

Abstract
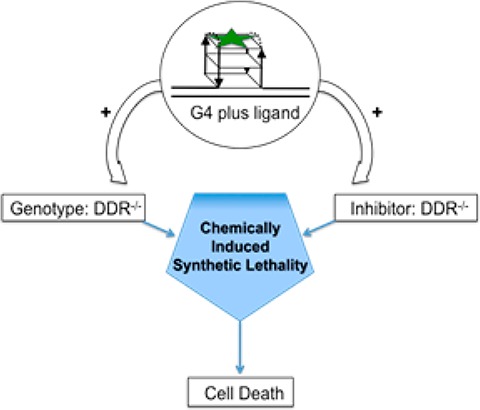
Synthetic lethality is a genetic concept in which cell death is induced by the combination of mutations in two sensitive genes, while mutation of either gene alone is not sufficient to affect cell survival. Synthetic lethality can also be achieved “chemically” by combination of drug-like molecules targeting distinct but cooperative pathways. Previously, we reported that the small molecule pyridostatin (PDS) stabilizes G-quadruplexes (G4s) in cells and elicits a DNA damage response by causing the formation of DNA double strand breaks (DSB). Cell death mediated by ligand-induced G4 stabilization can be potentiated in cells deficient in DNA damage repair genes. Here, we demonstrate that PDS acts synergistically both with NU7441, an inhibitor of the DNA-PK kinase crucial for nonhomologous end joining repair of DNA DSBs, and BRCA2-deficient cells that are genetically impaired in homologous recombination-mediated DSB repair. G4 targeting ligands have potential as cancer therapeutic agents, acting synergistically with inhibition or mutation of the DNA damage repair machinery.
G-quadruplexes (G4s) are structures that form from guanine-rich nucleic acid sequences via the formation of guanine tetrads stabilized by a central, coordinating cation.1 These stable noncanonical structures have been hypothesized to exist in nature, and there is evidence linking G4s to biological processes, including correlations from chemical biological studies.2 Computational analyses using predictive algorithms have suggested sites at which G4s may form in genomes,3 highlighting an over-representation at certain regulatory regions.4 The existence of G4s in living systems was first shown at the telomeres of ciliates,5 where the formation of such DNA structures was linked to a cell-cycle dependent phosphorylation of telomere binding proteins.6 More recently, we employed the small molecule G4-stabilizing ligand pyridostatin (PDS)7 to target G4s in the DNA of human cancer cells and explicitly mapped the functional response to sites in the genome by deep sequencing.8 Furthermore, we have visualized telomeric and nontelomeric G4s in human cancer cells using selective antibodies and demonstrated that PDS can trap DNA G4s in the cell nucleus.9 We have also mapped G4 structures in genomic DNA derived from human cancer cells.10 It is apparent that G4 structures exist in cells and can serve as targets for small molecules.
Treatment of cells with some G4-stabilizing small molecules can lead to the formation of DNA double strand breaks (DSBs).8,11 A single DSB is sufficient to kill a cell,12 and these lesions can be generally repaired by distinct repair systems: homologous recombination (HR) or nonhomologous end joining (NHEJ).13 Deficiencies or chemical inhibition of these pathways can usefully sensitize cells toward DNA damage induced by radiation exposure or topoisomerase II inhibition (e.g., etoposide).14
Chemically induced synthetic lethality is an attractive therapeutic strategy that exploits a genetically deficient disease state or its chemical inhibition combined with a complementary drug treatment, to enhance the overall inhibitory effect.15 For example, breast cancer cells carrying mutations in the gene BRCA2 are deficient in the HR repair pathway and are consequently particularly sensitive to chemical inhibitors of alternative DNA repair pathways.16 Brosh and co workers have also shown that small molecule inhibition of Werner syndrome helicase sensitizes cells to the G4 ligand telomestatin.17 Similarly, we showed that DSB formation induced by PDS treatment would be more pronounced in cells genetically deficient in, or chemically inhibited in, repair pathways.8 Thus, while DNA G4s are emerging as targets for cancer in their own right,18 these structures are potential targets for a chemically induced synthetic lethality strategy.
Herein, we investigate the potential of G4 ligands for the development of chemically induced synthetic lethality. Specifically, the antiproliferative effects of a PDS analogue exhibit synergy in either cells deficient in HR repair (BRCA2–/–) or in a system where NHEJ has been chemically inhibited.
Previously, we identified three G4 ligands from the PDS family, PDS, PDSI, and PDSK (Figure 1A, Table S1), as strong G4 stabilizers and potent inhibitors of cell proliferation.19 Using an impedance-based continuous cell-monitoring approach (see Supporting Information) we examined, in real time, the growth inhibition of human HT1080 fibrosarcoma cells, previously shown to be sensitive to the PDS family,19 in the presence of each ligand for up to 72 h. Of the three ligands, PDSI was found to exhibit the greatest growth inhibition (GI50: PDSI, 0.43 μM; PDS, 0.89 μM; PDSK, 3.15 μM; Figure 1B) and was therefore used in further studies.
Figure 1.

(A) Structures of pyridostatins: PDS, PDSI and PDSK. (B) Growth inhibition by PDS molecules in HT1080 cells (GI50 in μM). **P = 0.006, 1 way ANOVA. (C) Structure of DNA-PKcs inhibitor NU7441.
We next investigated DNA damage induced by PDSI in the presence of a complementary inhibitor 2-N-morpholino-8-dibenzothiophenyl-chromen-4-one (NU7441) that effectively targets the catalytic subunit of a DNA-dependent protein kinase (DNA-PKcs), which is required for the NHEJ mode of DNA repair (Figure 1C).20 We confirmed that PDSI induces a DNA damage response in HT1080 cells, as judged by an increase in phosphorylated-53BP121 and a dose dependent increase in apoptotic cell death (Figures S1–2). Co-treatment of PDSI with NU7441 resulted in a greater number of 53BP1 foci, suggesting that DNA damage induced by PDSI is exacerbated when a DNA repair inhibitor is present (Figure S1B).
To investigate this further, we systematically examined the combination effect on cell survival of HT1080 cells treated with PDSI and NU7441. Typically, cells were cultured in a matrix of compound concentrations ranging from 0 to 10 times the determined GI50 values (Table S1). Cell survival was measured at 72 h by an end-point assay (CellTiter-Glo) that uses cellular ATP as a substrate for a luciferase, where luminescence readings are proportional to the number of viable cells. Using this system we can investigate if the combination of the two agents acts synergistically; where synergism is defined as providing a greater response than the simple addition of single treatments. This end-point assay measures cell viability/survival and not cell growth; the values cannot be directly compared to the GI50 presented above.
Since PDSI and NU7441 have distinct targets (i.e., DNA G4 structure for PDSI and the DNA-PK protein for NU7441), we used the Bliss independence model for calculating additivity, Z = X + Y(1 – X), where Z represents the expected combined output, X the cell survival of PDSI alone, and Y the cell survival of NU7441 alone.22 The calculated additivity is arithmetically subtracted from the experimentally measured cell survival and plotted as percentage synergy on a resultant 3D response surface using the program SynergySurface (See Supporting Information, Figure 2A); a positive peak indicates a synergistic interaction, and a negative peak indicates antagonism. The combination of PDSI and NU7441 (Figure 2A) showed a very high level of synergy consistent with chemically induced synthetic lethality. The optimum concentrations for synergy were 2.5 μM PDSI and 0.3 μM NU7441 resulting in a 45% synergy. This compares favorably with other synthetic lethal studies, such as the VE-821 aminopyrazine ATR inhibitor and cisplatin (0.3–10 μM range with ∼45% synergy).23 VE-821 also acts synergistically with other DNA damaging agents including carboplatin, etoposide, and camptothecin at similar dose ranges to this study.23a We confirmed that a DNA damage response (DDR) was induced at 2 μM PDSI and 0.3 μM NU7441 by measuring increases in levels of nuclear 53BP1, indicative of DSB formation, and of nuclear RAD51, indicating HR induction, by laser-scanning cytometry (Figure 2B,C, Table S3) after 24 h treatment. Treatment with PDSI or NU7441 alone increased levels of nuclear 53BP1 by up to 1.4-fold, whereas synergistic combinations led to a greater increase of 2.1-fold (Figure 2B, Table S3). Likewise, combination of PDSI and NU7441 resulted in a 30-fold increase of nuclear RAD51 levels compared to single treatment of NU7441 and a 1.5-fold increase relative to PDSI alone, which is greater than a solely additive response (Figure 2C, Table S3). NU7441 does not promote HR-mediated repair in the absence of PDSI, in agreement with a study that revealed potential inhibition of HR by NU7441.24
Figure 2.

Synergistic growth inhibition of cancer cells using PDSI and NU7441. (A) 3D response surface for the combination of PDSI with NU7441 in HT1080 cells. (B) Nuclear 53BP1 and (C) RAD51 in HT1080 cells treated with PDSI (2 μM) and NU7441 (300 nM) for 24 h. Error bars indicate SEM. (D) Growth inhibition by PDSI and NU7441 in isogenic HCT116 WT or BRCA2–/– cancer cells. Binary treatment was NU7441 (1 μM) with a range of PDSI concentrations (binary GI50). All GI50 data quoted in μM. Significance compared to PDSI (unpaired t test, one tail, *P < 0.05, **P < 0.01). (E,F) 3D response surfaces for the combination of PDSI with NU7441 in HCT116 WT cells or BRCA2–/– cells. (G) Nuclear 53BP1 and (H) RAD51 in HCT116 WT and BRCA2–/– cells treated with PDSI (2 μM) and NU7441 (150 nM) for 24 h. Dashed lines in C/H denote level of additive response. Details of statistical significance between pair wise combinations, for panels B, C, G, and H is illustrated in Table S3. Data in Panels A, E, and F were measured using an end point luminescence assay. Data in panel D were measured by continuous cell monitoring.
To complement the chemically induced synthetic lethality described above, we evaluated the effect of a G4 ligand in cells genetically lacking a vital component of the DNA damage repair machinery. BRCA2 is a key gene implicated in many cancers and is clinically treated using synthetic lethal rationales with PARP inhibitors.16 We reasoned that BRCA2-deficient cells would be more sensitive to G4 ligands than wild-type (WT) cells. PDSI and/or NU7441 were therefore applied to a pair of isogenic HCT116 colon carcinoma cells carrying either the WT BRCA2+/+ gene or where both copies have been removed by homologous recombination (BRCA2–/–).25 In BRCA2–/– cells, PDSI induced a greater inhibition of cell growth compared to WT (GI50: 1.8 vs 3.8 μM, Figure 2D, Table S2) and NU7441 had a similar response (GI50: 2.2 vs 4.8 μM); thus confirming that cells deficient in DNA repair are more sensitive to PDSI. Combination treatments also gave a synergistically greater cell death than WT cells as indicated by I values (I < 1 for synergy, I = 1 for additivity, and I > 1 for antagonism, Figure S3).26 This was seen in both BRCA2–/– and WT cells (binary GI50: 0.28 vs 1.7 μM, respectively). Importantly, the growth of BRCA2–/– cells in the presence of combinations of PDSI and NU7441 is severely compromised (Figure S4).
Using the 3D growth response analysis at 72 h as described above, we further explored this synergy using a matrix of concentrations (Figure 2E,F). These data indicate that the absence of HR in the BRCA2–/– cells potentiates the synergistic effects of PDSI and NU7441. We further confirmed a substantially larger increase in nuclear RAD51 levels in BRCA2–/– cells following treatment with PDSI or in combination with NU7441, compared to untreated and WT cells (Figure 2H, Table S3).
In conclusion, we have demonstrated that a G4 ligand can induce synthetic lethality in cancer cells either by exploiting the inherent HR DSB repair deficiency in BRCA2–/– cells or by accompanying a second inhibitor that targets a similarly vital DSB repair mechanism (NHEJ). G4 ligands have considerable potential as efficacious antiproliferative agents when used as part of a rational synergistic strategy.
Acknowledgments
We thank Cancer Research U.K. for program funding and core support.
Supporting Information Available
All experimental details and supplementary tables and figures cited above. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Davis J. Angew. Chem., Int. Ed. 2004, 43, 668. [DOI] [PubMed] [Google Scholar]
- a Bochman M. L.; Paeschke K.; Zakian V. A. Nat. Rev. Genet. 2012, 13, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Di Antonio M.; Rodriguez R.; Balasubramanian S. Methods 2012, 57, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bugaut A.; Balsubramanian S. Nucleic Acids Res. 2012, 40, 4727. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Siddiqui-Jain A.; Grand C. L.; Bearss D. J.; Hurley L. H. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 11593. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rankin S.; Reszka A. P.; Huppert J.; Zloh M.; Parkinson G. N.; Todd A. K.; Ladame S.; Balasubramanian S.; Neidle S. J. Am. Chem. Soc. 2005, 127, 10584. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Cogoi S.; Xodo L. E. Nucleic Acids Res. 2006, 34, 2536. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kumari S.; Bugaut A.; Huppert J. L.; Balasubramanian S. Nat. Chem. Biol. 2007, 3, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Huppert J. L.; Balasubramanian S. Nucleic Acids Res. 2005, 33, 2908. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Todd A. K.; Johnston M.; Neidle S. Nucleic Acids Res. 2005, 33, 2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert J. L.; Balasubramanian S. Nucleic Acids Res. 2007, 35, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffitzel C.; Berger I.; Postberg J.; Hanes J.; Lipps H. J.; Pluckthun A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K.; Juranek S.; Simonsson T.; Hempel A.; Rhodes D.; Lipps H. J. Nat. Struct. Mol. Biol. 2008, 15, 598. [DOI] [PubMed] [Google Scholar]
- Rodriguez R.; Müller S.; Yeoman J. A.; Trentesaux C.; Riou J.-F.; Balasubramanian S. J. Am. Chem. Soc. 2008, 130, 15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R.; Miller K. M.; Forment J. V.; Bradshaw C. R.; Nikan M.; Britton S.; Oelschlaegel T.; Xhemalce B.; Balasubramanian S.; Jackson S. P. Nat. Chem. Biol. 2012, 8, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Nat. Chem. 2013, 5, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E. Y. N.; Beraldi D.; Tannahill D.; Balasubramanian S. Nat. Com. 2013, 4, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douarre C.; Mergui X.; Sidibe A.; Gomez D.; Alberti P.; Mailliet P.; Trentesaux C.; Riou J.-F. Nucleic Acids Res. 2013, 41, 3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ward J. F. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95. [DOI] [PubMed] [Google Scholar]; b Iliakis G. Bioessays 1991, 13, 641. [DOI] [PubMed] [Google Scholar]
- a Kanaar R.; Hoeijmakers J. H.; van Ghent D. C. Trends Cell Biol. 1998, 8, 483. [DOI] [PubMed] [Google Scholar]; b Rothkamm K.; Kruger I.; Thompson L. H.; Lobrich M. Mol. Cell. Biol. 2003, 23, 5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggo P. A.; Caldecott K.; Pidsley S.; Banks G. R. Cancer Res. 1989, 49, 7057. [PubMed] [Google Scholar]
- Kaelin W. G. Jr. Nat. Rev. Cancer 2005, 5, 689. [DOI] [PubMed] [Google Scholar]
- Farmer H.; McCabe N.; Lord C. J.; Tutt A. N. J.; Johnson D. A.; Richardson T. B.; Santarosa M.; Dillom K. J.; Hickson I.; Knights C.; Martin N. M. B.; Jackson S. P.; Smith G. C. M.; Ashworth A. Nature 2005, 434, 917. [DOI] [PubMed] [Google Scholar]
- Aggarwal M.; Sommers J. A.; Shoemaker R. H.; Brosh R. M. Jr. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S.; Hurley L. H.; Neidle S. Nat. Rev. Drug Discovery 2011, 10, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S.; Sanders D. A.; Di Antonio M.; Matsis S.; Riou J.-F.; Rodriguez R.; Balasubramanian S. Org. Biomol. Chem. 2012, 10, 6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yhao Y.; Thomas H. D.; Batey M. A.; Cowell I. G.; Richardson C. J.; Griffin R. J.; Calvert H.; Newell D. R.; Smith G. C. M.; Curtin N. J. Cancer Res. 2006, 66, 5354. [DOI] [PubMed] [Google Scholar]
- Schultz L. B.; Chehab N. H.; Malikzay A.; Halazonetis T. D. J. Cell. Biol. 2000, 151, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bliss C. I. Ann. Appl. Biol. 1939, 26, 585. [Google Scholar]; b Jonker D. M.; Visser S. A. G.; van der Graaf P. H.; Voskuyl R. A.; Danhof M. Pharmacol. Therapeut. 2005, 106, 1. [DOI] [PubMed] [Google Scholar]
- a Reaper P. M.; Griffiths M. R.; Long J. M.; Charrier J.-D.; MacCormick S.; Charlton P. A.; Golec J. M. C.; Pollard J. R. Nat. Chem. Biol. 2011, 7, 428. [DOI] [PubMed] [Google Scholar]; b Charrier J.-D.; Durrant S. J.; Golce J. M. C.; Kay D. P.; Knegtel R. M. A.; MacCormick S.; Mortimore M.; O’Donnell M. E.; Pinder J. L.; Reaper P. M.; Rutherford A. P.; Wang P. S. H.; Young S. C.; Pollard J. R. J. Med. Chem. 2011, 54, 2320. [DOI] [PubMed] [Google Scholar]
- Tavecchio M.; Munck J. M.; Cano C.; Newell D. R.; Curtin N. J. Cancer Chemother. Pharmacol. 2012, 69, 155. [DOI] [PubMed] [Google Scholar]
- Xu H.; Xian J.; McKinney S.; Vire E.; Wong J.; Tong R.; Wei V.; Kouzarides T.; Caldas C.; Aparicio S.. Oncogene 2013, submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Amrein L.; Loignon M.; Goulet A.-C.; Dunn M.; Jean-Claude B.; Aloyz R.; Panasci L. J. Pharmacol. Exp Ther. 2007, 321, 848. [DOI] [PubMed] [Google Scholar]; b Berenbaum M. C. Cancer Res. 1992, 52, 4558. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.