

Abstract
Molecular dynamics, coupled with fluorescence data for charged dipeptides of tryptophanyl glutamic acid (Trp-Glu), reveal a detailed picture of how specific conformation effects fluorescence. Fluorescence emission spectra and time-resolved emission measurements have been collected for all four charged species. Molecular dynamics simulations 20 to 30 ns in length have also been carried out for the Trp-Glu species as simulation provides aqueous-phase conformational data that can be correlated with the fluorescence data. The calculations show that each dipeptide species is characterized by a similar set of six, discrete Chi 1, Chi 2 dihedral angle pairs. The preferred Chi 1 angles---60°, 180° and 300°---play the significant role in positioning the terminal amine relative to the indole ring. A Chi 1 angle of 60° results in the arching of the backbone over the indole ring, and no interaction of the ring with the terminal amine. Chi 1 values of 180° and 300° result in an extension of the backbone away from the indole ring, and a NH3 cation-π interaction with indole. This interaction is believed responsible for charge transfer quenching. Two fluorescence lifetimes and their corresponding amplitudes correlate with the Chi 1 angle probability distribution for all four charged Trp-Glu dipeptides. Fluorescence emission band maxima are also consistent with the proposed pattern of terminal amine cation quenching of fluorescence.
Introduction
The amino acid residue, tryptophan, fluoresces at a distinct wavelength that is convenient for the study of solution-phase proteins. However, this simplicity belies the difficulty of spectral interpretation. Fluorescence emission maxima shifts, band shapes, quantum yields and lifetimes respond to changes in solution and protein environment, but an understanding of the pattern of response has been enigmatic [1–4].
Recently, progress has been made in understanding the energy landscape of the responsible chromophore, indole. Various double resonance spectroscopic studies of tryptophan model compounds and dipeptides in the gas phase have enabled the linking of spectroscopic details to individual conformers and strongly suggested the presence of nonradiative energy dissipation mechanisms [5–9]. High resolution photo fragment translational spectroscopy of indole has confirmed the contribution of the dissociative state, 1 πσ*, to UV-induced photophysics where NH bond breaking occurs [10]. A theoretical modeling of energy dissipative states[11] has revealed at least five singlet electronic states for the neutral, gas-phase dipeptide, N-acetyl tryptophan methyl amide, including two nonradiative states located on the peptide backbone: a locally-excited state and a charge transfer state. A backbone hydrogen bond acts as a device for the dissipation of the excited state via charge transfer, especially for the C7 conformer, a seven-membered, hydrogen bonded ring structure of the backbone that is the equivalent of a γ-turn [11]. All these studies of the tryptophan molecule, while extremely valuable, are in the gas phase, which is not a physiological environment.
In the presence of an electric field such as that provided by water, the photophysics of the indole chromophore are governed by the electric field sensitive states, 1La (radiative) and 1πσ* (nonradiative). Solvation of gas phase tryptophan and tryptophan dipeptide cations results in conformational change and extensive photodissociation spectral changes[12–14]. Thus relaxation mechanisms of nonsolvated, neutral, gas-phase molecules may diverge from those for solution phase, charged tryptophan species. Solution conformation of the backbone is also expected to diverge from that observed in the gas phase as γ-turns are not common in aqueous phase proteins.
We have chosen to examine the dipeptide, tryptophanyl glutamic acid in the solution phase and in its different charged states that evolve as pH increases: protonated at the terminal amine (total molecular charge = 1+, pH 1.5); in the zwitterion state, which features, in addition, a negative charge at the terminal carboxylic acid (total molecular charge = 0, pH 3.5); additionally deprotonated at the glutamic carboxylic acid (total molecular charge = 1−, pH 5.5) and deprotonated at the terminal amine cation (total molecular charge = 2−, pH 10.0). These dipeptide species will be referred throughout as Trp-Glu 1+, Trp-Glu 0, Trp-Glu 1− and Trp-Glu 2−, respectively. There are several reasons for choosing this molecular system. The relevant tryptophan photophysics takes place in biological systems where tryptophans are found in an aqueous phase interacting with other charged residues. While it is true that tryptophan residues do not frequently encounter an N-terminal amine cation in proteins, interaction with the charged amine groups of arginine and lysine are common [15]. Indeed, the cation-π interaction between tryptophan and arginine or lysine is a significant noncovalent force governing protein structure, including macromolecular assembly [16]. Thus, the role that the amine cation plays in controlling the emissive state of tryptophan is relevant to protein fluorescence. Edge-on carboxylate interaction with indole ring hydrogens is another noncovalent force governing residue interactions in proteins [17]. This interaction also plays a role in Trp-Glu dipeptide conformation, as discussed below.
The specific photophysical behavior of tryptophan that we seek to explain is the ~8 nm fluorescence emission shift to higher energy for Trp-Glu in the 1+, zwitterion and 1− charge state relative to the 2− charge state [18]. Our previous spectroscopic and molecular dynamics study of solution phase Trp-Gly and Gly-Trp zwitterions and anions linked a stretched out peptide backbone conformation, which places the terminal amine cation over the indole ring, with a fluorescence emission blue shift [19]. Here, molecular dynamics simulations 20 to 30 ns in length have been carried out for the four Trp-Glu species as simulation provides aqueous-phase data. The method used involves tracking all the relevant dihedral angles to locate favored conformations. Favored conformations were studied via quantum mechanical calculations; electrostatic fields and internal interactions of the dipeptides were extracted. Favored conformations for all dipeptide species are characterized by backbones with two C5, NH---O=C hydrogen bonded rings in all but two rotamers. This places the backbone in a β-sheet conformation, which is commonly found in proteins.
These calculations show that each dipeptide species is characterized by a similar set of six discrete Chi 1, Chi 2 dihedral angle pairs. Each dihedral angle pair for each dipeptide species has a particular associated probability. As there are six pairs of Chi 1, Chi 2 angles and typically two or three fluorescence lifetimes, it is clear that lifetime multiexponentiality is not in one-to-one correspondence with the number of rotamers. Instead, lifetimes appear related to the two possible backbone-indole conformations, one where the backbone stretches away from the ring and another where the backbone curls back over the ring. These are determined by the Chi 1 dihedral angle.
The conformation imposed by some of these dihedral angle pairs should lead to a charge transfer process between the indole ring and the terminal amine cation as the ring-cation distance is sufficiently close. Nonradiative processes result from the backbone charged group-indole ring interactions. As the pH changes, the charge on the Trp-Glu species changes, so similar Chi 1, Chi 2 angle pairs for different ions do not result in the same ring-charged group interactions. Also, charged group-ring electrostatic interactions restrict the transition from one conformation to another. Backbone groups can even become significantly immobilized in dipeptide species because of electrostatic interactions.
We have also measured the fluorescence lifetimes for all four Trp-Glu species, and have found correlations between fluorescence lifetimes and rotamer populations and their conformations. It is important to realize that the rotamers determine the backbone-indole ring electrostatic interactions, which in turn control nonradiative processes. These two factors cannot be considered independently when studying tryptophan fluorescence.
Computational Methods
DFT Calculations
Ab initio quantum mechanical calculations were carried out using the Gaussian 09 package [20], using the B3LYP density functional theory (DFT) method and 6-31++g(df,p) basis set. Implicit water was used with the Polarizable Continuum Model method. The calculations were done on four Intel 2.93 GHz quad-core Intel Core7 processors in the CUNY HPCC facilities, College of Staten Island, New York City. Each molecule’s geometry was optimized and checked for imaginary frequencies. Subsequent to this optimization, the excited and ground state orbitals were calculated, as well as the electronic state transitions and their oscillator strengths, which provided the electrostatic potential distribution of the ground state, the absorption spectra and the transition dipole moments. Lowest energy absorption transitions, dipole moments and oscillator strengths for the three Chi 1 rotamers of each Trp-Glu species are given in Table S1 in Supporting Information. Ground state quantum mechanics calculations for the three different Chi 1 angles revealed by molecular dynamics simulation were carried out in order to explain the favored angles and their stabilizing factors. Table S2 compares the Chi 1 angles obtained by ab initio calculations to the Gaussian mean value obtained via the molecular dynamics simulation. The largest Chi 1 angular difference of 11° is found for the first of the Trp-Glu 2− conformers, but this is well within the full-width-half-maximum for the Gaussian peak width of this conformer, 27°. Time constraints prevented running simulation on each of the ~104 possible starting geometries, so instead the lowest energy conformer for each of these angles was located and used for the starting geometry. The main goal of the quantum mechanical calculations was to determine the electrostatic potential distribution throughout the molecule, which is a good indicator of intramolecular interactions. Mulliken charges were used to check for consistency and accuracy against natural bond analysis results. These results explain how the calculated conformations are consistent with the quantum mechanics results and/or the fluorescence spectra, but do not explain the source of the molecular dynamics results themselves.
Visualization of the molecules, including the electrostatic potential fields, was done using Molekel 5.4.0.8 [21]. Distances between atoms participating in electrostatic interactions were measured with gOpenMol software [22].
Molecular Dynamics Simulation
Molecular dynamics calculations were carried out using GROMACS 4.5.3 software [23], with a cubic box size more than 1 nm past the molecular boundary and with ~1000 solvent water molecules. Periodic boundary conditions were applied in three dimensions. Simulations were run at room temperature and 1 atm pressure after equilibrating the system using both canonical (NVT) and isothermal-isobaric (NPT) systems. The standard leap-frog algorithm was used to integrate the equations of motion with a time step size of 0.5 fs, and a total simulation time of 20–30 ns, which ensured that all of Chi1 dihedral angle space was sampled. The OPLS-AA force field together with the suggested TIP-4P water model were chosen because tryptophan fluorescence is known to be significantly affected by solvent conditions (pH, ionic strength, and hydrophobicity), and the OPLS-AA force field is parameterized with a focus on accurate solvent properties[24]. In addition, the results of calculations using this force field were tested and optimized for the correct torsional angles of several dipeptides [25]; a RMS error of 0.15° for Chi 1 in the Phe-Phe dipeptide via-a-vis ab initio results is obtained. Comparison of calculated energy differences for homodipeptide conformers with experimental values yields an average deviations of 0.25 kcal/mol, in excellent agreement[25]. These factors make the OPLS-AA force field the preferred choice for our own Trp-Glu studies.
Fluorescence Measurements and Data Analysis
L-tryptophanyl glutamic acid (Trp-Glu) was purchased from Research Plus, Inc. (Barnegat, NJ) and used without further purification. Dipeptide concentration for fluorescence measurements was adjusted to 0.01 mM by dilution of a 1 mg/ml stock solution in deionized water adjusted to the appropriate pH: 1+ cation: pH 1.5, zwitterionic: pH 3.4, 1− anion: pH 5.5, 2− anion: pH 10.0.
Steady state fluorescence spectra were acquired on a PTI QM-4/206 SE Spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence. The excitation wavelength was 279 nm. A 5 nm band pass was selected; with a 1 nm scan interval and a 0.5 second time constant. Water baselines were collected and subtracted where necessary. Time-correlated measurements were acquired on a Horiba Fluorolog model FL-1000 fluorometer using 281 nm light-emitting diode excitation (Horiba, Inc., Edison, NJ), 20 nm spectral bandwidth. The instrument response was 1.47 ns full-width half maximum. Measurements were corrected for instrument response using a casein scattering suspension. The fluorescent count maximum was set to 20,000 for all measurements; slits were set to 5–7 nm.
The fluorescence decay, I(t) at time, t, can be described as a sum of exponentials:
| (1) |
Where Bi is the normalized preexponential, initial fluorescence intensity, and τi, is the corresponding fluorescence lifetime.
Lifetimes were calculated by iterative convolution following a proprietary method (Decay Analysis Software, DAS 6, Horiba, Inc.) of non-linear, least squares fitting χ2 (mis-matches between the data and fitted function). This matrix method is purported to yield fewer false minima than the Marquardt algorithm.
χ2 is defined as a sum over N data channels:
| (2) |
Where Y(t) is the fluorescence decay data, and σ(t) is its standard deviation [26].
The matrix method for obtaining the best fit to the data proceeds as follows. The best (lowest value) χ2 is given by a set of minimized fitting parameters A (background noise), B (amplitude scaling factor), τ (lifetime) and Δ (shift) as:
| (3) |
With w(t) = 1/σ(t)2 as the data weight. The best fit values for A and B, A′ and B′, are arrived at by derivation of Eq. 3 with respect to each parameter. Given estimated values of τ and Δ, A′ and B′ may be arrived at by inversion of the derivation matrix:
| (4) |
This is the Horiba IBH algorithm[27].
Global χ2 minimization (not to be confused with the Chi 2 dihedral angle discussed below) and a random distribution of residuals were the standards of criteria for goodness-of-fit.
Results
Steady-state and time-resolved fluorescence measurements
The subtle effect of pH, primary sequence and conformation on tryptophan fluorescence is observed in the steady state fluorescence emission spectra for tryptophanyl glutamic acid (Trp-Glu) dipeptide charged species as given in Figure 1. The emission maxima for the cationic (Trp-Glu 1+, bold line), zwitterionic (Trp-Glu 0, dashed line), and negatively charged zwitterionic (“pseudo-zwitterionic,” Trp-Glu 1−, solid line) states of the dipeptides are located at 343 nm, 343 nm and 347 nm, respectively, a wavelength 7–11 nm shorter than that of the anionic dipeptide, 354 nm (Trp-Glu 2−, dotted line). Spectral widths also vary where the full width half maximum for Trp-Glu 0 is 47 nm while that for all other dipeptide species is ~ 65 nm. Additionally, the Trp-Glu 1+ spectrum exhibits a small shoulder at 311 nm, which is due to weak Raman scattering from water.
Figure 1.

Normalized fluorescence spectra of the Trp-Glu dipeptides at 4 pH’s. Trp-Glu 1+, pH 1.5, (bold line); Trp-Glu 0, pH 3.5, (dashed line); Trp-Glu 1−, pH 5.5, (solid line); Trp-Glu 2−, pH 9.3, (dotted line).
Time-correlated single photon counting measurements yield distinct lifetimes and associated amplitudes for each of the dipeptides. The fluorescence decay, instrument response curve, fitted function and residuals for TrpGlu 2− are shown in Figure S1. Two lifetimes were found for each of the species studied, and are given in Table 1. Lifetime measurements for Trp-Glu 1+ and Trp-Glu 0 yield two short lifetimes. Lifetimes for Trp-Glu 1+ are 2 ns, with an amplitude of 35% and 1 ns, with an amplitude of 65%. Lifetimes for Trp-Glu 0 are similarly short: 3 ns and 1.5 ns, but the amplitudes are very different; that of the 1.5 ns lifetime is 94%. Lifetimes for Trp-Glu 1− are 1.8 ns with 90% amplitude, and a much longer lifetime of 6.9 ns with only 10% amplitude. The lifetime measurements for Trp-Glu 2− are unique in that one long lifetime of 7.4 ns has an amplitude of 95%; the second, minor lifetime of 2 ns has an amplitude of only 5.0%.
Table 1.
Dihedral Angles with Probabilities and Fluorescence Lifetimesa for Trp-Glu Charged Species
| Dipeptide species | Chi 2 | Chi 1 | Lifetime/ns (Amplitude/%) | Goodness of Fit | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Median Angle/degrees | P/% | Median Angle/degrees | P/% | ||||
|
| |||||||
| Trp-Glu 1+ pH 1.7 | 87 | 69.1 | 64 | 34.1 | 2.08 (35.3) | 1.30 | |
| 264 | 30.0 | 187 | 13.8 | 1.03 (64.7) | |||
| 286 | 51.7 | ||||||
|
| |||||||
| Trp-Glu0 pH 3.5 | 85 | 81.8 | 67 | 9.6 | 3.04 (6.28) | 1.27 | |
| 262 | 17.3 | 182 | 20.5 | 1.49 (93.7) | |||
| 289 | 69.5 | ||||||
|
| |||||||
| Trp-Glu 1− pH 5.8 | 88 | 54.8 | 78 | 7.8 | 6.90 (10.2) | 7.22 (5.2)b | 1.29 |
| 263 | 44.5 | 182 | 27.0 | 1.81 (89.8) | 1.95 (82.5) | ||
| 288 | 64.6 | 0.73 (12.3) | |||||
|
| |||||||
| Trp-Glu 2− pH 10.0 | 75 | 66.4 | 70 | 83.4 | 7.43 (95.0) | 10.17 (23.9)b | 1.31 |
| 235 | 8.5 | 181 | 1.8 | 2.11 (5.01) | 6.79 (71.6) | ||
| 268 | 24.5 | 291 | 14.5 | 1.35 (4.4) | |||
281 nm excitation; 345 nm emission for Trp-Glu1+, Trp-Glu 0; 350 nm emission for Trp-Glu 1−; 355 nm emission for Trp-Glu 2−.
From RF Chen et al. 1991 18
Dihedral angles: determining relevance to fluorescence
The indicator of any specific local conformation is best identified by a dihedral angle associated with that conformation. The dihedral angles most likely to affect any indole ring interactions are the Chi 1 and Chi 2 angles, which are defined in Figure 2. This is because Chi 1 and Chi 2 dictate the proximity of the backbone closest to the indole ring. Other possible dihedral angles that could affect the environment about the indole ring and therefore, its fluorescence, are those involving the peptide backbone alone. These are the Psi 1 (atoms 11–14, Fig. 2), Psi 2 (atoms 14–17,), and Phi 2 (atoms 13–16) dihedral angles.
Figure 2.
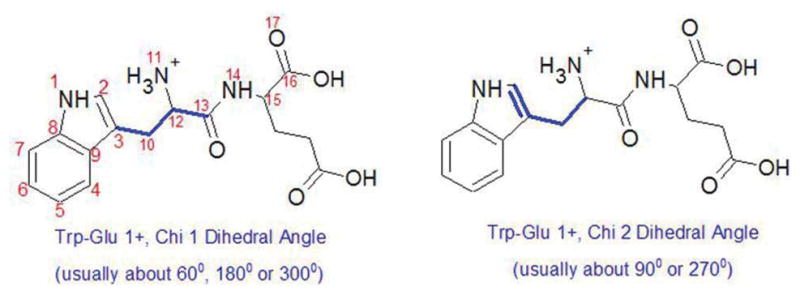
Atoms defining the dihedral angles in this study, numbered for convenience in the left structure. The Chi 1/Chi 2 dihedral angle is defined by the three bonds highlighted in bold line in the left/right structure. Psi 1 is the dihedral angle formed by atoms 11 to 14; Phi 2 is the angle formed by atoms 13 to 16; and Psi 2 is the angle formed by atoms 14 to 17.
The Psi 1 angle is essentially fixed at 150°
The Psi 1 angle for all Trp-Glu species is centered at ~ 150°, as shown in the angular probability graph in Fig. 3C, and remains in the same conformation throughout the simulations. Only Trp-Glu 2− (Fig. 3C, dotted line) has a slight probability of occurring at ~330°, 180° away from 150°. This small difference is unlikely to be the cause of the fluorescence emission spectral shift of Trp-Glu 2− (Fig. 1, dotted line) because the effect of this Psi 1 angle change is to replace one electronegative atom, the amide nitrogen, with another, the amide carbonyl oxygen. Otherwise, no pattern from the Psi 1 angle could be found between the species to account for observed fluorescence spectra or lifetime changes.
Figure 3.

Probability distribution of all angles. A. Chi 1; B. Chi 2; C. Psi 1; D. Psi 2; E. Phi 2. In all cases, the thick line represents Trp-Glu 1+ (30 ns simulation); long dash, Trp-Glu 0 (20 ns); thin line, Trp-Glu 1− (20 ns); short dash, Trp-Glu 2− (20 ns).
Two angles predominate for Psi 2
The Psi 2 angle probability for all Trp-Glu species, given in Fig. 3D, were similarly centered at 130° and/or ~320° for all species. During the course of the simulation, Psi 2 frequently switched between these two angles. Again there is an exception: Trp-Glu1+ remained at ~330° for almost the entire simulation (Fig. 3D, heavy solid line and Fig. S2). Because the Psi 2 angle here is basically a rotation of the terminal acid group, the Psi 2 angular preference can be explained. For all species this is a simple rotation of a COO− group, which has two equivalent conformers that are 180° apart. For the Trp-Glu1+ species, however, one oxygen is protonated, providing for very different electrostatic interactions between the positive -OH and indole ring on one hand and the negative --C=O and the indole ring on the other, as shown in Fig. 7A. Thus, the Trp-Glu1+ species are distinguished by a peculiar Psi 2 value, but Trp-Glu1+ fluorescence emission is not unique (Fig. 1). Psi 2 angle changes then cannot account for the observed fluorescence emission differences between the Trp-Glu dipeptides.
Figure 7.

Electrostatic potential surfaces of the indole plane for all Trp-Glu species. All conformers are arranged in order of increasing Chi 1 angle: 60°, 180°, 300°. A–C. Trp-Glu 1+, D–F. Trp-Glu 0, G–I. Trp-Glu 1−, J–L. Trp-Glu 2−. Significant electrostatic interactions are indicated by dashed line. Distances are given in the text. The scale at the bottom of each conformer is in atomic units, with red indicating negative charge and blue, positive charge.
Phi 2 angles are invariant
The Phi 2 angles are the same for all dipeptide species, and are characterized by a continual and rapid switching between ~220° and ~270° over the time of the simulation (Fig. 3E and Fig. S2). The limited Phi 2 motion can be explained through examination of molecular motion: if Phi 2 deviates significantly from 220–270°, the terminal carboxylate oxygens would sterically clash with the peptide oxygen (Data not shown). Although a curious phenomenon of the simulation, the Phi 2 angular distribution does not reflect the fluorescence changes between the dipeptide species (Fig. 1, Table 1).
Chi 2 places the backbone on either face of the indole ring
The Chi 2 dihedral angle is closest to the indole ring (Fig. 2, right). In all dipeptide species, it assumes values of ~ 90° and ~270° as shown in Fig. 3B, with 90° being slightly more favored. The Chi 2 angle describes the position of the planar indole ring relative to the rest of the molecule. Since 270° = −90°, the Chi 2 angles of 270° and 90° result in the backbone and Glu residue positioned either above or below the indole ring plane. As the simulated values of Chi 2 do not distinguish any single dipeptide species, Chi 2 rotamer values cannot be directly related to the observed fluorescence spectral differences (Fig. 1 and Table 1). Note that when Chi 2 = 90°, the terminal carboxylate may interact with the C2 hydrogen as shown in Fig. 7I. However, when Chi 2 = 270°, the terminal carboxylate interaction with the C4 hydrogen may occur (Fig. 7A).
Discrete Chi 1 angles define the terminal amine – indole interaction
The remaining dihedral angle, Chi 1 assumes only three values for all Trp dipeptide species, and the Chi 1 probability distribution peaks are found at: 64–78°, 181–187°, 286–291° (Fig. 3A). For sake of clarity, these preferred Chi 1 angle distributions will be referred to as 60°, 180°, and 300°. Each Chi 1 probability distribution peak is ~ 27° FWHM, and represents a range of possible angles. As thermal energy of a molecule at room temperature is ~0.6 kcal/mol, the experimental Chi 1 angle for dipeptide species will also vary.
Transitions between the three different Chi 1 angles were not frequent during the 20–30 ns simulations (For example, see Fig. S2). A Ramanchandran-like plot showing the six dihedral angle pairs, (Chi 1, Chi 2), adopted by the four Trp-Glu dipeptide ions, is given in Figure 6. Each Trp-Glu dipeptide species exhibits its own characteristic dihedral angle preferences. For Trp-Glu 1+ (Fig. 6A), only the (180°, 270°) pair is under populated. In the case of Trp-Glu 0 (Fig. 6B), the dihedral angle pair, (60°, 300°), is never visited, while the angle pair, (300°, 90°), are strongly favored. For Trp-Glu 1−, the dihedral angle pair, (60°, 90°) is not visited (Fig. 6C). The (Chi 1, Chi 2) angles for Trp-Glu 2−, given in Fig. 6D, are the most restricted as three dihedral angle pairs are sparsely populated: (180°, 90°), (300°, 90°) and (180°, 270°).
Figure 6.

Probability distribution of the dihedral angle pair, (Chi 1, Chi 2) for A. Trp-Glu 1+ B. Trp-Glu 0 C. Trp-Glu 1− D. Trp-Glu 2−. The scatter plot shows the Chi 1, Chi 2 pairs that describe the most common conformations. The linear plots along the axes show the probability distribution of the Chi 1 or Chi 2 angle throughout the simulations.
Chi 1 and Chi 2 angles change digitally over time
Examination of the 20–30 ns time course of simultaneous Chi 1, Chi 2 angle changes show their relative timing. One example of this is shown in Figure 4A, for Trp-Glu 1+. A Trp-Glu dipeptide ion typically maintains a particular Chi 1 angle up to ~ 8 ns before rapidly switching (within 1 ~ 50 fs) to one of the other two Chi 1 angles. There is no true interdependence of the Chi 1 and Chi 2 angle changes, but small changes in Chi 2 occur to accommodate a specific Chi 1 angle change or to facilitate its stability, as can be seen in Fig. 4A from about 17–21 ns. When Chi 2 ~90° and Chi 1 changes to 180°, Chi 2 accommodates this change by increasing to ~110°. In addition, some Chi 1 and Chi 2 transitions occur almost instantly, over the course of a few femtoseconds, and others occur somewhat more gradually, with the change taking 30–50 femtoseconds, as can be ascertained from Fig. 4A. The transitions between most Chi 1 and Chi 2 angles are common enough, but it is rare to have a transition between Chi 1~60° and Chi 1~300° without first having Chi 1~180°, as can be seen by examination of Fig. 4B. This is to be expected, because a direct transition between Chi 1~60° and Chi 1~300° would mean dragging the peptide backbone and glutamic acid residue across the π electrons of the indole ring.
Figure 4.

Molecular dynamics simulation Chi 1 and Chi 2 dihedral angle changes for the Trp-Glu 1+ rotamer. A. Chi 1 (black trace) and Chi 2 (red trace) angle changes as a function of time in picoseconds. B. Scatter plot, Chi 1 vs. Chi 2, where each angular transition is connected to the preceding and succeeding one.
Chi 1 preferences for Trp-Glu species
The dominant Chi 1 angle is 300° for Trp-Glu 1+, Trp-Glu 0, and Trp-Glu 1− whereas it is 60° for the Trp-Glu 2− species (Fig. 3A). Chi 1 dictates the relative orientation of the backbone to the indole ring, as illustrated in Figure 7. When Chi 1~180° or 300°, the backbone tends to be stretched out or oriented away from the indole ring, as illustrated by the molecular model for Trp-Glu 1+ (Fig. 7B) Trp-Glu 0 (Fig. 7E), Trp-Glu1− (Fig. 7H) or Trp-Glu 2− (Fig. 7K). However, when Chi 1~60°, the backbone tends to curl back over the indole, as shown by Fig. 7A, D, G, J.
Chi 1 determines NH3+ - indole interaction
The MD simulation shows that the Chi 1 angle tracks the interaction between the terminal amine cation H+ and the indole ring, as shown in Fig. 5. When Chi 1 is 180° or 300°, the distance between a terminal amine hydrogen and the center of the pyrrole ring of indole is ~3.5 Å, but when Chi is 60°, the distance increases to ~5.0 Å. As the van der Waals interaction cutoff radius is roughly 4 Å, Chi 1 defines the proximity of the terminal amine cation to the indole ring. As the terminal amine cation is known to participate in charge transfer a nonradiative process competing with fluorescence, Chi 1 is identified as a factor indirectly responsible for observed fluorescence differences.
Figure 5.
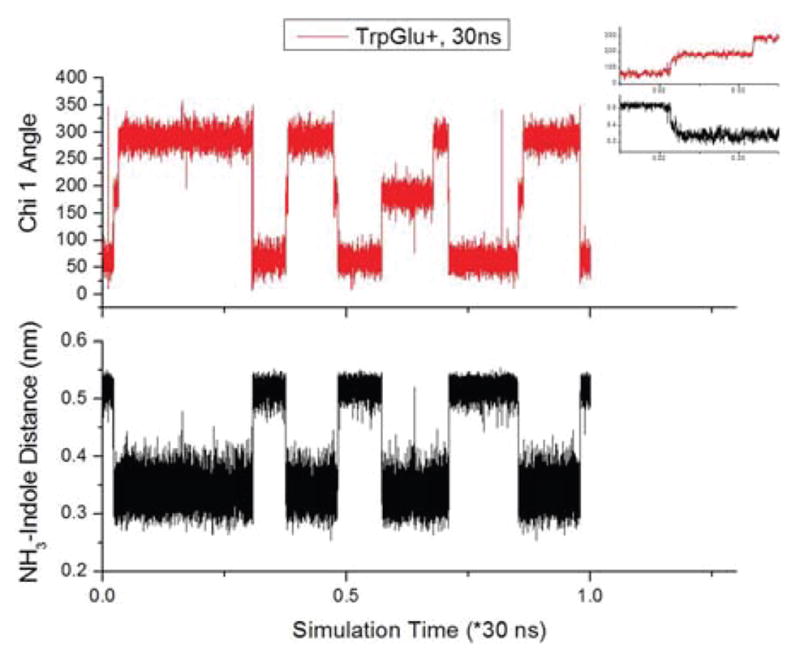
Correlation between the dihedral angle, Chi 1, for Trp-Glu+ (top) and the distance between the closest H of the terminal amine cation and the center of the pyrrole ring of indole (bottom). The inset (same axes and units) is a close-up of the tandem changes at the beginning of the MD simulation.
Relating Chi 1 angle simulation results to spectroscopic data
The scatter plot of Chi 1, Chi 2 dihedral angle pairs given in Figure 6 makes plain the rotamer preferences of each Trp-Glu species and the differences between them. While the Chi 1 angle, 300°, is favored by Trp-Glu 1+, Trp-Glu 0 and Trp-Glu 1−, the overwhelmingly favored Chi 1 angle by Trp-Glu 2− is 60° with a probability of 83% (Table 1). It is noteworthy that only Trp-Glu 2− avoids the Chi 1 = 180° dihedral angle (Fig. 6 and Table 1). The pattern of Chi1 dihedral angle preferences shown in Figure 6 and Table 1 can be matched to the pattern of steady state fluorescence emission maximum shifts (Fig. 1) and fluorescence lifetimes (Table 1) exhibited by the Trp-Glu ions. In both instances---theoretical and experimental---the Trp-Glu 2− species is distinguished. Experimentally, the Trp-Glu 2− emission maximum is at the longest wavelength, 354 nm (Fig. 1). One Trp-Glu 2− fluorescence lifetime is the longest of any here observed (7.43 ns) and has the highest amplitude (95%, Table 1). Theoretically, the Trp-Glu 2− species avoids the Chi 1 = 180° rotamer. Electrostatic potentials (Figure 7) bridge these data and provide explanation for the Chi 1 angular preferences, as discussed below.
Electrostatic interactions control conformer preference
The electrostatic potential for the three conformers of each Trp-Glu species is shown in Figure 7. Potentials are indicated for each atom in the plane of the indole ring. Potential for the methylene carbon attached to C3 is also included. Electrostatic interactions between atoms that are within 4 Å are indicated by a dashed line. In general, benzyl ring carbons, C4–C7 and the pyrrole nitrogen, N1, are negatively charged while charge on pyrrole ring carbons, C2 and C3, varies from rotamer to rotamer (Fig. 7 A–L). Interestingly, negative charge density can sometimes be found on the methylene carbon attached to C3 (Fig. 7 C, E, F, I, K, L). A check with natural bond order calculations (Data not shown) indicates that this negative charge density over the methylene carbon is not an artifact.
Molecules consisting of several joined aromatic rings, such as the indole studied here, are commonly depicted as having a uniform, negatively charged π electron cloud above and below the ring plane. Electrostatic calculations here show that the C8 and C9 atoms co-joining the pyrrole and benzyl rings carry a partial positive charge near the atomic plane (Fig. 7). Ultrafast electron diffraction study of indole reveals that the C8–C9 and C5–C6 bonds are slightly longer than others in the benzyl ring, giving the ring a ‘quinoid’ character [28]. Recent atomic force microscopy study of polycyclic aromatic hydrocarbons and fullerenes also show subtle bond order differences in conjoined rings [29]. The current, standard molecular representation of polyaromtics with alternating double bonds does not convey this subtlety in charge distribution. The positive charge on the C8 and C9 atoms is a fact that can impact on organic syntheses, as well as on stable conformations of molecules.
Backbone conformation
Examination of backbone conformation reveals that all but two rotamers (Fig. 7C and I) are characterized by two intrabackbone amide hydrogen bonds, each of which forms a C5 hydrogen bonded ring. The remaining two rotamers each have one C5 ring on their backbone (Fig. 7C and I). Amide hydrogen bond lengths vary slightly for each species: 1.98–2.26 Å for the Trp-Glu 1+, 1.87–2.08 Å for Trp-Glu 0, 1.87–2.14 Å for Trp-Glu 1− and 2.15–2.58 Å for Trp-Glu 2−. The C5 motif is found in β-sheets.
Numerous ring-backbone electrostatic interactions for Trp-Glu 1+ rotamers
Several electrostatic interactions stabilize the position of the backbone relative to the indole ring in the Trp-Glu 1+ rotamers (Fig. 7 A–C). As the C-terminal carboxyl group is protonated only in this species, electrostatic interactions between this group and the indole ring are unique. Thus for the 60° rotamer (Fig. 7A), the carboxyl – OH hydrogen bonds with ring C4 and C5 (distances: 4.5 Å).
The 60° rotamer for Trp-Glu1+ is also stabilized by the interaction of the negative, terminal carbonyl O with the slightly positive C8 and C9 atoms (distance: 3.8–3.9 Å). Another possible stabilization comes from the interaction of the peptide NH with the negatively charged C2 and C3 (distance: 3.0–3.4 Å). As for other Chi 1~60° rotamers, there is no interaction between the terminal amine group and the indole ring.
The Chi 1~180° rotamer for Trp-Glu 1+ is not heavily favored (Table 1), but significantly, it is stabilized by electrostatic interactions between the positive terminal NH3 group and the negative C2 and C3 atoms (distance: 2.4–2.6 Å, Fig. 7B). When Chi 1~180°, the Trp-Glu backbone for all species is stretched away from the indole ring.
The most likely Chi 1 angle for Trp-Glu 1+ is ~300° (Table 1, Fig. 7C). Once again, the positive NH3+ terminus hydrogen bonds with both C2 and C3 (distance: 2.4–3.2 Å). At the other end of the backbone, the terminal carboxyl oxygen hydrogen bonds to the hydrogen on C2 (distance: 2.4 Å). This last form of stabilization---where a large ring is formed between the backbone and the pyrrole---does not occur for any of the other Chi 1 angles. However, it is observed for Trp-Glu 0 (Chi 2 ~ 270°, Fig. 7F) and Trp-Glu 1− (Chi 2 ~ 90°, Fig. 7 I); and is the likely reason for the high probability of the Chi 1~300° conformation for all three species.
Trp-Glu 0 is also stabilized by terminal amine cation interactions
The propensity of the Trp-Glu 0 species to adapt the Chi 1~60° is much less than for Trp-Glu 1+ (Table 1). The electrostatic calculations show an interaction between the peptide NH and the negative C3 (distance: 3.1 Å, Fig. 7 D). Some additional stability may arise from a C4 hydrogen – carbonyl oxygen interaction (distance: 3.4 Å). There is also a favorable interaction between the terminal NH3 and the peptide carbonyl O (Not shown, distance: 2.1 Å). Trp-Glu 0 has a higher probability of adapting the Chi 1~180° conformation, shown in Fig. 7E. It is mainly stabilized by the attraction between the terminal NH3 cation and the negative C2 and C3 atoms (distance: 2.5–2.6 Å). As for Trp-Glu 1+, the most likely rotamer for Trp-Glu 0 is one where Chi 1~300° (Table 1, Fig. 7F). It is stabilized by terminal NH3 interactions with C2 and C3 (distance: 2.4–2.5 Å), and terminal carboxyl oxygen attraction to the C4 hydrogen (distance: 3.6 Å). Note that here, Chi 2 ~ 270° so that a ring is formed between the backbone and the benzyl ring. This rotamer is at somewhat lower energy than the (300°, 90°) rotamer for Trp-Glu 0.
Trp-Glu 1− conformer distribution follows that of Trp-Glu 0
The Trp-Glu 1− conformer has a small probability of a Chi 1~60° (Table 1), and this conformation is stabilized by hydrogen bonding between the peptide NH and the C2 and C3 ring carbons (distance: 3.0–3.3 Å, Fig. 7G).
The more likely Chi 1~180° rotamer (Fig. 7H) is stabilized by the terminal NH3 cation attraction to the C3 and C2 atoms (distance: 2.4–2.7 Å). Again the Chi 1~300° rotamer (Fig. 7I) is the most likely due to the terminal amine cation and terminal carboxyl anion interactions with the pyrrole ring. The standard stabilization for the Chi 1~300° rotamer is the proximity of the positive NH3 to the C3 and C2 atoms (distance: 2.4 Å, 3.2 Å, resp.), as in species discussed above. There is also an “edge-of-indole ring” interaction between the terminal carboxyl oxygen and the C2 hydrogen (distance: 2.2 Å), as observed for Trp-Glu 1+ in the 300° rotamer (Fig. 7C). The analogous interaction for the Trp-Glu 0, Chi 1~300° rotamer occurs with the C4 hydrogen (Fig. 7F). It is likely that for all the Trp-Glu, Chi 1~300° rotamers, both C4 and C2 hydrogens bond to the terminal carboxyl, but only the lowest energy conformer is used in the quantum mechanical calculation. The Chi 2 angle determines whether the C2 or C4 atom hydrogen bonds to the terminal carboxyl. When Chi 2 = 90°, the backbone swings along side of the pyrrole ring, facilitating carboxyl H-bonding to the C2 hydrogen. When Chi 2 = 270°, the backbone arches around to the benzyl ring, where carboxyl hydrogen bonding to C4 hydrogen occurs. The methylene group is again negatively charged (Fig. 7I). This phenomenon seems to be a redistribution of the π ring electron density towards the methylene group, with the positive NH3 group taking up some of the electron density from C2 and/or C3.
Trp-Glu2− conformers possess the fewest electrostatic interactions
The Trp-Glu 2− species is unlike all the others in its Chi 1 angle probability distribution because the greatest probability is for Chi 1~60°, not 300°. Oddly enough, the electrostatics show nothing in particular stabilizing this conformation (Fig. 7J).
After finding the lowest energy Chi 1~180° conformation in the molecular dynamics simulation and further optimizing the geometry using DFT, the Chi 1~180° rotamer for Trp-Glu 2− shows an unexpected Chi 2~10°. The most common and certainly the least sterically hindered Chi 2 should be ~ ± 90°, but for the Trp-Glu 2− species this is rejected when Chi 1~180°. If Chi 2 were the standard ~90°, the negative N of the NH2 would be situated above the negative C2 and C3 atoms of the indole. The hydrogens of the NH2 would be positioned close to the peptide oxygen, but there is no third amine hydrogen to interact with the π electrons, as for the other Trp-Glu species. The Chi 1~180° rotamer (Fig. 7K) gains some stability by the interaction of the terminal N with the positive C2 hydrogen (distance: 2.8 Å), but it seems that the sterically opposed structure caused by the Chi 2~10° angle results in a very low probability (Table 1) for this rotamer. Again, the negative charge on the methylene group is noted.
The Trp-Glu 2− Chi 1~300° rotamer (Fig. 7L) is stable when Chi 2~ ±90°. The positive H of the terminal NH2 interacts favorably with the negatively charged C2 atom (distance: 2.9 Å), but the electronegative N (distance: 3.3 Å), will repulse the C2 atom. The net effect of these electrostatics is to give the Chi 1~300° a higher probability than that of the 180° rotamer, yet to keep it from having as high a probability as the Chi 1~60° rotamer (Fig. 7J). The negative charge on the methylene group seems to be an extension of the ring π electron cloud.
Discussion
From a broad perspective, charges on the backbone and in some cases, the glutamate residue of the Trp-Glu dipeptide species control the number of noncovalent interactions with the indole ring and hinder conversion from one rotamer to another. These interactions are made explicit in the low energy conformers obtained by MD simulations (Fig. 7). Where the terminal amine is positively charged, backbone-indole ring noncovalent interactions are numerous (Fig. 7 A–I). Specifically, amine cation-π electron attractions are indicated in most rotamers (Fig. 7A–I). Additionally, terminal carboxylate, edge-on interaction with ring hydrogens are also indicated in at least three rotamers (Fig. 7C, F, I). For the Trp-Glu 2− species (Fig. 7 J–L), the negatively charged backbone is electrostatically repelled from the π electron cloud sandwiching the indole ring so backbone-indole ring interactions are sparse. As charge transfer mechanisms are associated with both diminished quantum yields and emission blue shifts for tryptophan, the blue emission shifts observed for the positively charged Trp-Glu species (Fig. 1) likely result from their backbone-indole ring electrostatic interactions. We note that an additional effect of the NH3 cation proximity to the indole ring in the stretched out rotamers is a partial shielding of the ring from solvent.
Discrete Chi 1 angles control backbone – indole interactions
The Chi 2 dihedral angle takes on two values for all Trp-Glu species, ~90° or ~270°, placing the backbone above or below the symmetric indole plane. The Chi 1 angle, however, is centered on three values: 60°, 180°, and 300° (Fig. 6) according to MD simulation. The 60° angle curls the backbone over the indole ring (Fig. 7A, D, G, J) with the shape and angles of the curled backbone depending on the backbone angles, Psi 1, Psi 2, and Phi 2. The N---C bond of the NH3 group is parallel to the indole ring plane. Amine hydrogens point away from the ring so there is no cation-π interaction. In contrast, when the Chi 1 angle is either ~180° or ~300°, the backbone is typically “stretched’ away from the indole ring (Fig. 7 B, C, E, K, L), with the overall backbone shape again determined by Psi 1, Psi 2, and Phi 2. For these Chi 1 angles, the NH3 group is situated right above C2 or C3 of the indole ring (Fig. 7B–C, E–F, H–I). This NH3+- ring interaction explains why the fluorescence emission maximum for Trp-Glu 2− (354 nm) is red shifted ca. 10 nm relative to the emission maxima for Trp-Glu 1+, Trp-Glu 0, and Trp-Glu 1−. The lack of charge on the Trp-Glu 2− terminal amine obviates quenching-associated fluorescence blue shifts. In the absence of electrostatic interaction between the backbone and indole ring, the Chi 1~60° conformer is favored.
Correlations between fluorescence lifetimes and rotamer populations
A broad pattern of correlation between the fluorescence lifetimes and Chi 1 dihedral angle probability distribution is found (Table 1). Two lifetimes have been obtained for each of the Trp-Glu species. For each species, the longer lifetime amplitude correlates roughly with the Chi 1~60° rotamer probability. Recall that the Chi 1~60° rotamer represents the backbone “curled” conformation (Fig. 7A, D, G, J) where the terminal amine cation does not interact with the indole ring, and charge transfer quenching is apparently minimized. The shorter lifetime amplitude is then related to the probability amplitudes for the Chi 1~180° and 300° rotamers where the backbone assumes a “stretched out” conformation (Fig. 7B–C, E–F, H–I, K–L) and the terminal amine cation is positioned close to the indole ring. Where MD simulation shows that the backbone prefers a “stretched out” conformation, as for Trp-Glu 1+, Trp-Glu 0 and Trp-Glu 1−, the shorter lifetime has the greater amplitude (64.7–89.8%). MD simulation shows that the Trp-Glu 2− species prefers the Chi 1 ~ 60° rotamer (Table, 1, 83.4% and Fig. 6D) where there is no backbone-indole ring interaction, and thus the longer lifetime for this species has the greater amplitude (95.0%). The pattern of fluorescence lifetimes and associated amplitudes is thus consistent with the conformational preferences of each of the Trp-Glu species, specifically with the Chi 1 dihedral angle preference, which controls the placement of the terminal amine cation with respect to the indole ring, and therefore controls charge transfer quenching.
Although the Chi 2 angle probabilities for the Trp-Glu 1+ (Table 1) suggest a correlation with its fluorescence lifetime amplitudes, examination of the progression of Chi 2 probabilities from Trp-Glu 1+ to Trp-Glu 2− shows that the correlation breaks down as negative charge increases on Trp-Glu.
Electrostatic Potential Surfaces
The electrostatic potential surfaces (Fig. 7) provide an insightful explanation for the favored conformations of these species. This data shows that the main stabilizing factor for conformations where Chi 1~180° or 300° is the noncovalent interaction of the positive NH3 with the partially negatively charged C2 or C3 atoms (Fig. 7B–C, E–F, H–I). This favorable interaction is only possible for Trp-Glu 1+, Trp-Glu 0, and Trp-Glu 1−, which is why their dominant conformation has Chi 1~180° or 300°. For all these species, Chi 1~300° is the more probable dihedral angle (Fig. 6A–C) because the negative terminal carbonyl attraction to either the positive C2 hydrogen (Fig. 7C, I) or the positive C4 hydrogen (Fig. 7F) adds additional stability.
Even the larger probability of the Chi 1~ 60° conformation for the Trp-Glu 1+ species relative to that for Trp-Glu 0 and Trp-Glu 1− (Table 1) can be rationalized in terms of backbone electrostatic interaction with the indole ring. The terminal carboxylic acid hydrogen of Trp-Glu 1+, absent in the other two species, has a favorable noncovalent interaction with the partially negative C5 and C6 atoms (Fig. 7A). It should be noted that the Trp-Glu 1+ emission band is broader than that of the Trp-Glu 0 and Trp-Glu 1− on the band red edge (Fig. 1), quite possibly because of the higher relative amount of the Chi 1~60° conformation in that species.
Indole Ring Resonance Extends to Methylene Group
The electrostatic calculations reveal an unexpected negative charge on the tryptophan methylene group (Fig. 7L), which exists even where the C3 atom has a more positive charge (Fig. 7C, F, I, K). Natural bond order calculations show a similar pattern of negative charge on the methylene group, adding reassurance that this exo-ring negative charge is not an artifact of the simulation (Data not shown). The methylene negative charge suggests a resonance structure that extends beyond and includes the indole ring. Calculations of electron density for 3-methyl indole show a negative charge density on the methyl group as well (Data not shown). Preliminary experiments that test for H-D exchange at the 3-methyl position are in progress. Results will be presented in a forthcoming paper.
Conclusions
Steady state fluorescence results for solution phase Trp-Glu ions show a red shift in the emission maximum only when the terminal amine is deprotonated. Fluorescence lifetime measurements reveal a more gradual lengthening in one of two lifetimes and associated amplitude with increasing Trp-Glu negative charge. 20 to 30 ns Molecular dynamics simulations of each Trp-Glu ion species suggest specific aqueous phase conformational changes and noncovalent interactions to account for the observed fluorescence data. Discrete pairs of Chi 1, Chi 2 angles control the interaction of the backbone with the indole ring. The role of Chi 2 is basically to position the backbone above or below the symmetric indole plane. Chi 1 is the factor determining noncovalent interactions between backbone charged groups and the indole ring. The most significant of these appears to involve the terminal amine cation, which is expected to participate in cation-π interaction with the indole ring and is believed responsible for charge transfer. Where this interaction is present (Trp-Glu 1+, Trp-Glu 0 and Trp-Glu1−), the emission maximum is blue-shifted (i.e., 343 −7 nm) and fluorescence lifetimes are shorter. Molecular dynamics simulations show that Chi 1 takes on three discrete values for all Trp-Glu ions. These are centered about 60°, 180° and 300°. The 60° rotamer induces the backbone to ‘curl’ back over the indole ring with the result that the terminal amine is not positioned over the ring. This Chi 1 angle dominates the Trp-Glu 2− simulation; emission is red-shifted, and a long lifetime of 7 ns dominates. The 180° and 300° Chi 1 angles cause the backbone to stretch away from the indole ring, such that the terminal amine cation is within hydrogen bonding distance of the indole. These Chi 1 angles are favored by Trp-Glu 1+, Trp-Glu 0 and Trp-Glu 1−. Emission is blue shifted for these species and fluorescence lifetimes are shorter. It is important to realize that amine cation-π interactions are a significant Arg or Lys residue interaction with Trp in proteins. Protein interfaces are enriched in aromatic amino acids, including Trp, and Arg, suggesting a role for cation- π interaction in macromolecular assembly [16].
Backbone charged group interactions may also restrict rotamer motion. For an indole buried in a confining protein matrix, rotamer motion may indeed be very restricted; but charged groups positioned about the indole will still determine radiative and nonradiative processes as shown here for the Trp-Glu dipeptide species. Where tryptophan is accessible, solvent relaxation will also play a role. Mapping of electrostatic potential for each Trp-Glu rotamer reveals subtle changes in ring electron density with backbone interaction. For all species, C8 and C9 atoms are relatively positively charged. Charge on the C2 and C3 atoms greatly varies from rotamer to rotamer. A partial negative charge is also found on the exo-ring methylene group in some rotamers, suggesting an extension of ring conjugation to this carbon.
Supplementary Material
Acknowledgments
This work was made possible by NIH grant 41744-00-02. We thank Prof. Mark Kobrak for advice given in review of the manuscript.
Contract/grant sponsor: the National Institutes of Health; contract/grant number: 41744-00-02.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Callis PR, Liu T. J Phys Chem B. 2004;108:4248–4259. [Google Scholar]
- 2.Chen Y, Barkley M. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 3.Peralta Conde A, Ovejas V, Montero R, Castano F, Longarte A. Chem Phys Letters. 2012;530:25–30. [Google Scholar]
- 4.Vivian JT, Callis PR. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dian BC, et al. J Chem Phys. 2002;117:10688–10702. [Google Scholar]
- 6.Dian BC, Longarte A, Zwier TS. J Chem Phys. 2003;118:2696–2706. [Google Scholar]
- 7.Haber T, Seefeld K, Engler G, Grimme S, Kleinermanns K. Phys Chem Chem Phys. 2008;10:2844–2851. doi: 10.1039/b718710f. [DOI] [PubMed] [Google Scholar]
- 8.Hunig I, Kleinermanns K. Phys Chem Chem Phys. 2004;6:2650–2658. [Google Scholar]
- 9.Hünig I, Seefeld KA, Kleinermanns K. Chem Phys Lett. 2003;369:173–179. [Google Scholar]
- 10.Nix MGD, Devine AL, Cronin B, Ashfold MNR. Physical Chemistry Chemical Physics. 2006;8:2610–2618. doi: 10.1039/b603499c. [DOI] [PubMed] [Google Scholar]
- 11.Shemesh D, Sobolewski AL, Domcke W. Phys Chem Chem Phys. 2010;12:4899–4905. doi: 10.1039/b927024h. [DOI] [PubMed] [Google Scholar]
- 12.Fujihara A, Matsumoto H, Shibata Y, Ishikawa H, Fuke K. J Phys Chem A. 2008;112:1457–1463. doi: 10.1021/jp709614e. [DOI] [PubMed] [Google Scholar]
- 13.Fujihara A, Noguchi N, Yamada Y, Ishikawa H, Fuke K. J Phys Chem A. 2009;113:8169–8175. doi: 10.1021/jp902451k. [DOI] [PubMed] [Google Scholar]
- 14.Mercier SR, et al. J Am Chem Soc. 2006;128:16938–16943. doi: 10.1021/ja065980n. [DOI] [PubMed] [Google Scholar]
- 15.Singh J, Thornton JM. Atlas of protein side-chain interactions. 1–2 Oxford University Press; New York, NY: 1991. [Google Scholar]
- 16.Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 17.Jackson MR, et al. J Phys Chem B. 2007;111:8242–8249. doi: 10.1021/jp0661995. [DOI] [PubMed] [Google Scholar]
- 18.Chen RF, Knutson JR, Ziffer H, Porter D. Biochemistry. 1991;30:5184–5195. doi: 10.1021/bi00235a011. [DOI] [PubMed] [Google Scholar]
- 19.Eisenberg A, Juszczak L. J Amino Acids. 2012;2012:10. doi: 10.1155/2012/73506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frisch MJ, et al. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 21.Varetto U. MOLEKEL 5.4.0.8. Swiss National Supercomputing Centre; Lugano, Switzerland: [Google Scholar]
- 22.Laaksonen LL. gOpenMol. Center of Scientific Computations; Espoo, FI: 1999. [Google Scholar]
- 23.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen W, Maxwell D, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 25.Kaminski G, Friesner R, Tirado-Rives J, Jorgensen W. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 26.Johnson M, Frasier S. In: Methods Enzymol. Hirs C, Timashef S, editors. Vol. 117. Elsevier; New York: 1985. pp. 301–342. [Google Scholar]
- 27.Anonymous. DAS 6 User’s Guide. Horiba, Inc; Edison, NJ: Part # J81119. [Google Scholar]
- 28.Park ST, Gahlmann A, He Y, Feenstra JS, Zewail AH. Angew Chem Int Ed. 2008;47:9496–9499. doi: 10.1002/anie.200804152. [DOI] [PubMed] [Google Scholar]
- 29.Gross L, et al. Science. 2012;337:1326–1329. doi: 10.1126/science.1225621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.