

Abstract

Photoinduced cross-linking (PIC) has become a powerful tool in chemical biology for the identification and mapping of stable or transient interactions between biomacromolecules and their (unknown) ligands. However, the value of PIC for in vitro and in vivo structural proteomics can be realized only if cross-linking reports accurately on biomacromolecule secondary, tertiary, and quaternary structures with residue-specific resolution. Progress in this area requires rigorous and comparative studies of PIC reagents, but despite widespread use of PIC, these have rarely been performed. The use of PIC to report reliably on noncovalent structure is therefore limited, and its potentials have yet to be fully realized. In the present study, we compared the abilities of three probes, phenyl trifluoromethyldiazirine (TFMD), benzophenone (BP), and phenylazide (PA), to record structural information within a biomolecular complex. For this purpose, we employed a self-assembled amyloid-like peptide nanostructure as a tightly and specifically packed model environment in which to photolyze the reagents. Information about PIC products was gathered using mass spectrometry and ion mobility spectrometry, and the data were interpreted using a mechanism-oriented approach. While all three PIC groups appeared to generate information within the packed peptide environment, the data highlight technical limitations of BP and PA. On the other hand, TFMD displayed accuracy and generated straightforward results. Thus TFMD, with its robust and rapid photochemistry, was shown to be an ideal probe for cross-linking of peptide nanostructures. The implications of our findings for detailed analyses of complex systems, including those that are transiently populated, are discussed.
Photoinduced covalent cross-linking (PIC) represents a powerful approach with which to probe biological systems and processes. At the level of interaction networks, cross-linking enables (unknown) interacting partners to be captured, for example, proteins that bind functionalized peptides,1−3 proteins that bind functionalized small molecules,4−6 small molecules that bind to functionalized proteins,7 proteins that bind to functionalized proteins,8−10 and functionalized proteins that oligomerize.11 Additionally, at the level of domains, residues, or even atoms, PIC can also provide information on secondary or tertiary structure.12−16 The use of PIC to interrogate complex biological systems, structures, and networks, both in vitro and in vivo, has increased in recent years, based on advances in site-specific incorporation of PIC groups into proteins (using total synthesis, semi-synthesis, or incorporation by the use of modified codons17) combined with enhancements in analytical methodology, specifically mass spectrometry (MS), that enable the products of cross-linking to be identified uniquely in residue-specific detail.14
Benzophenone (BP), phenylazide (PA) and trifluoromethyldiazirine (TFMD) are three commonly used PIC groups, to which various advantages and disadvantages have been ascribed. Diazirines are capable of rapid, indiscriminate reactions but tend to be synthetically and/or commercially less accessible. BP is capable of high-yielding PIC, but long irradiation times are often required and its excitation is reversible. PA generates a highly reactive nitrene that rearranges to an intermediate with strong reactivity preferences.13,18 Given their commercial availability, PA and BP have been employed widely in PIC studies of protein interactions, despite misgivings about their suitability for extracting molecular-level information. For BP the caveats include the potential for cross-linking chemistry to be determined by diffusion rather than supramolecular structure, while for PA cross-links can be templated by covalent preference rather than noncovalent structure.18,19 The Kd (affinity) and dynamics of different protein–protein/ligand or peptide interactions vary widely; hence different PIC reagents may exhibit differences in reactivity, product distribution, and yield depending on the system. In selecting appropriate PIC reagents, each reagent therefore needs to be compared, but surprisingly, despite the widespread use of cross-linking in the analysis of protein systems, comparative studies of these three PIC groups are rare.20−23 Several studies have compared cross-linking efficiency and/or yield,21−23 but to our knowledge, a comparison of the utility of these reagents for garnering per residue20 information on (supra)molecular structure has not been performed to date, leaving open the question as to the optimal reagent for analysis of protein interactions in structural detail.24 This requires consideration of the underlying mechanistic cross-linking chemistry and any reactive preferences25 unique to the PIC group in question. Such studies are critical if PIC is to be used to address modern challenges in proteomics, biotechnology, and structural molecular biology, which require the (conventional) ability of PIC to map pairwise interactions, married with a structural rationale for such interactions.
Recently, Schofield and co-workers reported a comparative study in which five PIC groups were compared on the basis of cross-linker reactivity (kinetics and yield) and, using proteolytic digestion, their abilities to report on the interaction of a ligand with human 2-oxoglutarate oxygenase.26 The authors observed that different PIC groups generate different cross-links and concluded that PIC group selection should be empirically guided or that such studies should employ a selection of different PIC groups. Our group27,28 has previously employed diazirines to study supramolecular organization in amyloid-like peptide nanostructures using ion mobility spectrometry (IMS)29,30 coupled with tandem mass spectrometry (MS/MS) to identify the cross-links formed in residue-specific detail. These studies showed that individual cross-links within peptide nanostructures can furnish information on conformation and intermolecular association within highly ordered amyloid-like assemblies of a model peptide (Aβ16–22).27,28 Simultaneously, Sinz and co-workers used diazirines to determine a β-turn preference for a peptide in solution.16 Given the rigid, close packing of individual peptides within amyloid-like nanostructures of Aβ16–22, these structures form an ideal system with which to systematically compare cross-links obtained for BP, PA, and diazirine (TFMD) within the same self-assembled structure. Here we use a unique Aβ16–22 supramolecular structure (the fibrillar form resulting from aggregation at pH 7.0)31 to illustrate the advantages and disadvantages of different PIC reagents (BP, PA, and TFMD) that should be considered when designing experiments for structural analysis of protein architectures and interacting surfaces. The results revealed potential technical limitations of PA and BP, while highlighting the favorable properties of TFMD. TFMD photolyzed as readily as PA and generated a product distribution that was easy to interpret. Importantly, TFMD formed only peptide–peptide intermolecular cross-links in the presence of supramolecular structure and light. The implications of these results for the study of biomolecular interactions using PIC are discussed, acknowledging the favorable properties of diazirines10 and the availability of technologies with which to incorporate them into biomolecules using chemical synthesis11 or genetic methods,32,33 notably TFMD-phenylalanine34 (employed in the present study). Taken together, our results highlight the potential of TFMD as a reagent with which to pursue in vitro and in vivo structural proteomics and complex systems research using PIC.
Results and Discussion
Synthesis and Nanostructure Characterization
We selected Aβ16–22 amyloid-like nanofibrils that form readily at pH 7.0 in vitro(31) as the scaffold for comparison of the utility of different PIC reagents, since this stable, closely packed, and robust structure provides an ideal environment in which to photolyze and compare all three PIC groups. Aβ16–22 is also a good model on account of its synthetic accessibility,28,31,35,36 its ability to self-assemble into different but uniquely defined nanostructures independent of amino acid substitutions, and its amenability to analysis by high-performance liquid chromatography (HPLC)35,36 and/or MS.28 Also of interest is its role as a mimetic of the biologically important Aβ peptides involved in Alzheimer’s disease.31,37,38 Assembly of Aβ16–22 is sensitive to variation in primary structure, but it is rare that destabilizing modifications abrogate aggregation.35,36,39 Rather, amino acid substitutions lead to subtle effects (e.g., on morphology or aggregation propensity of this peptide).31,35,36,40−42
Once incorporated within Aβ16–22 nanostructures, each PIC functionality was photolyzed using standard laboratory apparatus (6-W illuminator with 254- and 365-nm lamps). Products formed within the supramolecular structure (i.e., through supramolecular templating) were compared with those obtained in the absence of supramolecular structure (i.e., as a monomer in solution). Previously, Aβ16–22 modified with a TFMD group at Phe-20 (Aβ16–22-TFMD20) was shown to display self-assembly behavior similar to that of wild-type (WT) Aβ16–22.27 Presently, Aβ16–22 analogues with azido and benzoyl modifications at Phe-20 (termed Aβ16–22-PA20 and Aβ16–22-BP20, respectively) were prepared via Fmoc solid-phase peptide synthesis (Figure 1, Supplementary Tables ESI 1 and 2 and Supplementary Figures ESI 1–5).
Figure 1.

Structures of Aβ16–22 and photoreactive analogues.
Peptides were incubated under aggregation-promoting conditions (0.4 mM peptide, aqueous solution, buffered at pH 7.0, 15 days, 4 °C, quiescent). Sedimentation-HPLC43 revealed that all peptides aggregated substantially (mole fraction >75%, see Supplementary Table ESI 3 and Supplementary Figures ESI 7–10 for details). Mixed samples containing WT Aβ16–22 spiked with photoreactive analogues (ratio 4:1, WT:modified) also yielded aggregates in which the modified peptide co-assembled with the WT peptide (see below). All aggregates were analyzed further using negative-stain transmission electron microscopy (TEM) (Figure 2 and Supplementary Table ESI 4), allowing higher-order supramolecular structure44 (e.g., fibrils versus nanotubes) and sample homogeneity to be evaluated. All assemblies were fibrous, indicating that no gross perturbations to the Aβ16–22 supramolecular structure had resulted from the substitutions made to primary structure.
Figure 2.

TEM of nanostructures formed upon incubation of Aβ16–22 and its photoreactive analogues at 4 °C for 15 days. (a–c) Fibers formed from stock solutions of Aβ16–22, Aβ16–22-PA20, or Aβ16–22-BP20, respectively. (d–f) Fibers formed when a stock solution of Aβ16–22 was spiked (ratio 4:1) with Aβ16–22-TFMD20, Aβ16–22-PA20, or Aβ16–22-BP20, respectively (scale bars = 100 nm).
Cross-linking and Analysis of Monomeric Peptides
To study PIC in the absence of supramolecular structure, we irradiated peptides that had been diluted into hexafluoroisopropanol (HFIP), a solvent in which the peptides are monomeric.27 Molecularly dissolved peptides were irradiated (254 or 365 nm) for varying amounts of time (typically 0, 5, or 60 min) and analyzed using liquid chromatography (LC) interfaced to an ion trap mass spectrometer. The choice of wavelength was guided by experiment (see UV–vis spectra in Supplementary Figures ESI 5 and ESI 6) and, where necessary, by relevant information from the literature. The results of all photolysis experiments are summarized in Table 1. WT Aβ16–22 did not photolyze at either wavelength on the time scale of a typical photolysis reaction (Supplementary Figure ESI 11). Irradiation of Aβ16–22-TFMD20 at 365 nm generated a distribution of adducts as described previously.27 Presently, it was noteworthy that a significant quantity of the diazirine remained at 5 min, but that this had been consumed by 60 min. The dominant photolysis product was assigned as a hexafluoroisopropyl ether (m/z 1142; change in mass, Δm, = +140), formed via insertion of singlet carbene and/or solvolysis of photoisomerised Aβ16–22-TFMD20. Other major products were assigned as a water adduct (m/z 992; Δm = −10) and cyclized monomer (m/z 974; Δm = −28) (Supplementary Figure ESI 12). The cyclized monomer was identifed as a macrolactone, as evidenced by the formation of an aminolysis product (m/z 1033; Δm = +59) upon reaction with n-propylamine.
Table 1. Summary of Results for All Photolysis Experiments.
| peptide |
||||||
|---|---|---|---|---|---|---|
| phase | λ (nm) | t (min) | Aβ16–22 | Aβ16–22TFMD20 | Aβ16–22PA20 | Aβ16–22BP20 |
| solution | 254 | 5 | NDa | ND | partial photolysis; appearance of HFIP adduct and other insertion products | no changeb |
| 60 | no change | ND | photolysis nearing completion; HFIP adduct is the dominant product; homodimer observed | ND | ||
| 365 | 5 | ND | partial photolysis; appearance of HFIP adduct and cyclized monomer | ND | no change | |
| 60 | no change | complete photolysis; HFIP adduct is the dominant product | ND | no change | ||
| homoaggregate | 254 | 60 | ND | ND | complete photolysis; putative oxidation products dominate | ND |
| 365 | 60 | no change28 | complete photolysis; peptide insertion products dominate | ND | homodimer observed in dark control | |
| heteroaggregate | 254 | 60 | NAc | ND | heterodimer observed | ND |
| 365 | 60 | NA | heterodimer observed | ND | heterodimer observed in dark control | |
ND = no data.
All changes reported are relative to a dark control.
NA = not applicable.
Photoloysis of Aβ16–22-PA20 at 254 nm (i.e., near its λmax) occurred on a time scale similar to that of Aβ16–22-TFMD20 at 365 nm but generated more products (Supplementary Figure ESI 13). As with Aβ16–22-TFMD20, the major product (m/z 1075; Δm = +140) was an HFIP adduct. This product could be one of three isomeric structures: an O-substituted hydroxylamine (formed via a singlet nitrene), a hemiaminal (via a triplet nitrene),45 or a substituted azepine (formed via a 1,2-didehydroazepine). A second major ion (m/z 939) was assigned as an oxidation product corresponding to a monomer having lost N2 and gained two oxygen atoms (putative nitro compound). Significantly, in contrast to the results obtained for Aβ16–22-TFMD20, covalent dimerization was observed. The clearest evidence was m/z 992, which indicates dimerization through one intermolecular cross-link with an additional HFIP insertion. Clearly, formation of dimeric products in HFIP (i.e., a solvent that inhibits molecular association) is cause for concern, because it suggests PA can form intermolecular cross-links independently of supramolecular templating.
For Aβ16–22-BP20, we were unable to observe the carbonyl n→π* transition (for which λmax typically falls in the range 330–350)46 that generates the reactive intermediate (BP triplet). This absence is explained by some unusual solvent effects, which were the subject of a recent report by Lewandowska-Andralojc et al.(47) HFIP induces a blue shift of the weak n→π* absorption, causing it to become hidden under the tail of the strong aromatic π→π* absorption (see Supplementary Figure ESI 5). Although the authors were unable to determine the precise position of λmax when the HFIP content was above 80% (v/v), they were still able to observe a proportion of BP’s n→π* triplet spectroscopically when the sample was irradiated at 340 nm. On this basis, our 365-nm UV source should have been effective in generating the reactive BP triplet (noting this is routinely the case).6 However, when the experiment was performed, no changes were observed in the sample, even after 60 min of irradiation (Supplementary Figure ESI 14). A brief (5 min) irradiation at 254 nm was also ineffective in initiating cross-linking (see Supplementary Figure ESI 15).
Cross-linking and Analysis of Aggregated Peptides
Subsequent experiments focused on the PIC of peptide aggregates. Each aggregate was irradiated, isolated by centrifugation, and then disaggregated in HFIP as detailed previously.27 Analysis of photolysis products was conducted using LC–MS, and where signals needed to be resolved further, using the separative capabilities of IMS–MS on a Waters Synapt HDMS mass spectrometer. The results obtained for homopolymeric Aβ16–22-TFMD20 aggregates (i.e., samples comprising only one peptide monomer) assembled at 4 °C were indistinguishable from those reported previously (Supplementary Figure ESI 16).27 The major photolysis products were peptide insertion products (m/z 974; Δm = −28), a water adduct (m/z 992; Δm = −10), a water-quenched covalent dimer (doubly charged m/z 984; Δm = −28 for the first monomer and −10 for the second), and an HFIP adduct (m/z 1142; Δm = +140). We previously identified the HFIP adduct as resulting from isomerization of TFMD to the diazoisomer which in the presence of mildly acidic HFIP gives a reactive carbocation that is quenched by HFIP.27 Note that m/z 984 had not appeared when HPLC-purified Aβ16–22-TFMD20 was photolyzed in HFIP (i.e., no intermolecular cross-links formed in the absence of supramolecular structure). Photolysis of heteropolymeric Aβ16–22·Aβ16–22-TFMD20 aggregates (i.e., samples comprising WT and modified monomers in a 4:1 ratio) generated a product distribution (Figure 3) containing a covalent heterodimer (doubly charged m/z 935; Δm = −28). Previous MS/MS analyses27 of the various photolysis products enabled cross-link positions to be elucidated, including intramolecular cross-linking between TFMD-Phe-20 and Glu-22 (identified by post-PIC aminolysis) and an intermolecular cross-link from TFMD-Phe-20 to Lys-16, which was consistent with an in-register antiparallel β-sheet structure.
Figure 3.
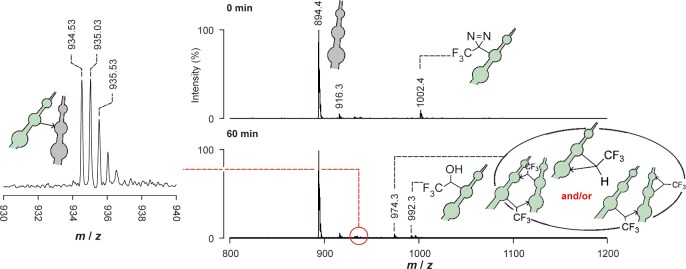
Photolysis of aggregated Aβ16–22·Aβ16–22-TFMD20 (365 nm) monitored by MS showing product distribution observed after 60 min; doubly charged heterodimer peaks from IMS–MS analysis are presented on the left. Calculated m/z for the monoisotopic, doubly charged heterodimer is 934.50; note the observed 0.5 m/z unit spacing of the isotope shifts indicating doubly charged ions.
The product distribution obtained upon photolysis of homopolymeric Aβ16–22-PA20 aggregates (Figure 4a) was more complicated than that obtained for Aβ16–22-TFMD20. The products were not well resolved by LC–MS, so the sample was reanalyzed using IMS–MS. The IMS–MS 3D driftscope plot (Figure 4b) reveals a distribution of monomers, dimers, and trimers (n = 1, z = 1; n = 2, z = 2; and n = 3, z = 3; where n is the number of monomers from which the species is composed and z is the ion charge) with the same m/z (m/z ∼908; cf. data obtained when Aβ16–22-PA20 had been photolyzed in HFIP solution). As in diazirine-mediated PIC, this distribution is characteristic of intermolecular and/or intramolecular cross-linking with loss of 1 × N2 per monomer (Figure 4c). Interestingly, a HFIP adduct (m/z 1075) also appeared in the present experiment; this must have formed by solvolysis of a photolysis product during the HFIP disaggregation step of analysis. To examine PA cross-linking in more detail, we interrogated the structure of the cyclized monomer (singly charged m/z 908) using IMS–MS/MS. Just as for cyclized Aβ16–22-TFMD20, a discontinuous series of b ions resulting from single amide cleavages (Roepstorff–Fohlman–Biemann nomenclature)48,49 indicated that an intramolecular cross-link had formed between the PIC functionality (PA) and Glu-22 (Figure 4d). This cross-link is consistent with the peptide adopting a β-strand conformation. Other major species resolved by IMS–MS were oxidation products that had lost one molecule of nitrogen per monomer (Δm = −28) and gained one or two oxygen atoms (Δm = +16 or +32, respectively). As a result, PA gives rise to substantially more complex reaction products compared with those of TFMD. The observed product distributions are consistent with the work of others, where products arising from the photolysis of PA in aerated media have been characterized.50,51 In terms of PIC methodology, a key consideration is whether the oxidation products react further to form cross-links. The literature is conflicting: in a report by Waddell and co-workers, nitro derivatives of PA were considered benign products that do not react photochemically or otherwise,51 whereas in a PIC study by Escher and Schwyzer, an inhibitor containing 4-nitro-l-phenylalanine was unexpectedly observed to cross-link photochemically to α-chymotrypsin.52 Additionally, where PIC mechanisms are concerned, it is well-known that triplet phenylnitrene can dimerize via an azo cross-link, and that azoxy cross-links are also possible. The extent to which these reactions should contribute to cross-linking in an aerated medium is unclear from the literature, with reported yields being variable.50,51,53
Figure 4.

(a) Photolysis of aggregated Aβ16–22-PA20 (254 nm), monitored by LC–MS. Assigned products are depicted in cartoon form. (b) IMS–MS 3D-driftscope plot (m/z versus drift time, tD, versus square-root-scale intensity) showing 2D separation of photolysis products, where n is the number of monomers from which the species is composed and z is the ion charge (a full mass spectrum is given on the left vertical axis). (c) Mass spectra from vertical slices of the IMS–MS plot (i.e., 2D mass spectra for m/z at selected narrow range of tD), featuring singly charged monomers (n = z = 1) and doubly charged dimers (n = z = 2). Assignments are depicted in cartoon form. (d) Sequencing of cyclized Aβ16–22-PA20 from MS/MS data. Red dotted lines are used to highlight the absence of b6 and b7 and hence the intramolecular cross-link position.
Irradiation of the mixed aggregate (Aβ16–22·Aβ16–22-PA20) yielded a product distribution similar to that from irradiation of Aβ16–22-PA20 alone (Supplementary Figure ESI 17), except that a covalent heterodimer (doubly charged m/z 901) was also formed. Due to the added complexity of the spectra and the fact that dimers were obtained by cross-linking of monomeric Aβ16–22-PA20 in HFIP (see above), these products were not analyzed further.
Irradiation of homopolymeric Aβ16–22-BP20 aggregates for 60 min at 365 nm did not cause changes to the sample (Figure 5 and Supplementary Figure ESI 18), and accordingly, nor did irradiation of aggregated Aβ16–22·Aβ16–22-BP20. As with the findings described earlier (i.e., no reactivity upon irradiation in the presence of HFIP), the results observed could arise for a number of reasons; it could be that insufficient BP triplet was generated or, alternatively, that an excited state was formed but was unable to react. Analysis of the control sample revealed putative intermolecular cross-links formed in the dark (Supplementary Figure ESI 18). Specifically, a weak signal centered on m/z 989 (i.e., ∼9 m/z units less than the m/z of monomeric Aβ16–22-BP20) indicated dimerization with loss of water, which was confirmed using IMS–MS (Figure 5a). There are two explanations: (i) intermolecular cross-linking occurred via BP triplet with subsequent acid-catalyzed dehydration of the cross-link,54 or (ii) cross-linking occurred via condensation of ground-state BP with the Lys ε-amine of a second monomer. Given that cross-linking did not take place during irradiation, the latter explanation (i.e., imine formation in the dark) is plausible, especially considering that imine formation by 4-benzoyl-l-phenylalanine has been observed by others.55 To investigate whether the observed imine cross-links were specific, the supernatant of a centrifuged assembly mixture was examined for evidence of m/z 990. If imine formation is templated within an aggregate, cross-linked units should not appear in the solution phase. The fact that the supernatant did not contain m/z 990 suggests imine formation was templated. This dimer also appeared for the solubilized Aβ16–22·Aβ16–22-BP20 aggregate, alongside a heterodimer, in which the BP group of Aβ16–22-BP20 had apparently condensed with the Lys ε-amine of Aβ16–22 (Figure 5b). The imine dimers of Aβ16–22-BP20 and of Aβ16–22·Aβ16–22-BP20 are structurally analogous to the covalent heterodimer formed from Aβ16–22·Aβ16–22-TFMD20, which was found to contain a cross-link between position 20 of Aβ16–22-TFMD20 and Lys-16 of Aβ16–22.
Figure 5.

(a) IMS–MS analyses of solubilized aggregates of (i) Aβ16–22-BP20 and (ii) Aβ16–22·Aβ16–22-BP20, neither of which had been irradiated. The z-axis is a log intensity scale. (b) Mass spectra of isolated doubly charged dimers. These spectra show the ions under the highlighted portions of the IMS–MS plots. Assignments are depicted in cartoon form, along with m/zcalcd for [M + 2H]2+ and m/zobsd.
Conclusions
In summary, we have used amyloid-like nanostructures of Aβ16–22 to evaluate the effects of TFMD, PA, and BP groups on peptide aggregation, irrespective of the substitution introduced, and the ability of these different cross-linkers to report on their supramolecular environments via PIC. None of TFMD, PA, or BP impaired the ability of Aβ16–22 to form nanostructures when incorporated at residue Phe-20, and in all cases self-assembled fibrillar structures with similar morphology were formed. Major differences were observed, however, in the PIC experiments. TFMD photolyzed readily, generating a simple distribution of products, for which detailed analysis was straightforward and interpretation of supramolecular structure was possible,27 (although care should be taken to account for products resulting from decomposition of TFMDs linear diazoisomer under acidic conditions, as reported previously). Importantly, TFMD generated only intermolecular peptide–peptide cross-links in the presence of supramolecular structure. PA also yielded structural information via the formation of inter- or intramolecular cross-links. However, control experiments revealed that cross-linking was not determined solely by supramolecular templating. Finally, BP did not form photolysis products under the conditions employed but did form intermolecular imine cross-links in the dark. Thus, in this model system, PA and BP demonstrated the potential to generate inaccuracies, while TFMD more closely resembled an ‘ideal’ PIC reagent. We note that modified tRNA/tRNA synthetase pairs have been developed for the incorporation of diazirine-based amino acids using recombinant expression,10,33,34 while the synthesis of diazirine-modified amino acids (Phe, Trp, Leu, Met, Lys, and Pro) is readily achievable,2,3,27,32,33,56 ensuring that diazirines can be incorporated into any peptide or protein of choice. Furthermore, the carbene resulting from TFMD excitation, which has a picosecond lifetime,57 represents the most reactive of the PIC groups in common use, opening the door to mechanistic studies on dynamic complex systems.
More broadly, PIC is most powerful where it can be used to provide molecular level information, either within a supramolecular structure or between interacting protein partners of complexes, be these either long-lived or transient. The approach employed here, making use of state-of-the art MS techniques, permits a large number of PIC products to be analyzed and allows cross-linking sites to be uniquely identified. These can then be interpreted in the context of chemical reactivity and noncovalent structure. The peptide nanostructure used in this work highlights the ability of PIC using TMFD-Phe to report on molecular recognition, even for structures that involve dense packing of side chains. By photolyzing PIC reagents in the presence of a high concentration of intermolecular interactions (i.e., within a peptide nanostructure), we were able to observe these cross-linking reactions very clearly, and our findings have direct implications for future PIC analyses of biological amyloids and other complexes. In moving to more dynamic systems, such as protein–protein interfaces, the chemical rationale for choosing a PIC reagent should be no different. However, factors such as cross-linker yield and rate will also be relevant; in this work the PIC reagent as one of the seven amino acids in the sequence represents around 20% of the mass of the sample for homopolymeric aggregates and around 5% of the mass of the sample for heteropolymeric aggregates. When a protein system is used (200–400 amino acids), the mass of cross-linker will be proportionally much lower, making it harder to study cross-links by MS and/or by gel electrophoresis. Thus, a cross-linker optimized to capture the interacting partner (e.g., slower rate of cross-linking but high yielding) might be preferred to one that is optimized to capture structural information (e.g., indiscriminate reactivity capturing key noncovalent contacts, but also other species due to the dynamics of the interaction under study). Further, preferred chemical reactivity of the PIC group may not bias the outcome of a PIC experiment, and in some cases, such reactivity may even be desired (e.g., where a binding site contains an amino acid with which the PIC group preferentially reacts). Thus, empirically guided PIC selection or use of multiple PIC groups simultaneously will mitigate against erroneous structural interpretation of a cross-linking study. PIC analysis of biomolecular complexes remains a challenging endeavor, but one that can reward the investigator with otherwise inaccessible information. We anticipate that the findings reported herein should aid the choice of reagent, the interpretation of information, and therefore the study of complex dynamic biomolecular interactions and systems using PIC.
Methods
Peptides were prepared by automated solid-phase peptide synthesis using a CEM Liberty peptide synthesizer operated without microwave irradiation and purified by reverse phase HPLC. Nanostructures were prepared by dilution of a 20 mM peptide stock solution in DMSO with buffer to 0.4 mM final concentration, followed by incubation at ambient temperature (4 °C), in the dark, without agitation, for 15 days. Nanostructures were characterized using TEM with uranyl acetate staining. Photolysis reactions were carried out at ambient temperature using a 6 W ultraviolet lamp (254 or 365 nm) and analyzed after 5 and/or 60 min. For solution-phase studies, the peptide stock was diluted with HFIP and irradiated immediately. For experiments with nanostructures, aliquots of the assembly mixture were gently homogenized prior to irradiation. Irradiated nanostructures were then isolated by centrifugation and treated with HFIP to effect dissolution prior to LC–MS, IMS–MS, and IMS–MS/MS analyses. For full experimental details, see the Supporting Information.
Acknowledgments
This work was supported by the European Research Council [ERC-StG-240324]. We thank the British Biotechnology and Biological Sciences Research Council (BBSRC) for a studentship for G.W.P. and C. M. Huscroft (School of Chemistry HPLC Service, University of Leeds). The Synapt HDMS was purchased using funds from the BBSRC Research Equipment Initiative (BB/E012558/1).
Supporting Information Available
Synthetic methods and characterization, methods for assembly and characterization of nanostructures, and additional MS analyses and methodology. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawaguchi Y.; Tanaka G.; Nakase I.; Imanishi M.; Chiba J.; Hatanaka Y.; Futaki S. (2013) Identification of cellular proteins interacting with octaarginine (R8) cell-penetrating peptide by photo-crosslinking. Bioorg. Med. Chem. Lett. 23, 3738–3740. [DOI] [PubMed] [Google Scholar]
- Srinivas N.; Jetter P.; Ueberbacher B. J.; Werneburg M.; Zerbe K.; Steinmann J.; Van der Meijden B.; Bernardini F.; Lederer A.; Dias R. L. A.; Misson P. E.; Henze H.; Zumbrunn J.; Gombert F. O.; Obrecht D.; Hunziker P.; Schauer S.; Ziegler U.; Käch A.; Eberl L.; Riedel K.; DeMarco S. J.; Robinson J. A. (2010) Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013. [DOI] [PubMed] [Google Scholar]
- Nakashima H.; Hashimoto M.; Sadakane Y.; Tomohiro T.; Hatanaka Y. (2006) Simple and versatile method for tagging phenyldiazirine photophores. J. Am. Chem. Soc. 128, 15092–15093. [DOI] [PubMed] [Google Scholar]
- Sarathi Addy P.; Saha B.; Pradeep Singh N. D.; Das A. K.; Bush J. T.; Lejeune C.; Schofield C. J.; Basak A. (2013) 1,3,5-Trisubstituted benzenes as fluorescent photoaffinity probes for human carbonic anhydrase II capture. Chem. Commun. 49, 1930–1932. [DOI] [PubMed] [Google Scholar]
- Brigham J. L.; Perera B. G. K.; Maly D. J. (2013) A hexylchloride-based catch-and-release system for chemical proteomic applications. ACS Chem. Biol. 8, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth B. P.; Wilson D. J.; Nelson K. M.; Boshoff H. I.; Barry C. E.; Aldrich C. C. (2012) Development of a selective activity-based probe for adenylating enzymes: Profiling MbtA involved in siderophore biosynthesis from Mycobacterium tuberculosis. ACS Chem. Biol. 7, 1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunbeck A.; Huber T.; Abrol R.; Trzaskowski B.; Goddard W. A.; Sakmar T. P. (2012) Genetically encoded photo-cross-linkers map the binding site of an allosteric drug on a G protein-coupled receptor. ACS Chem. Biol. 7, 967–972. [DOI] [PubMed] [Google Scholar]
- Uchinomiya S.; Nonaka H.; Wakayama S.; Ojida A.; Hamachi I. (2013) In-cell covalent labeling of reactive His-tag fused proteins. Chem. Commun. 49, 5022–5024. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy M.; Dugan A.; Nwokoye A.; Fung Y.-H.; Lancia J. K.; Majmudar C. Y.; Mapp A. K. (2011) Caught in the act: Covalent cross-linking captures activator–coactivator interactions in vivo. ACS Chem. Biol. 6, 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Lin S.; Song X.; Liu J.; Fu Y.; Ge X.; Fu X.; Chang Z.; Chen P. R. (2011) A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat. Chem. Biol. 7, 671–677. [DOI] [PubMed] [Google Scholar]
- Vila-Perelló M.; Pratt M. R.; Tulin F.; Muir T. W. (2007) Covalent capture of phospho-dependent protein oligomerization by site-specific incorporation of a diazirine photo-cross-linker. J. Am. Chem. Soc. 129, 8068–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci C.; Reale S.; De Angelis F. (2013) Photoactivable amino acid bioisosteres and mass spectrometry: Snapshots of in vivo 3D protein structures. ChemBioChem 14, 181–183. [DOI] [PubMed] [Google Scholar]
- Preston G. W.; Wilson A. J. (2013) Photo-induced covalent cross-linking for the analysis of biomolecular interactions. Chem. Soc. Rev. 42, 3289–3301. [DOI] [PubMed] [Google Scholar]
- Paramelle D.; Miralles G.; Subra G.; Martinez J. (2013) Chemical cross-linkers for protein structure studies by mass spectrometry. Proteomics 13, 438–456. [DOI] [PubMed] [Google Scholar]
- Morris J. L.; Reddington S. C.; Murphy D. M.; Jones D. D.; Platts J. A.; Tippmann E. M. (2013) Aryl azide photochemistry in defined protein environments. Org. Lett. 15, 728–731. [DOI] [PubMed] [Google Scholar]
- Kölbel K.; Ihling C. H.; Sinz A. (2012) Analysis of peptide secondary structures by photoactivatable amino acid analogues. Angew. Chem., Int. Ed. 51, 12602–12605. [DOI] [PubMed] [Google Scholar]
- Hancock S. M.; Uprety R.; Deiters A.; Chin J. W. (2010) Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J. Am. Chem. Soc. 132, 14819–14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley H. (1983) Photogenerated Reagents in Biochemistry and Molecular Biology, Vol. 12, Elsevier, Amsterdam. [Google Scholar]
- Brunner J. (1993) New photolabeling and cross-linking methods. Annu. Rev. Biochem. 62, 483–514. [DOI] [PubMed] [Google Scholar]
- Jiao C. Y.; Alves I. D.; Point V.; Lavielle S.; Sagan S.; Chassaing G. (2010) Comparing lipid photo-cross-linking efficacy of penetratin analogues bearing three different photoprobes: dithienyl ketone, benzophenone, and trifluoromethylaryldiazirine. Bioconjugate Chem. 21, 352–359. [DOI] [PubMed] [Google Scholar]
- Tate J. J.; Persinger J.; Bartholomew B. (1998) Survey of four different photoreactive moieties for DNA photoaffinity labeling of yeast RNA polymerase III transcription complexes. Nucleic Acids Res. 26, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber P. J. A.; Beck-Sickinger A. G. (1997) Comparison of the photochemical behavior of four different photoactivatable probes. J. Pept. Res. 49, 375–383. [DOI] [PubMed] [Google Scholar]
- Kapfer I.; Jacques P.; Toubal H.; Goeldner M. P. (1995) Comparative photoaffinity labeling study between azidophenyl, difluoroazidophenyl, and tetrafluoroazidophenyl derivatives for the GABA-gated chloride channels. Bioconjugate Chem. 6, 109–114. [DOI] [PubMed] [Google Scholar]
- Mann M.; Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261. [DOI] [PubMed] [Google Scholar]
- Raimer B.; Lindel T. (2013) Photoactivation of (p-methoxyphenyl)(trifluoromethyl)diazirine in the presence of phenolic reaction partners. Chem.—Eur. J. 19, 6551–6555. [DOI] [PubMed] [Google Scholar]
- Bush J. T.; Walport L. J.; McGouran J. F.; Leung I. K. H.; Berridge G.; van Berkel S. S.; Basak A.; Kessler B. M.; Schofield C. J. (2013) The Ugi four-component reaction enables expedient synthesis and comparison of photoaffinity probes. Chem. Sci. 4, 4115–4120. [Google Scholar]
- Preston G. W.; Radford S. E.; Ashcroft A. E.; Wilson A. J. (2012) Covalent cross-linking within supramolecular peptide structures. Anal. Chem. 84, 6790–6797. [DOI] [PubMed] [Google Scholar]
- Smith D. P.; Anderson J.; Plante J.; Ashcroft A. E.; Radford S. E.; Wilson A. J.; Parker M. J. (2008) Trifluoromethyldiazirine: an effective photo-induced cross-linking probe for exploring amyloid formation. Chem. Commun. 5728–5730. [DOI] [PubMed] [Google Scholar]
- Uetrecht C.; Rose R. J.; van Duijn E.; Lorenzen K.; Heck A. J. R. (2010) Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 39, 1633–1655. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J.-E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. (2009) Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbach J. J.; Ishii Y.; Antzutkin O. N.; Leapman R. D.; Rizzo N. W.; Dyda F.; Reed J.; Tycko R. (2000) Amyloid fibril formation by Aβ16–22, a seven-residue fragment of the Alzheimer’s beta-amyloid peptide, and structural characterization by solid state NMR. Biochemistry 39, 13748–13759. [DOI] [PubMed] [Google Scholar]
- Suchanek M.; Radzikowska A.; Thiele C. (2005) Photo-leucine and photo methionine allow identification of protein-protein interactions in living cells. Nat. Methods 2, 261–268. [DOI] [PubMed] [Google Scholar]
- Chou C.; Uprety R.; Davis L.; Chin J. W.; Deiters A. (2011) Genetically encoding an aliphatic diazirine for protein photocrosslinking. Chem. Sci. 2, 480–483. [Google Scholar]
- Tippmann E. M.; Liu W.; Summerer D.; Mack A. V.; Schultz P. G. (2007) A genetically encoded diazirine photocrosslinker in Escherichia coli. ChemBioChem 8, 2210–2214. [DOI] [PubMed] [Google Scholar]
- Senguen F. T.; Lee N. R.; Gu X. F.; Ryan D. M.; Doran T. M.; Anderson E. A.; Nilsson B. L. (2011) Probing aromatic, hydrophobic, and steric effects on the self-assembly of an amyloid-beta fragment peptide. Mol. BioSys. 7, 486–496. [DOI] [PubMed] [Google Scholar]
- Senguen F. T.; Doran T. M.; Anderson E. A.; Nilsson B. L. (2011) Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-beta 16–22 self-assembly. Mol. BioSys. 7, 497–510. [DOI] [PubMed] [Google Scholar]
- Qiang W.; Yau W.-M.; Luo Y.; Mattson M. P.; Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Guo Z. H.; Yau W. M.; Mattson M. P.; Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Pingali S. V.; Jogalekar A. S.; Snyder J. P.; Thiyagarajan P.; Lynn D. G. (2008) Cross-strand pairing and amyloid assembly. Biochemistry 47, 10018–10026. [DOI] [PubMed] [Google Scholar]
- Chaudhary N.; Nagaraj R. (2011) Impact on the replacement of Phe by Trp in a short fragment of A beta amyloid peptide on the formation of fibrils. J. Pept. Sci. 17, 115–123. [DOI] [PubMed] [Google Scholar]
- Inouye H.; Gleason K. A.; Zhang D.; Decatur S. M.; Kirschner D. A. (2010) Differential effects of Phe19 and Phe20 on fibril formation by amyloidogenic peptide Aβ16–22 (Ac-KLVFFAE-NH2). Proteins: Struct., Funct. Bioinf. 78, 2306–2321. [DOI] [PubMed] [Google Scholar]
- Mehta A. K.; Lu K.; Childers W. S.; Liang Y.; Dublin S. N.; Dong J. J.; Snyder J. P.; Pingali S. V.; Thiyagarajan P.; Lynn D. G. (2008) Facial symmetry in protein self-assembly. J. Am. Chem. Soc. 130, 9829–9835. [DOI] [PubMed] [Google Scholar]
- O’Nuallain B.; Thakur A. K.; Williams A. D.; Bhattacharyya A. M.; Chen S. M.; Thiagarajan G.; Wetzel R. (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 413, 34–74. [DOI] [PubMed] [Google Scholar]
- Serpell L. C.; Sunde M.; Benson M. D.; Tennent G. A.; Pepys M. B.; Fraser P. E. (2000) The protofilament substructure of amyloid fibrils. J. Mol. Biol. 300, 1033–1039. [DOI] [PubMed] [Google Scholar]
- Sydnes M. O.; Doi I.; Ohishi A.; Kuse M.; Isobe M. (2008) Determination of solvent-trapped products obtained by photolysis of aryl azides in 2,2,2-trifluoroethanol. Chem. Asian J. 3, 102–112. [DOI] [PubMed] [Google Scholar]
- Castro G.; Blanco S.; Giordano O. (2000) UV spectral properties of benzophenone. Influence of solvents and substituents. Molecules 5, 424–425. [Google Scholar]
- Lewandowska-Andralojc A.; Hug G. L.; Hörner G.; Pedzinski T.; Marciniak B. (2012) Unusual photobehavior of benzophenone triplets in hexafluoroisopropanol. Inversion of the triplet character of benzophenone. J. Photochem. Photobiol. A: Chem. 244, 1–8. [Google Scholar]
- Roepstorff P.; Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601–601. [DOI] [PubMed] [Google Scholar]
- Biemann K. (1988) Contributions of mass spectrometry to peptide and protein structure. Biomed. Environ. Mass Spectrom. 16, 99–111. [DOI] [PubMed] [Google Scholar]
- Ishikawa S.; Tsuji S.; Sawaki Y. (1991) Structure and reactivity of nitroso oxides. J. Am. Chem. Soc. 113, 4282–4288. [Google Scholar]
- Go C. L.; Waddell W. H. (1983) Evolution of photooxidation products upon irradiation of phenyl azide in the presence of molecular oxygen. J. Org. Chem. 48, 2897–2900. [Google Scholar]
- Escher E.; Schwyzer R. (1974) p-Nitrophenylalanine, p-azidophenylalanine, m-azidophenylalanine, and o-nitro-p-azido-phenylalanine as photoaffinity labels. FEBS Lett. 46, 347–350. [DOI] [PubMed] [Google Scholar]
- Abramovitch R. A.; Challand S. R. (1972) Reaction of triplet aryl nitrenes and azides with molecular oxygen. J. Chem. Soc., Chem. Commun. 964–966. [Google Scholar]
- Dorman G.; Prestwich G. D. (1994) Benzophenone photophores in biochemistry. Biochemistry 33, 5661–5673. [DOI] [PubMed] [Google Scholar]
- Young T. S.; Young D. D.; Ahmad I.; Louis J. M.; Benkovic S. J.; Schultz P. G. (2011) Evolution of cyclic peptide protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 11052–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartmann T.; Lindel T. (2013) l-Phototryptophan. Eur. J. Org. Chem. 2013, 1649–1652. [Google Scholar]
- Zhang Y.; Burdzinski G.; Kubicki J.; Platz M. S. (2008) Direct observation of carbene and diazo formation from aryldiazirines by ultrafast infrared spectroscopy. J. Am. Chem. Soc. 130, 16134–16135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.