

Abstract
Direct functionalization of a variety of quinones with several boronic acids has been developed. This scalable reaction proceeds readily at room temperature in an open flask using inexpensive reagents: catalytic silver(I) nitrate in the presence of a persulfate co-oxidant. The scope with respect to quinones is broad with a variety of alkyl- and arylboronic acids undergoing efficient cross-coupling. The mechanism is presumed to proceed through a nucleophilic radical addition to the quinone with in situ reoxidation of the resulting dihydroquinone. This method has been applied to complex substrates including a steroid derivative and a farnesyl natural product.
The quinone moiety possesses electron and proton transfer properties that are essential to nearly every living organism.1 This privileged class of structures pervades the realms of chemistry, material science, nanotechnology, and medicine.2 They are embedded in several natural products2b including several sesquiterpenes,3 the kinamycins4 and terphenylquinones.5 Additionally, there are several drugs and therapeutic leads that contain the quinone subunit.6 Arylated quinones possess unique visual and electronic properties that make them useful in photosynthesis7 and appealing structures to the dye industry.8 Quinones also play a key role in cell respiration.9 It is somewhat surprising therefore that methods for the direct functionalization of quinones are sparse.
Several common methods for C–C bond formation on α,β-unsaturated carbonyls (such as the addition of organometallics and Heck coupling) fail on quinones due to their unique electronic properties.2a When these methods do succeed, often a reoxidation step is necessary to revert to the quinone oxidation state. These difficulties have prompted chemists to employ other modes of functionalization including prehalogenation of the quinone followed by a palladium-catalyzed cross-coupling (2→1, Figure 1A),10 or radical coupling on the unfunctionalized quinone (3→1). However, halogenated quinones are often hard to access due to chemo- and regioselectivity issues during halogenation reactions. The ability of quinones to act as ligands and oxidants to transition metals such as palladium poses additional challenges to cross-coupling approaches.11 Alternative strategies employing protected halo-dihydroquinones followed by deprotection12 have proven both lengthy and inefficient as have Diels–Alder/functionalization/retro Diels–Alder sequences.13 In the case of the radical coupling approach, alkyl radicals successfully add to quinones,14 but cannot be generated via common methods such as alkyl halides/tin-hydrides due to competitive reduction of the quinone substrate.2a Aryl radicals also react with quinones,15 but generally can only be accessed through aryldiazonium salts which can be difficult to synthesize, are unstable, and have been shown to be explosive. For this reason, a more general method for the functionalization of quinones is in demand.
Figure 1.

(A) Traditional access to functionalized quinones. (B) Initial findings with respect to arylboronic acid reactivity.
Several groups have made notable advances in the direct C–H functionalization of quinones in recent years. An early communication from Itahara showed that palladium(II) acetate could mediate the coupling of benzoquinone to an arene cosolvent at reflux.16 Engler used stoichiometric titanium to couple arenes bearing a cyclopropylsilicon moiety to quinones.17 Palladium has also been used to couple arylchlorides to benzo- and naphthoquinones.18 Csákÿ and Molina developed a method to directly arylate naphthoquinones with arylboronic acids in the presence of palladium/copper catalysis followed by reoxidation with FeCl3.19 Similarly, Demchuk showed that naphthoquinones could be functionalized with electron-rich aryltrifluoroborates under rhodium catalysis and elevated temperatures.20 Most recently, Renaud has accomplished the coupling of customized catecholboranes to quinones followed by reoxidation.21 Notably, arylboronic acids have recently been coupled to electron-deficient olefins in excess via a presumed radical mechanism with stoichiometric manganese(III) acetate (no quinone examples given).22 Inspired by our recent discovery of the silver-catalyzed addition of arylboronic acids to protonated heterocycles,23 and given the inherent reactivity of quinones with radicals,1,2 it was reasoned that the same reactive intermediate generated from the boronic acid precursors could interact with other electrophiles such as quinones. In addition, the oxidizing conditions used to generate radicals from boronic acids would serve the dual role of reoxidizing the dihydroquinonesgenerated generated from the radical addition step.
This prediction was actualized when 1,4-benzoquinone was exposed to tolylboronic acid (1.5 equiv) in the presence of catalytic silver(I) nitrate (0.2 equiv) and potassium persulfate (3.0 equiv). Within three hours, the reaction was complete and delivered the monoarylated quinone 4 in 72% yield after isolation (Figure 1B). This reaction, which is operationally trivial to perform, is run under air at room temperature, is scalable, and exhibits a broad substrate scope (vide infra). Since the inherent reactivity of the quinone is utilized, no prefunctionalization is required. The affordability of the reaction is dictated by the price of the coupling partners, as the catalyst (silver(I) nitrate) and stoichiometric oxidant (potassium persulfate) are very cheap (<$1.00 and <$0.01 per gram, respectively), a notable advantage over existing methods that require expensive oxidants and transition metal catalysts. The mono selectivity of these reactions is also notable: no bis-arylation was seen on quinone under these reaction conditions, even with the slight excess of boronic acid that is necessary to drive the reaction to completion.
It was found that the scope with respect to the boronic acid coupling partner was expansive, allowing for efficient reactivity with aryl- and alkylboronic acids (Table 1). Arylboronic acids with alkyl substitution at any position in the ring reacted well under the standard conditions (4, 6–11). A broad range of electronic properties were tolerated from electron-rich aryl ethers (16–24) to electron-withdrawn arylnitro- (29), arylcyano (25), aryltrifluoromethyl (26), and arylsulfate (27) functionalities. Halogenation at ortho-, meta-, and para- positions was compatible with the reaction conditions (12 – 17), including sensitive aryliodides (which would be incompatible to many transition-metal-based methods, and could serve as further functional handles). Several other functionalities were tolerated, including esters (28), ketones (30), and silanes (31). Alkylboronic acids also provided coupling products in decent to fair yields for both linear (34, 36), branched (35), and cyclic (37, 38) systems. It should be noted that the preparation of 34–38 from the corresponding alkyl Grignard or organolithium reagents failed to deliver products. Some cases where the yields were moderate are attributed to the highly unstable nature of the substituted quinone products (e.g. 25, 29, 39).
Table 1.
Scope of the coupling of alkyl- and arylboronic acids to 1,4-benzoquinone.a
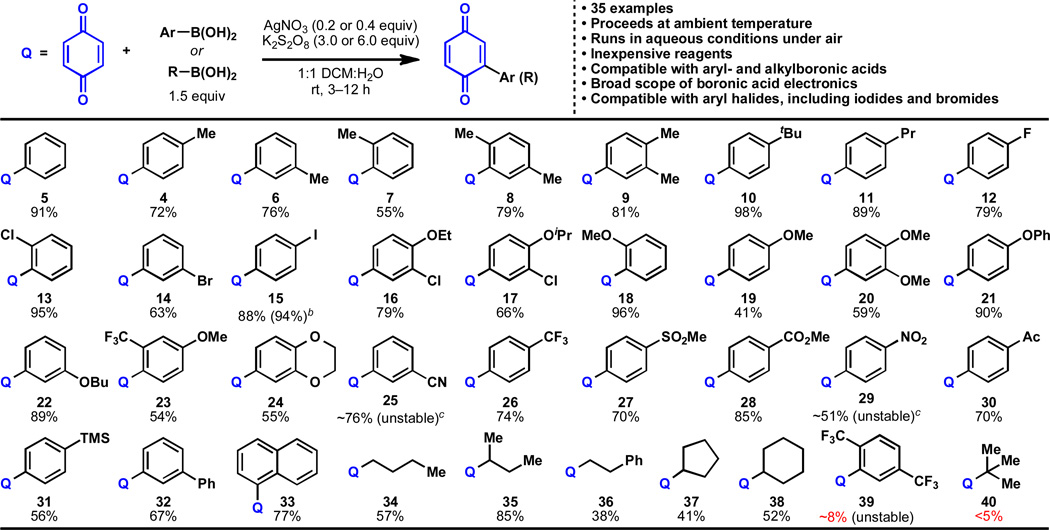 |
Benzoquinone (0.25 mmol), boronic acid (0.375 mmol), AgNO3 (0.05 mmol), K2S2O8 (0.75 mmol), 23 °C, 3–12 h; isolated yields of chromatographically and spectroscopically pure products yields displayed, unless otherwise noted. (NH4)2S2O8 is also suitable as an oxidant.
Yield of the reaction performed on gram-scale with no organic solvent (see Figure 2).
Products were unstable to several isolation conditions, but proved relatively stable in solution after extraction. See Supporting Information for more details.
Despite the high level of generality, there are a few limitations with regards to substrate scope. Very electron-poor arylboronic acids (e.g. 39) and very hindered alkylboronic acids (e.g. 40) reacted poorly. Benzylboronic acids were subject to oxidative decomposition under the reported conditions and failed to deliver product. Finally, vinylboronic acids failed to deliver appreciable amounts of products.
The reaction proved fairly general for quinone derivatives when reacted with p-tolylboronic acid (Table 2). Halogenation and additional oxidation were tolerated (41, 44, 45), as well as benzannulation of the quinone (46). Methyl substitution on the quinone resulted in decreased yield (42, 43). The slightly unpredictable yields with regards to quinone scope are undoubtedly due to the electronic effects that substitution can embark upon the quinone (a part of its hallmark reactivity, and the reason that mono selectivity is observed in Table 1).2
Table 2.
Scope of quinones for the arylation with p-tolylboronic acid.a
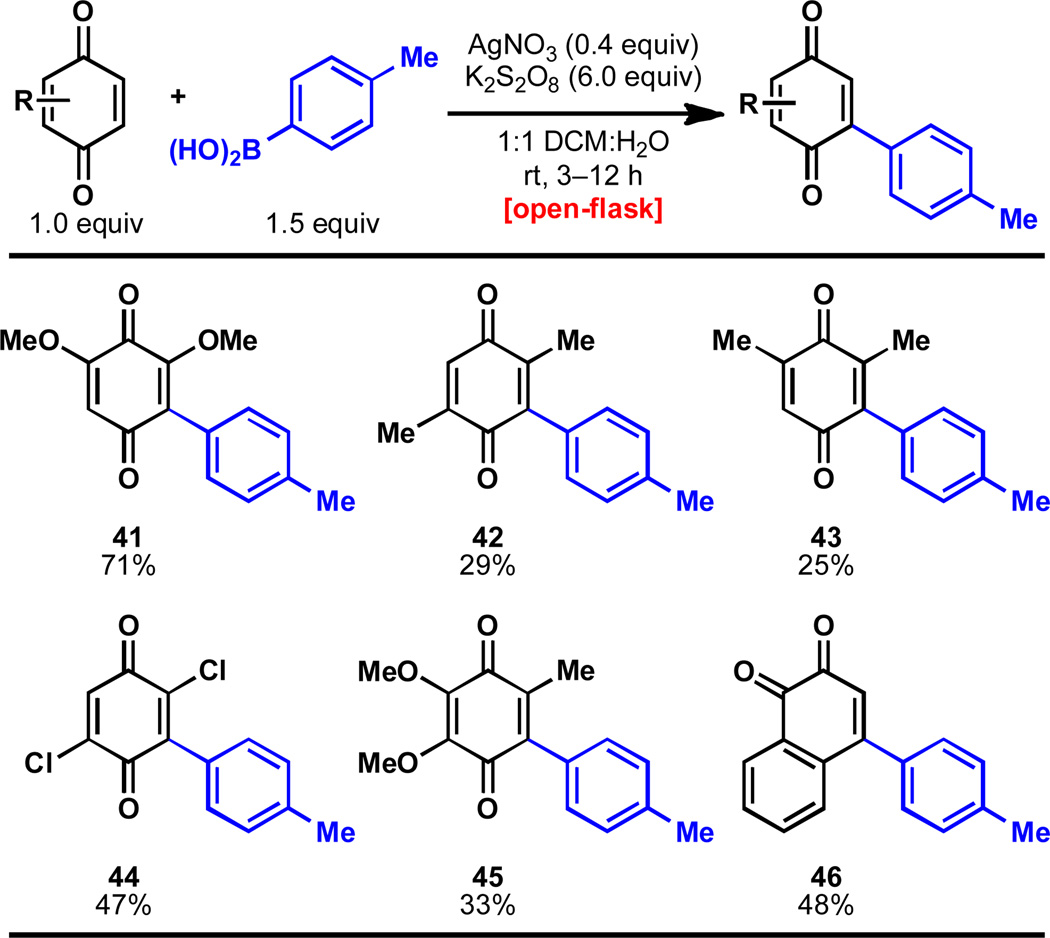 |
Quinone (0.25 mmol), p-tolylboronic acid (0.375 mmol), AgNO3 (0.05 mmol), K2S2O8 (0.75 mmol), 23 °C, 3–12 h; isolated yields of chromatographically and spectroscopically pure products displayed. For reactions over 3 h, a second addition of AgNO3 (0.05 mmol) and K2S2O8 (0.75 mmol) were added. (NH4)2S2O8 is also s uitable as an oxidant.
Two other notable variations of this mode of reactivity are shown in Figure 2. Aryltrifluoroborates reacted with identical efficiency under the standard conditions, albeit with a slightly lower rate, as demonstrated by the reaction of potassium phenyltrifluoroborate with 1,4-benzoquinone in 95% yield. Additionally, it was found that the organic layer of the biphasic solvent system could be omitted on large scale with no impact on yield, as demonstrated by the gram-scale reaction of 4-iodophenylboronic acid with 1,4-benzoquinone in 94% yield with no organic solvent. This is notable for both the scalability and environmental compatibility of the reported conditions.
Figure 2.

Exploration of Molander salts and purely aqueous conditions.
Perhaps the most striking demonstration of the utility of this reaction is displayed in the modification of large molecules of biological interest. Figure 3A shows the functionalization of estrone boronic acid (47)24 with 1,4-benzoquinone, delivering the estrone-benzoquinone hybrid 48 in 51% yield under the standard conditions. This reaction proceeds at room temperature without requiring any protection of the ketone unit on the steroid backbone. An example of allylboronic acid reactivity is shown in Figure 3B with the synthesis of the natural product 2-farnesyl-1,4-benzoquinone (51).25 The pinacolboronate of farnesol (49) was first converted into its Molander salt 50 (necessitated by the sluggish reactivity of the boronate ester itself). The crude Molander salt was then subjected to the standard conditions to directly attach a 1,4-benzoquinone in 58% yield. Some isomerization of the olefin closest to the boronate center in farnesol was observed during this reaction, supporting an allyl radical intermediate.
Figure 3.

Applications to natural products and natural product hybrids.
Despite the prevalence of quinones in several fields of science and technology, universal access to several quinone derivatives has been hampered by a lack of general methodologies relating to their synthesis. In fact, many of the products reported in this communication represent new chemical entities. The direct C–H arylation/alkylation of quinones reported herein proceeds at ambient temperature in an open flask, is scalable, can be run without organic solvents when desired, demonstrates broad scope, and is highly chemoselective. This powerfully simple chemical avenue to aryl- and alkylquinones will allow for facile and affordable access to these structural motifs, thus filling an important gap in the capabilities of organic synthesis.
Supplementary Material
Acknowledgement
We are grateful to Dr. Raju Mohan (Exelixis) for a generous donation of boronic acids. Financial support for this work was provided by the NIH/NIGMS (GM-073949), the NSF (predoctoral fellowship for I.B.S.), Japan Society for the Promotion of Science (JSPS, post-doctoral fellowship for Y.F.), the Spanish MICINN (predoctoral grant for V.D.), Amgen, and Bristol-Myers Squibb (unrestricted research support).
Footnotes
Supporting Information Available: Detailed experimental procedures, copies of all spectral data and full characterization. This material is available via the Internet at http://pubs.acs.org.
References
- 1.For a recent review, see: Nowicka B, Kruk J. Biochimica et Biophysica Acta. 2010;1797:1587. doi: 10.1016/j.bbabio.2010.06.007.
- 2.(a) Patai S, Rappoport Z, editors. The Chemistry of the Quinonoid Compounds Vol. 2, Parts 1 and 2. New York: Wiley; 1988. [Google Scholar]; (b) Thomson RH. Naturally Occuring Quinones IV. London: Blackie Academic; 1997. [Google Scholar]
- 3.Marcos IS, Conde A, Moro RF, Basabe P, Diez D, Urones JG. Mini-Reviews Med. Chem. 2010;7:230. [Google Scholar]
- 4.(a) Gould SJ. Chem. Rev. 1997;97:2499. doi: 10.1021/cr9600215. [DOI] [PubMed] [Google Scholar]; (b) Marco- Contelles J, Mokina MT. Curr. Org. Chem. 2003;7:1433. [Google Scholar]; (c) Kumamoto T, Ishikawa T, Omura S. Yuki Gosei Kagaku Kyokaishi. 2004;62:49. [Google Scholar]
- 5.(a) Cali V, Spatafora C, Tringali C. Studies in Nat. Prod. Chem. 2003;29:263–307. [Google Scholar]; (b) Liu J-K. Chem. Rev. 2006;106:2209. doi: 10.1021/cr050248c. [DOI] [PubMed] [Google Scholar]
- 6.For recent reviews, see: Koyama J. Recent Patents Anti-Infect. Drug Discov. 2006;1:113. doi: 10.2174/157489106775244073. Verma RP. Anti-Cancer Agents Med. Chem. 2006;6:489. doi: 10.2174/187152006778226512. Babula P, Adam V, Havel L, Kizek R. Ceska Slovens. Farm. 2007;56:114. Ferreira ICFR, Vaz JA, Vasconcelos MH, Martins A. Mini-Reviews Med. Chem. 2010;10:424. doi: 10.2174/1871520611009050424. Dandawate PR, Vyas AC, Padhye SB, Singh MW, Baruah JB. Mini-Reviews Med. Chem. 2010;10:436. doi: 10.2174/138955710791330909.
- 7.Cramer WA, Crofts AR. Electron and proton transport. In: Govindjee, editor. Photosynthesis, Vol 1. New York: Academic Press; 1982. pp. 387–467. [Google Scholar]
- 8.Bechtold T. Natural Colorants – quinoid, anthraquinoid, and naphthoquinoid dyes. In: Bechtold T, Mussak R, editors. Handbook of Natural Colorants. New York: Wiley; 2009. pp. 151–182. [Google Scholar]
- 9.Price CE, Driessen AJM. Biochimica et Biophysica Acta. 2010;1803:748. doi: 10.1016/j.bbamcr.2010.01.019. and references therein. [DOI] [PubMed] [Google Scholar]
- 10.For selected palladium-catalyzed couplings to halogenated quinones, see: Best WM, Sims CG, Winslade M. Aust. J. Chem. 2001;54:401. Lana EJL, Carazza F, de Oliveira RA. Helv. Chim. Acta. 2004;87:1825. Gan X, Jiang W, Wang W, Hu L. Org. Lett. 2009;11:589. doi: 10.1021/ol802645f.
- 11.For recent reviews, see: Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. Sun C-L, Li B-J, Shi Z-J. Chem. Commun. 2010;46:677. doi: 10.1039/b908581e.
- 12.For a paper with several examples of functionalization of protected dihydroquinones followed by reoxidation, see: Jung Y-S, Joe B-Y, Cho SJ, Konishi Y. Bioorg. Med. Chem. Lett. 2005;15:1125. doi: 10.1016/j.bmcl.2004.12.029.
- 13.For a recent example, see: Mehta G, Babu Khan T. Tetrahedron Lett. 2010;51:6590.
- 14.For original reports using carboxylic acids and Minisci conditions, see: Jacobsen N, Torssell K. Justus Liebegs Ann. Chem. 1972;763:165. Jacobsen N, Torssell K. Acta Chem Scan. 1973;27:3211. For Barton conditions, see: Barton DHR, Bridon D, Zard SZ. Tetrahedron. 1987;43:5307. For initial reports of the Mn(III) mediated addition of 1,3-dicarbonyls, see: Chuang C-P, Wang S-F. J. Chin. Chem. Soc. 1997;44:217. Chuang C-P, Wang S-F. Tetrahedron. 1998;54:10043. Jiang M-C, Chuang C-P. J. Org. Chem. 2000;65:5409. doi: 10.1021/jo991947q. For alkylation with trialkylboranes, see: Hawthorne MF, Reintjes M. J. Am. Chem. Soc. 1964;86:951. Hawthorne MF, Reintjes M. J. Am. Chem. Soc. 1965;87:4585. Bieber LW, Rolim Neto PJ, Generino RM. Tetrahedron Lett. 1999;40:4473. For reaction with acyl radicals, see: Lin Z-Y, Chen Y-L, Lee C-S, Chuang C-P. Eur. J. Org. Chem. 2010:3876.
- 15.For reviews on the Meerwein arylation, see: Rondestvedt CS., Jr Org. React. 1960;11:189. Rondestvedt CS., Jr Org. React. 1976;24:225. Galli C. Chem. Rev. 1988;88:765. Weis CD. Dyes Pigm. 1988;9:1. Heinrich MR. Chem. Eur. J. 2009;15:820. doi: 10.1002/chem.200801306.
- 16. Itahara T. Chem. Ind. 1982;16:599. Itahara T. J. Org. Chem. 1985;50:5546. Application to quinone library: de Oliveira RA, Carazza F, da Silva Pereira MO. Synth. Commun. 2000;30:4563.
- 17.Engler TA, Reddy JP. J. Org. Chem. 1991;56:6491. [Google Scholar]
- 18.Singh PK, Rohtagi BK, Khanna RN. Synth. Commun. 1992;22:987. [Google Scholar]
- 19.Molina MT, Navarro C, Moreno A, Csákÿ AG. Org. Lett. 2009;11:4938. doi: 10.1021/ol902084g. [DOI] [PubMed] [Google Scholar]
- 20.Demchuk OG, Michal Pietrusiewicz K. Synlett. 2009;7:1149. [Google Scholar]
- 21.Lüthy M, Darmency V, Renaud P. Eur. J. Org. Chem. 2010 In Press. [Google Scholar]
- 22.Dickschat A, Studer A. Org. Lett. 2010;12:3972. doi: 10.1021/ol101818k. [DOI] [PubMed] [Google Scholar]
- 23.Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J. Am. Chem. Soc. 2010;132:13194. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed V, Liu Y, Silvestro C, Taylor SD. Bioorg. Med. Chem. 2006;14:8564. doi: 10.1016/j.bmc.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 25.Pena LA, Avella E, Puentes de Diaz AM. Rev. Colomb. Quim. 2000;29:25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.