control wells were used to identify mutant phenotypes consistent with changes in the regulation of nucleolar size.
Essential yeast strain collection
Most control wells in the essential collection are overgrown (in that they contain a high cell number per well) (fig. S2, A and B, an agglomeration of wild-type wells in the upper right quadrant) due to the incubation at 37°C. Such agglomeration poses a considerable problem during regression analysis and biases fitting for the rest of the control wells in the lower quadrants of the plot. Therefore, we aggregated the control wells and performed weighted nonlinear regression modeling. The weights were constructed to compensate for agglomeration of control wells and computed as exp(tx – ty), where tx denotes the transformed data point x coordinate and ty is a y coordinate. The goodness-of-fit analysis confirmed that the variability in a data set is well accounted for by the regression model yielding adjusted R2 = 0.6283. The model was then applied to compute Δ distances for all control wells and to all data in the essential collection. The wild-type boundary wells were used to estimate thresholds for identification of genes regulating nucleolar size. Wells characterized with Δ ∈ (−∞; −0.475]∪[1.298; ∞) define a set of genes with nucleolar size defects.
Nonessential yeast strain collection
To identify the subset of genes that control nucleolar size, a linear weighted regression was used because the correlation between cell number in a well and nucleolar area is much less pronounced. The coefficient extracted from the regression model that describes the relationship between the normalized nucleolar area and the number of cells in a well equals 0.94. The interval of distances used to determine mutant phenotypes was: (−∞; −2.42]∪[2.08; ∞). Note that these values were scaled to the boundaries of the essential collection for the purpose of pooling all SGA nucleolar size data in table S1.
Overall, this analysis miscategorized only 14 of 388 (~4%) wild-type measurements in the essential collection as mutant phenotypes, thus demonstrating that we have established a robust and high-stringency discovery pipeline for nucleolar size phenotypes.
Drosophila cell–based RNAi screen
The Drosophila RNAi screen was carried out at the DRSC (http://www.flyrnai.org/) (78). A total of 0.25 μg of dsRNA per wellwas arrayed in 384-well PerkinElmer Opera compatible plates. Serum-starved cellswere incubated with the dsRNA for 1 hour, and subsequently, fetal bovine serum in Schneider’s medium was added to a final concentration of 10%. After 96 hours, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline with 0.1% Triton X-100 for 30 min. A standard staining protocol was used to label the nucleolus [anti-fibrillarin antibody (1:555, EnCor Biotechnology)], the outline of the cell [Alexa Fluor 488–coupled phalloidin (1:1000, Life Technologies)], and DNA [DAPI (1:1000)]. The plates were imaged with an automated high-throughput confocal microscope (PerkinElmer Evotec Opera). Z-axis stacks were recorded for seven positions in each well with a 60× objective. To capture the maximum size of the nucleolus in one cell layer, images were collapsed into one plane using a maximum intensity projection function. Segregation, detection, and measurement of the imaged objects were done using a Definiens software pipeline. All mean values for nucleolar area (per well) on a plate were normalized to the mean of pooled, dedicated wells with RNAi targeting GFP (control) on the same plate (note: we did not observe a correlation between the number of cells per well and the area of the nucleoli). Subsequently, z scores were calculated for genes expressed in KC cells and used for downstream analyses such as GO term enrichment or complex analysis. A z score above or below ±2 was used as a cutoff for hit selection (note: z scores >7 are not displayed in Fig. 3B but are listed in table S4; data are available in table S4 and at http://www.flyrnai.org/).
TALO8 minichromosome purification
The TALO8 minichromosome purification was done as previously described (35, 36) with minor modifications. The small size of the minichromosome (~2 kb) and the average presence of ~50 copies per cell ensure a high ratio of promoter-associated proteins relative to other chromatin-associated proteins. We introduced circular minichromosomes containing a selectable marker (TRP1), an origin of replication (ARS1), tandem repeats of lactose operators (LacO), and the promoter region of the 35S rDNA into a host strain expressing a FLAG-LacI fusion protein from a constitutive promoter. Using these strains, each purification was done using a 10 liters of yeast culture grown overnight at 30°C to mid-log phase (OD600nm, 0.6 to 0.8). Whole-cell extracts were made using a cryogenic grinder (Mixer Mills MM 400, Retsch) with the following setting: five cycles each at 30 Hz with a 1-min break in between followed by freezing the sample in liquid nitrogen. After resuspending the cell powder in the appropriate buffer volume, the procedure was carried out following the original protocol. Purified minichromosomes were analyzed by agarose gel electrophoresis, and proteins were visualized by silver staining on a 10% (v/v) SDS polyacrylamide gel and Western blot. Finally, the eluates were purified (trichloroacetic acid precipitation) and submitted for MS analysis [Taplin Mass Spectrometry Core Facility (https://taplin.med.harvard.edu)]. Data are available in table S4 and Raw images S1 to S4.
RNA extraction and qRT-PCR analysis
For qRT-PCR, RNA was isolated from yeast cells using standard laboratory techniques, treated with DNase (TURBO DNA-free Kit, Ambion), and 1 μg of RNA was reverse-transcribed using random hexamers (SuperScript III First-Strand Kit, Invitrogen). The resulting complementary DNA (cDNA) was analyzed in triplicate and quantified by comparison to a standard curve, composed of 10-fold serial dilutions of wild-type cDNA using the Stratagene Mx3000P according to the manufacturer’s instructions. Shown is the mean and SD for at least three independent experiments. Primers used for qRT-PCR amplification are listed in table S9, and primer pairs produced only one amplification band (140 to 250 bp) when tested by conventional RT-PCR. The specificity of individual qRT-PCR products was assessed by melting curve analysis. Control reactions (that is, PCR amplification of total RNA without RT) were performed in parallel. The signal at regions of interest was normalized to PMA1. For plotting the data, combined data were normalized to wild-type values.
Total RNA extractions of Drosophila KC cells (3 days after dsRNA treatment), third instar wandering larvae, and adult flies were performed with the TRIzol reagent (Life Technologies) following the manufacturer’s recommendations. For extraction of RNA out of whole larvae and adults, five animals per genotype were homogenized in TRIzol using a Bullet Blender (Next Advance). After RNA purification, 1 μg of RNAwas reverse-transcribed and used in the qRT-PCR as described above. qPCR primers used for quantifying pre-rRNA in Drosophila map to the internal transcribed spacer 1 and were designed after a probe used by Fichelson et al. (50) [values were normalized to α-tubulin, rpl32, GAPDH, or nuclear fallout (CG33991); used primers are listed in table S9].
Chromatin immunoprecipitation
ChIPs were performed as previously described (37), with minor modifications. Briefly, 100 ml of yeast culture was grown in YPD to mid-log phase (OD600nm, 0.6 to 0.8), cross-linked in 1% (v/v) formaldehyde for 30 min, and quenched with 0.115 M glycine. Cells were then lysed by bead beating, and the chromatin fraction was sheared to 200- to 500-bp fragments using a Bioruptor sonicator (Diagenode) (twelve 30-s pulses, with a 90-s break between pulses). For immunoprecipitations, 5 μl of anti-myc antibody A14 (Santa Cruz Biotechnology) or 1 μl of anti–histone H3 antibody (Abcam) was incubated overnight at 4°C with the chromatin extracts (500 μg of soluble lysate in 1 ml of lysis buffer) and then coupled for 3 to 5 hours at 4°C to 50 μl of 50% (w/v) Protein G–Sepharose 4 Fast Flow (GE Healthcare Life Sciences). After washing, samples were eluted, and cross-linking was reversed at 65°C overnight. Samples were subjected to proteinase K digestion and phenol-chloroform extraction, and the DNA was then precipitated. For input samples, cross-linking was reversed for 5 μg of soluble lysate in parallel with the immunoprecipitation samples described above. ChIP DNA was quantified by real-time PCR, using a Stratagene MX3000P. qPCR was done in triplicate for each primer set using a standard curve that was established by serial 10-fold dilutions of a representative input DNA from wild type. To determine the specificity of enrichment of the tagged proteins Hir1-myc and Hir2-myc, respectively, DNA was immunoprecipitated using an anti-myc antibody (A14; Santa Cruz Biotechnology), and the corresponding untagged control samples were included in each ChIP experiment. For determining the occupancy of histone H3, DNA was immunoprecipitated using an anti–histone H3 antibody (Abcam). The specificity of histone H3 binding was controlled by a “no-antibody” precipitation from the same chromatin extracts. The primer sequences are listed in table S9.
Immunohistochemistry
Staining of yeast cells was performed by growing cells into log phase and subsequent fixation with 5% formalin for 1 hour. Thereafter, cells were washed in a 1.2 M sorbitol, 0.1 M KPi (pH 7.5) buffer. After a 40-min zymolyase treatment, cells were plated on a poly-lysine–coated slide (1 mg/ml) and treated with methanol and acetone. After washing and blocking steps, cells were incubated with the primary antibody overnight, and then subsequent steps were carried out following a standard staining protocol. Staining of Drosophila salivary glands and ovaries was performed with standard methods as previously described in (52). The size of the nucleolus was quantified by imaging z-axis stacks through nuclei in third instar salivary gland cells, followed by a maximum intensity projection and a subsequent measurement of the area using the ImageJ software package. For statistical analysis, unless otherwise stated, two-tail unpaired Student’s t test was applied.
GO term and complex enrichment analysis
For the protein complex enrichment, we used COMPLEAT, a protein complex–based enrichment analysis tool to analyze the S. cerevisiae SGA screen and D. melanogaster RNAi data (34). COMPLEAT uses comprehensive protein complex resources generated for D. melanogaster and S. cerevisiae by (i) compiling literature-based protein complexes and (ii) predicting protein complexes from PPI networks. The resources consist of 6703 and 7713 protein complexes for S. cerevisiae and D. melanogaster, respectively, which cover almost 70% of proteins in S. cerevisiae and 50% of Drosophila proteins. For both S. cerevisiae and D. melanogaster data, we run the tool without preselecting hits (that is, complete genome-wide data were used as input data). The tool maps the distance (Δ) (S. cerevisiae) or z scores (D. melanogaster) of individual proteins or genes from the screen data sets to the complexes, and then a complex score is determined by calculating the interquartile mean:
where n denotes the number of proteins in the complex, and xi is the score (Δ or z score) of the ith protein in the complex.
The complex score directly reflects whether the complex is a positive or negative regulator of nucleolar size and preserves the magnitude of the Δ and the z score of the individual components. Furthermore, a P value is computed to estimate the significance of complex scores as compared to simulations of 1000 random complexes of the same size. The enriched complexes were visualized using the Cytoscape network visualization software.
For the comparison across D. melanogaster and S. cerevisiae complexes, we used COMPLEAT to identify commonly required complexes that regulate nucleolar size in both species. First, we mapped the z score from the D. melanogaster RNAi data to the S. cerevisiae ortholog genes using DIOPT, an ortholog mapping tool (http://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) (79). In case of one-to-many ortholog relationships (that is, a Drosophila gene mapped to multiple yeast genes), we used DIOPT scores to select the best ortholog match. Next, we performed the complex enrichment analysis on these data (RNAi data mapped from D. melanogaster to S. cerevisiae) using yeast complex resource. This step enables direct comparison of enriched complexes from the SGA screen and RNAi data sets because both enrichment analyses use the yeast complex resource. Similarly, we performed enrichment analysis using the Drosophila complex resource, where the yeast deletion screen data were mapped to Drosophila orthologous genes.
To perform a GO term enrichment, positive and negative nucleolar size regulators in D. melanogaster were analyzed using the fly genome as a background for GO term enrichment tests using the DAVID Bioinformatics Resources. S. cerevisiae data were analyzed in a similar way. The portion of the genome not covered by the screen was subtracted from the background set. Besides the enrichment analysis using the S. cerevisiae–positive and S. cerevisiae–negative hits, we also analyzed the yeast genes not covered in the SGA to identify the gene groups underrepresented in the yeast data set. Enriched GO terms were compared either directly or through immediate parent terms across positive and negative hits within a species or between D. melanogaster and S. cerevisiae data sets. Heat maps display the absolute value of the natural log–converted P values.
All analyses were performed in R environment (v. 2.15) and Perl (scripts are available upon request).
Optical density, flow cytometry, and cell size analysis
Cell growth rate was determined by online optical density measurement. Briefly, loss-of-function mutant strains harboring a KanMX-mediated deletion were subcultured from log-phase growth into optical-bottom 96-well plates containing 100 μl of fresh low-fluorescence medium. Cells were incubated at 30°C with intermittent shaking, and OD600nm was measured every 15 min for 35 hours using a GENios Plate Reader (Tecan). Growth rate was determined by building a linear model from 13 optical density measurements (3.25 hours) selected from mid-log phase growth.
Cell cycle distribution was determined by flow cytometry. Cells were grown at 30°C with shaking to mid-log phase in low-fluorescence medium in 24-well blocks containing a single borosilicate glass bead to aid mixing. Cells (100 μl) were ethanol-fixed, treated serially with RNase A and Proteinase K, and stained with SYTOX Green (Invitrogen). Fluorescence measurements were recorded using an LSR II flow cytometer (Becton Dickinson). A total of 50,000 single-cell measurements were recorded per strain. 1c and 2c DNA populations were analyzed using ModFit LT 3.3 (Verity Software House). Ploidy was determined by modal DNA content compared to a haploid reference. The ratio of G1:G2 cells is shown.
Cell size distribution in mid-log phase was determined directly from the cultures used to measure cell cycle distribution, above. One milliliter of culture was diluted into isotonic buffer, sonicated briefly, and a volume equivalent to 100 μl of original culture was measured through a calibrated 50-μm aperture on a Coulter Counter Z2 channelizer (Beckman Coulter Particle Characterization). Median cell volumes are shown.
Supplementary Material
Acknowledgments
We wish to thank B. Loppin, the Bloomington Drosophila Stock Center, the DRSC, the Vienna Drosophila RNAi Center, and the NIG for reagents; A. Kloimwieder for help with establishing the yeast nucleolus query strain; S. Mohr, Q. Gilly, and I. Flockhart for advice during the Drosophila RNAi screen; T. Tsukiyama for providing reagents and advice for the TALO8 method; S. Mohr for critically reading the manuscript; and all laboratory members of the Perrimon and Winston laboratories for discussions.
Funding: R.A.N. was supported by a European Molecular Biology Organization long-term fellowship and a Human Frontier Science Program long-term fellowship. T.G. was supported by a research fellowship (GR 3823/1-1) from German Research Foundation (DFG). This work was supported by RO1-GM067761, which supports the DRSC; R01-DK088718 and P01-CA120964 (N.P.); and RO1-GM045720 (F.W.). SGA screening and high-throughput imaging of yeast arrays were supported by grants MOP-97939 and MOP-275194 from the Canadian Institutes of Health Research to B.A. and Charles Boone. K.F. was supported by a PGS-A award from the National Sciences and Engineering Research Council and an Ontario Graduate Scholarship. N.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author contributions: R.A.N., T.G., F.W., and N.P. conceived and designed the study. R.A.N. performed the Drosophila RNAi screen with the help of M.B. and the yeast SGA screen with the help of K.F. R.A.N. performed the Drosophila HIR complex experiments. T.G. performed the yeast HIR complex experiments and the TALO8 purification. A.A.S., A.V., Y.H., and R.A.N. performed the bio-informatics analyses. R.A.N., T.G., F.W., B.A., and N.P. wrote the manuscript, and all authors discussed the results and edited the manuscript.
Competing interests: The authors declare that they have no competing interests.
Materials and data availability: The RNAi screen data are available at http://www.flyrnai.org/. The TALO8 MS data are shown in Supplementary table S4 and Raw images S1 to S4.
REFERENCES AND NOTES
- 1.Pianese G. Beitrag zur Histologie und Aetiologie des Carcinoms. Histologische und experimentelle Untersuchungen. Beitr Pathol Anat. 1896;142:1. [Google Scholar]
- 2.Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 3.Pich A, Chiusa L, Margaria E. Prognostic relevance of AgNORs in tumor pathology. Micron. 2000;31:133–141. doi: 10.1016/s0968-4328(99)00070-0. [DOI] [PubMed] [Google Scholar]
- 4.Boisvert FM, van Koningsbruggen S, Navascués J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 5.Prober DA, Edgar BA. Growth regulation by oncogenes—New insights from model organisms. Curr Opin Genet Dev. 2001;11:19–26. doi: 10.1016/s0959-437x(00)00151-9. [DOI] [PubMed] [Google Scholar]
- 6.Rudra D, Warner JR. What better measure than ribosome synthesis? Genes Dev. 2004;18:2431–2436. doi: 10.1101/gad.1256704. [DOI] [PubMed] [Google Scholar]
- 7.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 8.Tsang CK, Bertram PG, Ai W, Drenan R, Zheng XF. Chromatin-mediated regulation of nucleolar structure and RNA Pol I localization by TOR. EMBO J. 2003;22:6045–6056. doi: 10.1093/emboj/cdg578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arabi A, Wu S, Ridderstråle K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 10.Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 11.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 12.Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–1253. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Neumüller RA, Betschinger J, Fischer A, Bushati N, Poernbacher I, Mechtler K, Cohen SM, Knoblich JA. Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature. 2008;454:241–245. doi: 10.1038/nature07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumüller RA, Knoblich JA. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009;23:2675–2699. doi: 10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Y, Lu B. Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 2011;25:2644–2658. doi: 10.1101/gad.171959.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, Huser N, Proffitt C, Bliesath J, Haddach M, Schwaebe MK, Ryckman DM, Rice WG, Schmitt C, Lowe SW, Johnstone RW, Pearson RB, McArthur GA, Hannan RD. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, Lim JK, Von Hoff D, Anderes K, Rice WG. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 18.Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, Andrews B, Boone C. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomycespombe. Methods Enzymol. 2010;470:145–179. doi: 10.1016/S0076-6879(10)70007-0. [DOI] [PubMed] [Google Scholar]
- 19.Claypool JA, French SL, Johzuka K, Eliason K, Vu L, Dodd JA, Beyer AL, Nomura M. Tor pathway regulates Rrn3p-dependent recruitment of yeast RNA polymerase I to the promoter but does not participate in alteration of the number of active genes. Mol Biol Cell. 2004;15:946–956. doi: 10.1091/mbc.E03-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahajan PB. Modulation of transcription of rRNA genes by rapamycin. Int J Immunopharmacol. 1994;16:711–721. doi: 10.1016/0192-0561(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 21.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- 22.Powers T, Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaragoza D, Ghavidel A, Heitman J, Schultz MC. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol Cell Biol. 1998;18:4463–4470. doi: 10.1128/mcb.18.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, Vandersluis B, Bellay J, Devit M, Fleming JA, Stephens A, Haase J, Lin ZY, Baryshnikova A, Lu H, Yan Z, Jin K, Barker S, Datti A, Giaever G, Nislow C, Bulawa C, Myers CL, Costanzo M, Gingras AC, Zhang Z, Blomberg A, Bloom K, Andrews B, Boone C. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol. 2011;29:361–367. doi: 10.1038/nbt.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oakes M, Nogi Y, Clark MW, Nomura M. Structural alterations of the nucleolus in mutants of Saccharomyces cerevisiae defective in RNA polymerase I. Mol Cell Biol. 1993;13:2441–2455. doi: 10.1128/mcb.13.4.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mann C, Buhler JM, Treich I, Sentenac A. RPC40, a unique gene for a subunit shared between yeast RNA polymerases A and C. Cell. 1987;48:627–637. doi: 10.1016/0092-8674(87)90241-8. [DOI] [PubMed] [Google Scholar]
- 28.Moorefield B, Greene EA, Reeder RH. RNA polymerase I transcription factor Rrn3 is functionally conserved between yeast and human. Proc Natl Acad Sci USA. 2000;97:4724–4729. doi: 10.1073/pnas.080063997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto RT, Nogi Y, Dodd JA, Nomura M. RRN3 gene of Saccharomyces cerevisiae encodes an essential RNA polymerase I transcription factor which interacts with the polymerase independently of DNA template. EMBO J. 1996;15:3964–3973. [PMC free article] [PubMed] [Google Scholar]
- 30.Dasgupta A, Sprouse RO, French S, Aprikian P, Hontz R, Juedes SA, Smith JS, Beyer AL, Auble DT. Regulation of rRNA synthesis by TATA-binding protein-associated factor Mot1. Mol Cell Biol. 2007;27:2886–2896. doi: 10.1128/MCB.00054-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teixeira MT, Dujon B, Fabre E. Genome-wide nuclear morphology screen identifies novel genes involved in nuclear architecture and gene-silencing in Saccharomyces cerevisiae. J Mol Biol. 2002;321:551–561. doi: 10.1016/s0022-2836(02)00652-6. [DOI] [PubMed] [Google Scholar]
- 32.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 33.Clemente-Blanco A, Mayán-Santos M, Schneider DA, Machín F, Jarmuz A, Tschochner H, Aragón L. Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature. 2009;458:219–222. doi: 10.1038/nature07652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinayagam A, Hu Y, Kulkarni M, Roesel C, Sopko R, Mohr SE, Perrimon N. Protein complex–based analysis framework for high-throughput data sets. Sci Signal. 2013;6:rs5. doi: 10.1126/scisignal.2003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akiyoshi B, Nelson CR, Ranish JA, Biggins S. Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev. 2009;23:2887–2899. doi: 10.1101/gad.1865909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unnikrishnan A, Gafken PR, Tsukiyama T. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol. 2010;17:430–437. doi: 10.1038/nsmb.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, Moazed D. Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev. 2003;17:2162–2176. doi: 10.1101/gad.1108403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97:245–256. doi: 10.1016/s0092-8674(00)80734-5. [DOI] [PubMed] [Google Scholar]
- 39.Strunnikov A. Cdc14p regulates condensin binding to rDNA. Cell Cycle. 2009;8:1114. doi: 10.4161/cc.8.8.8469. [DOI] [PubMed] [Google Scholar]
- 40.Cosentino GP, Schmelzle T, Haghighat A, Helliwell SB, Hall MN, Sonenberg N. Eap1p, a novel eukaryotic translation initiation factor 4E-associated protein in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4604–4613. doi: 10.1128/mcb.20.13.4604-4613.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD. RSC, an essential, abundant chromatin-remodeling complex. Cell. 1996;87:1249–1260. doi: 10.1016/s0092-8674(00)81820-6. [DOI] [PubMed] [Google Scholar]
- 42.Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N. Heidelberg Fly Array Consortium, Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- 43.Cui Z, DiMario PJ. RNAi knockdown of Nopp140 induces. Minute-like phenotypes in Drosophila. Mol Biol Cell. 2007;18:2179–2191. doi: 10.1091/mbc.E07-01-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grewal SS, Evans JR, Edgar BA. Drosophila TIF-IA is required for ribosome synthesis and cell growth and is regulated by the TOR pathway. J Cell Biol. 2007;179:1105–1113. doi: 10.1083/jcb.200709044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulkarni MM, Booker M, Silver SJ, Friedman A, Hong P, Perrimon N, Mathey-Prevot B. Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods. 2006;3:833–838. doi: 10.1038/nmeth935. [DOI] [PubMed] [Google Scholar]
- 46.Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443:359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- 47.Reichow SL, Hamma T, Ferré-D’Amaré AR, Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. doi: 10.1093/nar/gkl1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Nues RW, Granneman S, Kudla G, Sloan KE, Chicken M, Tollervey D, Watkins NJ. Box C/D snoRNP catalysed methylation is aided by additional pre-rRNA base-pairing. EMBO J. 2011;30:2420–2430. doi: 10.1038/emboj.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, Russell J, Lee SC, Zomerdijk JC. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J. 2009;28:854–865. doi: 10.1038/emboj.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fichelson P, Moch C, Ivanovitch K, Martin C, Sidor CM, Lepesant JA, Bellaiche Y, Huynh JR. Live-imaging of single stem cells within their niche reveals that a U3snoRNP component segregates asymmetrically and is required for self-renewal in Drosophila. Nat Cell Biol. 2009;11:685–693. doi: 10.1038/ncb1874. [DOI] [PubMed] [Google Scholar]
- 51.Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet. 2008;40:476–483. doi: 10.1038/ng.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neumüller RA, Richter C, Fischer A, Novatchkova M, Neumüller KG, Knoblich JA. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell. 2011;8:580–593. doi: 10.1016/j.stem.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amin AD, Vishnoi N, Prochasson P. A global requirement for the HIR complex in the assembly of chromatin. Biochim Biophys Acta. 2012;1819:264–276. doi: 10.1016/j.bbagrm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 54.Green EM, Antczak AJ, Bailey AO, Franco AA, Wu KJ, Yates JR, III, Kaufman PD. Replication-independent histone deposition by the HIR complex and Asf1. Curr Biol. 2005;15:2044–2049. doi: 10.1016/j.cub.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osley MA, Lycan D. Trans-acting regulatory mutations that alter transcription of Saccharomyces cerevisiae histone genes. Mol Cell Biol. 1987;7:4204–4210. doi: 10.1128/mcb.7.12.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prochasson P, Florens L, Swanson SK, Washburn MP, Workman JL. The HIR corepressor complex binds to nucleosomes generating a distinct protein/DNA complex resistant to remodeling by SWI/SNF. Genes Dev. 2005;19:2534–2539. doi: 10.1101/gad.1341105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu H, Kim UJ, Schuster T, Grunstein M. Identification of a new set of cell cycle-regulatory genes that regulate S-phase transcription of histone genes in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:5249–5259. doi: 10.1128/mcb.12.11.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman KB, Karns LR, Lutz KA, Smith MM. Histone H3 transcription in Saccharomyces cerevisiae is controlled by multiple cell cycle activation sites and a constitutive negative regulatory element. Mol Cell Biol. 1992;12:5455–5463. doi: 10.1128/mcb.12.12.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Osley MA, Gould J, Kim S, Kane MY, Hereford L. Identification of sequences in a yeast histone promoter involved in periodic transcription. Cell. 1986;45:537–544. doi: 10.1016/0092-8674(86)90285-0. [DOI] [PubMed] [Google Scholar]
- 60.Kirov N, Shtilbans A, Rushlow C. Isolation and characterization of a new gene encoding a member of the HIRA family of proteins from Drosophila melanogaster. Gene. 1998;212:323–332. doi: 10.1016/s0378-1119(98)00143-7. [DOI] [PubMed] [Google Scholar]
- 61.Loppin B, Bonnefoy E, Anselme C, Laurençon A, Karr TL, Couble P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437:1386–1390. doi: 10.1038/nature04059. [DOI] [PubMed] [Google Scholar]
- 62.Bonnefoy E, Orsi GA, Couble P, Loppin B. The essential role of Drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS Genet. 2007;3:1991–2006. doi: 10.1371/journal.pgen.0030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aït-Ahmed O, Bellon B, Capri M, Joblet C, Thomas-Delaage M. The yemanuclein-α: A new Drosophila DNA binding protein specific for the oocyte nucleus. Mech Dev. 1992;37:69–80. doi: 10.1016/0925-4773(92)90016-d. [DOI] [PubMed] [Google Scholar]
- 64.Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, Vershilova E, Pirrotta V, Kadonaga JT, Lusser A, Fyodorov DV. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087–1090. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- 66.Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- 67.Schneiderman JI, Orsi GA, Hughes KT, Loppin B, Ahmad K. Nucleosome-depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc Natl Acad Sci USA. 2012;109:19721–19726. doi: 10.1073/pnas.1206629109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferreira-Cerca S, Pöll G, Kühn H, Neueder A, Jakob S, Tschochner H, Milkereit P. Analysis of the in vivo assembly pathway of eukaryotic 40S ribosomal proteins. Mol Cell. 2007;28:446–457. doi: 10.1016/j.molcel.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 69.Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010;8:e1000522. doi: 10.1371/journal.pbio.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laferté A, Favry E, Sentenac A, Riva M, Carles C, Chédin S. The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev. 2006;20:2030–2040. doi: 10.1101/gad.386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thiry M, Lafontaine DL. Birth of a nucleolus: The evolution of nucleolar compartments. Trends Cell Biol. 2005;15:194–199. doi: 10.1016/j.tcb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 74.Russell J, Zomerdijk JC. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci. 2005;30:87–96. doi: 10.1016/j.tibs.2004.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grummt I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17:1691–1702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- 76.McStay B, Grummt I. The epigenetics of rRNA genes: From molecular to chromosome biology. Annu Rev Cell Dev Biol. 2008;24:131–157. doi: 10.1146/annurev.cellbio.24.110707.175259. [DOI] [PubMed] [Google Scholar]
- 77.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 78.Flockhart IT, Booker M, Hu Y, McElvany B, Gilly Q, Mathey-Prevot B, Perrimon N, Mohr SE. FlyRNAi.org—The database of the Drosophila RNAi screening center: 2012 update. Nucleic Acids Res. 2012;40:D715–D719. doi: 10.1093/nar/gkr953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.