

This paper presents detailed kinetic assays for three activities, cyclic phosphodiesterase, polynucleotide kinase, and ligase, of the trifunctional plant tRNA ligase AtRNL. Comparison with a fungal tRNA ligase, KlaTrl1, shows important functional distinctions.
Keywords: 2′,3′ cyclic phosphodiesterase; RNA ligase; RNA repair; polynucleotide kinase
Abstract
Plant and fungal tRNA ligases are trifunctional enzymes that repair RNA breaks with 2′,3′-cyclic-PO4 and 5′-OH ends. They are composed of cyclic phosphodiesterase (CPDase) and polynucleotide kinase domains that heal the broken ends to generate the 3′-OH, 2′-PO4, and 5′-PO4 required for sealing by a ligase domain. Here, we use short HORNA>p substrates to determine, in a one-pot assay format under single-turnover conditions, the order and rates of the CPDase, kinase and ligase steps. The observed reaction sequence for the plant tRNA ligase AtRNL, independent of RNA length, is that the CPDase engages first, converting HORNA>p to HORNA2′p, which is then phosphorylated to pRNA2′p by the kinase. Whereas the rates of the AtRNL CPDase and kinase reactions are insensitive to RNA length, the rate of the ligase reaction is slowed by a factor of 16 in the transition from 10-mer RNA to 8-mer and further by eightfold in the transition from 8-mer RNA to 6-mer. We report that a single ribonucleoside-2′,3′-cyclic-PO4 moiety enables AtRNL to efficiently splice an otherwise all-DNA strand. Our characterization of a fungal tRNA ligase (KlaTrl1) highlights important functional distinctions vis à vis the plant homolog. We find that (1) the KlaTrl1 kinase is 300-fold faster than the AtRNL kinase; and (2) the KlaTrl1 kinase is highly specific for GTP or dGTP as the phosphate donor. Our findings recommend tRNA ligase as a tool to map ribonucleotides embedded in DNA and as a target for antifungal drug discovery.
INTRODUCTION
Plant and fungal tRNA ligases, exemplified by Arabidopsis thaliana AtRNL and Saccharomyces cerevisiae Trl1 (SceTrl1), are essential agents of informational and stress response pathways involving the repair of programmed tRNA and mRNA breaks with 2′,3′-cyclic phosphate and 5′-OH ends. Plant and fungal tRNA ligases perform three distinct RNA repair reactions: (1) The 2′,3′-cyclic phosphate (>p) end is hydrolyzed to a 3′-OH,2′-PO4 by a cyclic phosphodiesterase (CPDase); (2) the 5′-OH end is phosphorylated by an NTP-dependent polynucleotide kinase; and (3) the 3′-OH,2′-PO4 and 5′-PO4 ends are sealed by an ATP-dependent RNA ligase to form an unconventional 2′-PO4, 3′-5′ phosphodiester at the splice junction (Konarska et al. 1982; Greer et al. 1983; Schwartz et al. 1983; Pick et al. 1986; Pick and Hurwitz 1986; Englert and Beier 2005). AtRNL and SceTrl1 are composed of three separable catalytic domains: a C-terminal CPDase module belonging to the “2H” phosphoesterase superfamily; a central kinase module of the P-loop phosphotransferase superfamily; and an N-terminal ligase domain that belongs to the covalent nucleotidyltransferase superfamily (Fig. 1A; Apostol et al. 1991; Sawaya et al. 2003; Wang and Shuman 2005; Wang et al. 2006).
FIGURE 1.

One-pot assay for monitoring the AtRNL end-healing and sealing reactions. (A) Arabidopsis tRNA ligase is composed of an N-terminal ligase module, a central 5′ kinase module, and a C-terminal CPDase module, depicted as a cartoon model at the top of the panel. The site of covalent adenylylation at the ligase active site, the P-loop motif and aspartate general base at the kinase active site, and the two HxT motifs that comprise the CPDase active site are highlighted. AtRNL mutants with inactivating alanine substitutions in each of the three catalytic modules were reacted with a 3′ 32P-labeled 10-mer HORNA>p substrate (depicted at the bottom of the panel with the 32P-label denoted by •). Reaction mixtures (20 µL) containing 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 20 nM 10-mer HORNA>p, and 1 µM AtRNL (wild type or mutant as specified) were incubated for 5 min at 22°C. Individual reaction components were included (+) or omitted (−) as specified. The reactions were quenched with an equal volume of 90% formamide, 30 mM EDTA. The products were analyzed by electrophoresis through a 20% polyacrylamide gel containing 7 M urea in TBE. An autoradiograph of the gel is shown. The position and identities of the radiolabeled RNA substrate and the various healed or sealed products are indicated on the left and right. (B,C) Order and kinetic profile of the splicing reactions under single-turnover conditions. A reaction mixture (200 µL) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 10 mM MgCl2, 0.1 mM ATP, 20 nM 10-mer HORNA>p substrate, and 1 µM AtRNL was incubated at 22°C. Aliquots (20 µL) were removed at the times specified and quenched immediately with an equal volume of 90% formamide, 30 mM EDTA. The time 0 sample was withdrawn and quenched prior to adding AtRNL. The products were resolved by urea-PAGE. An autoradiogram of the gel is shown in panel C. The levels of the healed HORNA2′p and pRNA2′p intermediates and the ligated circle product were quantified by scanning the gel with a Fujix BAS2500 imager and are plotted as a function of time in panel B. Each datum is the average of three separate time-course experiments ±SEM. The data were fit by nonlinear regression in Prism to a sequential three-step reaction pathway shown at right in panel B. The apparent rate constants for the CPD, kinase, and ligase reactions are indicated.
The CPDase and kinase reactions are collectively referred to as “end-healing” (Schwer et al. 2004). Healing provides the proper termini for end sealing, which is dependent on the 2′-PO4 moiety. The sealing phase of tRNA splicing requires ATP and proceeds via three adenylate transfer steps. First, the ligase domain reacts with ATP to form a covalent ligase-(lysyl-Nζ)-AMP intermediate and displace pyrophosphate. Adenylylation occurs at a conserved lysine (Lys152 in AtRNL) (Fig. 1A) within a signature KxxG motif. Second, ligase transfers AMP to the 5′-PO4 RNA terminus to form an RNA-adenylate intermediate (AppRNA). Third, ligase directs the attack of the 3′-OH on AppRNA to form the splice junction and displace AMP.
Despite the importance of tRNA splicing in plant and fungal physiology, little was known about the kinetics of the RNA repair reactions catalyzed by tRNA ligases or how the specificity for sealing 2′-PO4 ends is enforced. Focusing initially on AtRNL, we aimed to close this knowledge gap, by determining the rates of end-healing and sealing under single-turnover conditions. We exploited linear 20-mer RNA substrates with different terminal structures and a collection of AtRNL mutants to dissect individual steps or subsets of steps along the RNA repair pathway (Remus and Shuman 2013). The linear RNAs used for those studies mimic an excised tRNA intron, which is efficiently sealed by AtRNL to form a circular RNA product (Englert and Beier 2005; Nandakumar et al. 2008).
We found that the isolated CPDase reaction was faster than the kinase and ligase reactions, suggesting that the 2′,3′>p // 5′-OH break is converted to a 3′-OH,2′-PO4 // 5′-OH prior to its conversion by the kinase to a 3′-OH,2′-PO4 // 5′-PO4 that can be joined by the ligase module. The rates of sealing of the half-healed HORNA2′p and fully healed pRNA2′p (0.056 sec−1) repair intermediates were similar to one another and to the rate of the complete HORNA>p repair pathway (0.048 sec−1), implying that the kinase and ligase reactions are jointly rate limiting. Whereas the ligase is strictly dependent on ATP as the nucleotide substrate for end sealing, the kinase readily used ATP, GTP, CTP, UTP, or dATP as the phosphate donor (Remus and Shuman 2013). Thus, the AtRNL kinase is not fastidious with respect to the nucleobase or the pentose sugar of the NTP.
We gained the first mechanistic insights into the 2′-PO4 specificity of ligation by showing that AtRNL readily seals a preadenylylated AppRNAOH substrate with a 3′-OH,2′-OH end. The similar rates of AppRNA2′p and AppRNAOH sealing indicated that the 2′-PO4 moiety makes little contribution to the phosphodiester synthesis step (step 3) of the ligase reaction. Rather, 2′-PO4 specificity of AtRNL is enforced at the level of RNA 5′ adenylylation (step 2), whereby the conversion of pRNAOH to AppRNAOH and ligated RNA in the presence of ATP was several hundredfold slower than the sealing of pRNA2′p under the same reaction conditions. We proposed that the strategy employed by AtRNL for discriminating 2′-PO4 vs. 2′-OH ends provides a quality control checkpoint to ensure that only purposeful RNA breaks are sealed and to avoid nonspecific capping of 5′-PO4 ends (Remus and Shuman 2013).
Here, we extend the analysis of plant tRNA ligase to address the following questions: (1) What is the minimal RNA substrate for end-healing and sealing? (2) How do decrements in RNA chain length affect the kinetics of the healing and sealing steps? and (3) Can AtRNL splice a 5′-OH DNA substrate with a single ribonucleoside-2′,3′-cyclic-PO4 end?
We also analyze the kinetics and substrate requirements of an exemplary fungal tRNA ligase, Kluyveromyces lactis Trl1 (KlaTrl1). Fungal Trl1 enzymes are potential therapeutic targets, because their structures and biochemical mechanisms are unique compared to the RtcB-type tRNA repair systems elaborated by metazoans, archaea, and many bacteria (Popow et al. 2011; Tanaka and Shuman 2011; Tanaka et al. 2011; Chakravarty et al. 2012; Englert et al. 2012; Das et al. 2013a; Desai et al. 2013). Indeed, mammalian proteomes have no discernable homologs of the sealing domain of fungal tRNA ligase. These features highlight the sealing components of tRNA ligases as untapped targets for the discovery of new anti-fungals. Even more attractive is the prospect of inhibiting multiple enzymatic steps in the fungal tRNA splicing pathway, which would reduce the risk of the emergence of resistance. For this scenario to be plausible, the case must be made that the CPDase or kinase modules of fungal Trl1 enzymes have “drugable” properties.
In the present study, we were especially interested in the nucleotide substrate specificity of the KlaTrl1 kinase module, in light of prior studies indicating that SceTrl1 preferentially uses GTP as the phosphate donor for its kinase reaction in vitro and in vivo (Westaway et al. 1993; Sawaya et al. 2003). We show that KlaTrl1 is highly selective for GTP or dGTP as the phosphate donor, and we exploit nucleotide analogs to elucidate the atomic determinants of nucleobase specificity. Kinetic analysis reveals that (1) the fungal kinase is much faster than the homologous plant kinase, and (2) the ligation steps are rate limiting for the joining of linear RNAs by KlaTrl1. Thus, fungal and plant tRNA ligases have distinctive substrate specificities and kinetic properties when executing the same chemical reactions.
RESULTS
One-pot assay for monitoring the AtRNL end-healing and sealing reactions
Previously, we employed a 3′ 32P-labeled 20-mer HORNA>p substrate to monitor the progress of the AtRNL splicing reaction. A technical limitation to this approach was the inability to separate by urea-PAGE the input 20-mer HORNA>p substrate from the HORNA2′p product of the CPDase reaction; consequently, the measurement of CPDase activity entailed an additional (and cumbersome) step in which the reactants and products were digested with RNase T1 to yield 3′ 32P-labeled tetra-nucleotides with 2′,3′-cyclic-PO4 or 2′-PO4 ends that could be resolved by PAGE (Remus and Shuman 2013). Here, in order to assay the healing and sealing pathway of AtRNL in a one-pot format, we used a 10-mer HORNA>p substrate with a single radiolabel between the 3′-terminal and penultimate nucleosides (Fig. 1A). The 10-mer HORNA>p substrate (20 nM) was reacted for 5 min with a 50-fold molar excess of recombinant wild-type AtRNL in the presence of 10 mM Mg2+ and 0.1 mM ATP. To limit the reaction to just the CPDase step, we either omitted Mg2+ and/or ATP, or replaced wild-type AtRNL with mutated versions K700A-S701A or D726A that are devoid of kinase activity (Remus and Shuman 2013). To limit the reaction to just the kinase step, we used the CPDase-defective T1001A mutant (Remus and Shuman 2013). To allow end-healing but preclude the ligation reaction, we used the K152A mutant. The reaction products were analyzed by urea-PAGE and visualized by autoradiography (Fig. 1A).
In the reactions limited to the CPDase, the HORNA>p substrate was hydrolyzed to form a more rapidly migrating HORNA2′p species (Fig. 1A). When the ligation reaction was prevented by the K152A mutation, the HORNA>p substrate was converted into a fully healed pRNA2′p species that migrated ahead of the HORNA2′p CPDase reaction product (Fig. 1A). The CPDase-defective T1001A mutant performed the 5′ phosphorylation step, leading to the formation of a pRNA>p species that could not be ligated. In contrast, wild-type AtRNL converted the 10-mer HORNA>p substrate into a circular product, migrating more rapidly than the healed pRNA>p species, via intramolecular end joining (Fig. 1A). Thus, we can resolve the healed intermediates and sealed products of the AtRNL splicing pathway by using a short RNA substrate.
Order and kinetic profile of the AtRNL reactions under single-turnover conditions
The temporal progress of the reaction of 1 µM AtRNL with 20 nM 10-mer HORNA>p substrate is shown in Figure 1C. The substrate was sequentially converted into HORNA2′p and pRNA2′p intermediates en route to the ligated circle product. The distribution of labeled RNAs is plotted in Figure 1B, with each datum being the average of three separate kinetic experiments (±SEM). The data were fit by nonlinear regression in Prism to a sequential three-step mechanism depicted in Figure 1B, whereby the reaction order is first CPDase, then kinase, then ligase. The single-turnover rate constants were 0.10 ± 0.005 sec−1 for the CPDase, 0.049 ± 0.0015 sec−1 for the kinase; and 0.035 ± 0.0011 sec−1 for the ligase (Fig. 1B). The rate of the AtRNL ligation step with the 10-mer substrate HORNA>p (0.035 sec−1) was similar to the rate determined previously with a 20-mer HORNA>p (0.056 sec−1) (Remus and Shuman 2013).
Effects of RNA size decrements on the kinetics of end-healing and sealing
It was of interest to determine the effects of shortening the HORNA>p substrate on the outcomes and kinetics of the component steps of the AtRNL splicing pathway, with the aim of identifying minimized substrates for each enzymatic module. We began by shortening the 10-mer substrate to an 8-mer, removing the 5′ dinucleotide but otherwise maintaining the nucleotide sequence (Fig. 2A). Reactions of wild-type AtRNL and kinase-defective, ligase-defective, or CPDase-defective AtRNL mutants with the 3′ 32P-labeled 8-mer HORNA>p yielded half-healed HORNA2′p or pRNA>p derivatives or a fully healed pRNA2′p that were resolvable by urea-PAGE (Fig. 2A). The wild-type enzyme effected only partial conversion of pRNA2′p to circular product during the 5 min reaction (Fig. 2A), suggesting that the length decrement might impact sealing selectively.
FIGURE 2.

Reaction of AtRNL with an 8-mer HORNA>p substrate. (A) Wild-type AtRNL and the indicated mutants were reacted with a 3′ 32P-labeled 8-mer HORNA>p substrate (depicted at the bottom of the panel with the 32P-label denoted by •). Reaction mixtures (20 µL) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 20 nM 8-mer HORNA>p, and 1 µM AtRNL (wild-type or mutant as specified) were incubated for 5 min at 22°C. The reactions were quenched with formamide, EDTA, and the products were analyzed by urea-PAGE. An autoradiograph of the gel is shown. The position and identities of the radiolabeled RNA substrate and the various healed or sealed products are indicated on the left and right. (B,C) Single-turnover kinetics. A reaction mixture (250 µL) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 10 mM MgCl2, 0.1 mM ATP, 20 nM 8-mer HORNA>p substrate, and 1 µM AtRNL was incubated at 22°C. Aliquots (20 µL) were removed at the times specified and quenched immediately with formamide, EDTA. The time 0 sample was withdrawn and quenched prior to adding AtRNL. The products were resolved by urea-PAGE. An autoradiogram of the gel is shown in panel B. The levels of the healed HORNA2′p and pRNA2′p intermediates and the ligated circle product were quantified by scanning the gel and are plotted as a function of time in panel C. Each datum is the average of three separate time-course experiments ±SEM. The data were fit by nonlinear regression in Prism to a sequential three-step reaction pathway shown at right in panel B. The apparent rate constants for the CPD, kinase, and ligase reactions are indicated.
The kinetic profile of the single turnover reaction of wild-type AtRNL with the 8-mer HORNA>p is shown in Figure 2B. The RNA distribution was quantified and plotted vs. time in Figure 2C. Whereas the data fit well to a sequential mechanism of CPDase → kinase → ligase steps, the rates of the component reactions were differentially affected by shortening the RNA substrate. The CPDase rate constant for the 8-mer was 0.073 ± 0.0048 sec−1, similar to the rate of 0.1 sec−1 for the 10-mer. The kinase rate constant for the 8-mer was 0.015 ± 0.00044 sec−1, which was threefold slower than the kinase reaction for the 10-mer. The instructive finding was that the rate constant for intramolecular ligation of the healed 8-mer was 0.0022 ± 0.00008 sec−1, which was 16-fold slower than the rate of intramolecular ligation of the 10-mer. This rate decrement suggests a conformational constraint on the simultaneous occupancy of the binding sites for the reactive 3′-OH/2′-PO4 and 5′-PO4 ends within the ligase domain of AtRNL when the distance between the RNA ends is shortened.
We put this idea to the test by reacting AtRNL with a 3′ 32P-labeled 6-mer HORNA>p substrate. The time course of the single-turnover reaction is shown in Figure 3A. Again, we saw sequential conversion of the 6-mer HORNA>p to half-healed HORNA2′p and healed pRNA2′p intermediates, but the outcome of the ligation phase was the formation of a slowly migrating linear dimer RNA, via intermolecular ligation, rather than the circular RNA product formed with the 8-mer and 10-mer HORNA>p substrates. The kinetic profile is quantified in Figure 3B and well fit to the three-step sequential mechanism. The apparent rate constant of 0.17 ± 0.01 sec−1 for the CPDase reaction with the 6-mer was similar to (indeed, slightly faster than) the CPDase rate of 0.1 sec−1 on the 10-mer. Thus, a 6-mer HORNA>p exemplifies a minimized kinetically effective substrate for the AtRNL CPDase under single-turnover conditions. In the same vein, the kinase rate constant did not diminish in the transition to 6-mer (0.018 ± 0.00031 sec−1) from 8-mer (0.015 sec−1). In contrast, the rate constant of 0.00026 ± 0.000008 sec−1 for intermolecular ligation of the healed 6-mer was eightfold slower than the rate of circularization of the healed 8-mer, and 130-fold slower than the rate of circularization of the 10-mer. Thus, RNA length strongly influences the outcome and kinetics of the ligation phase of the AtRNL splicing pathway.
FIGURE 3.

Reaction of AtRNL with a 6-mer HORNA>p substrate. A reaction mixture (250 µL) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 10 mM MgCl2, 0.1 mM ATP, 20 nM 6-mer HORNA>p substrate (depicted at the bottom of panel A with the 32P-label denoted by •), and 1 µM AtRNL was incubated at 22°C. Aliquots (20 µL) were removed at the times specified and quenched immediately with formamide, EDTA. The time 0 sample was withdrawn and quenched prior to adding AtRNL. The products were resolved by urea-PAGE. An autoradiogram of the gel is shown in panel A. The levels of the healed HORNA2′p and pRNA2′p intermediates and the ligated dimer product were quantified by scanning the gel and are plotted as a function of time in panel B. Each datum is the average of three separate time-course experiments ±SEM. The data were fit by nonlinear regression in Prism to a sequential three-step reaction pathway shown at right in panel B. The apparent rate constants for the CPD, kinase, and ligase reactions are indicated.
A single ribonucleoside 2′,3′-cyclic-PO4 enables AtRNL to splice DNA
AtRNL and fungal Trl1 enzymes are essential components of RNA repair pathways in vivo. Their 2′-PO4 requirement for end joining mandates that the reactive 3′-OH terminus be a ribonucleoside. To our knowledge, the extent of the RNA requirement for productive and kinetically vigorous splicing has not been determined. A simple way to address this issue is to query whether any or all of the nucleotides of a spliceable polynucleotide substrate for AtRNL (exclusive of the 3′ nucleotide) can be replaced by deoxynucleotides. This approach has been fruitful in delineating the minimal RNA content required for other RNA repair enzymes, including T4 RNA ligase 2 (Nandakumar and Shuman 2004), the RNA terminal 2′-O-methyltransferase Hen1 (Jain and Shuman 2011), the RNA 3′-terminal cyclase RtcA (Das and Shuman 2013), and the RNA 3′-PO4/5′-OH ligase RtcB (Das et al. 2013a).
Here, we tested whether AtRNL could splice a HOD17R>p substrate, consisting of 17 deoxynucleotides and a single 32P-labeled 3′-terminal ribonucleoside-2′,3′-cyclic-PO4 (Fig. 4). The substrate was prepared by templated enzymatic ligation of a cold HOD17ROH strand, 5′-d(CATATCCGTGTCGCCCT)(rC), to a 5′ 32P-labeled 10-mer DNA strand, 5′-pATTCCGATAG, followed by digestion of the isolated D17RpD10 strand with RNase A to yield HOD17R3′p, which was then converted to HOD17R>p by RtcA (Das and Shuman 2013).
FIGURE 4.

AtRNL efficiently splices a 5′-OH DNA with a ribonucleoside-2′,3′-cyclic-PO4 terminus. A reaction mixture (160 µL) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 2 mM DTT, 2 mM MnCl2, 100 µM ATP, 1 µM AtRNL, and 20 nM 3′ 32P-labeled 18-mer HOD17Rp substrate (shown with the 3′ ribonucleoside shaded and the 32P-label denoted by •) was incubated at 22°C. Aliquots (20 µL) were withdrawn at the times specified and quenched immediately with an equal volume of 90% formamide, 30 mM EDTA. The time 0 sample was withdrawn and quenched prior to adding AtRNL. The products were analyzed by urea-PAGE. An autoradiograph of the gel is shown in the top panel. The position of the linear substrate (HOD17R>p) is indicated on the left; the positions of the healed intermediate (pD17R2′p) and the ligated circle product are indicated on the right. The kinetic profile of the intramolecular end joining reaction is plotted in the bottom panel. Each datum in the graph is the average of three separate experiments ±SEM. A nonlinear regression curve fit of the data to a single exponential is shown.
The HOD17R>p substrate was reacted with AtRNL under single-turnover conditions, and the products were analyzed by urea-PAGE (Fig. 4, top panel). We detected the formation of a healed intermediate pD17R2′p at early time points en route to a more rapidly migrating circular end-product. (Note that the initial HOD17R2′p intermediate was not resolvable from the input substrate by the PAGE procedure.) A plot of the extent of ligation as a function of time is shown in Figure 4, the bottom panel. The data were fit by nonlinear regression to a single exponential with a rate constant of 0.027 ± 0.02 sec−1 for the composite splicing reaction. This value for an all-DNA substrate with a single ribonucleoside-2′,3′>p compares favorably with the rate constant of 0.048 sec−1 for the composite splicing reaction of an all-RNA 20-mer HORNA>p substrate (Remus and Shuman 2013). We conclude that the terminal ribonucleoside is the sole RNA specificity determinant for polynucleotide ligation by plant tRNA ligase. The implications of this finding for practical applications of tRNA ligases will be discussed.
Distinctive end-healing properties of K. lactis tRNA ligase
An earlier comparison of the specific activities of the isolated healing (kinase-CPDase) and sealing (ligase) domains of SceTrl1 and AtRNL showed that the plant sealing module was more active in vitro than the yeast homolog, whereas the yeast healing domain was more active than the plant homolog, when each was paired with an excess of its own partner domain (Nandakumar et al. 2008). Moreover, a 1:1 cross-species mixture of the yeast healing and plant sealing domains displayed 25-fold higher specific activity in tRNA splicing activity than a reciprocal 1:1 combination of the plant healing and yeast sealing domains. These initial results hinted at the existence of distinctive properties of one or both enzymatic activities of the yeast kinase-CPDase module. One reported difference is that the AtRNL kinase uses any common NTP as a phosphate donor, whereas the SceTrl1 kinase prefers GTP (Westaway et al. 1993; Remus and Shuman 2013). Indeed, Greer and colleagues discovered that the SceTrl1 kinase was 100-fold more efficient with GTP vs. ATP, as reflected in the yield of pRNA product as a function of NTP concentration (Westaway et al. 1993).
Here, we aimed to analyze the kinetics and substrate specificity of end-healing by Kluveromyces lactis tRNA ligase. KlaTrl1 is an 813-aa polypeptide with 45% amino acid sequence identity to SceTrl1. We produced the C-terminal kinase-CPDase domain, KlaTrl1-(385–813), in E. coli and purified the 49-kDa recombinant protein (Supplemental Fig. S1A). We reacted 1 µM KlaTrl1-(385–813) with 20 nM 3′ 32P-labeled 10-mer HORNA>p substrate and a divalent cation for 1 min, in the absence of added NTP or in the presence of 100 µM ATP, GTP, CTP, or UTP. The reactions were quenched with EDTA, and the products were analyzed by urea-PAGE. Absent an added NTP, the HORNA>p was hydrolyzed quantitatively by the CPDase to a HORNA2′p species that migrated ahead of the input substrate (Fig. 5A). Inclusion of 100 µM GTP or UTP elicited the formation of the fully healed product, pRNA2′p, in quantitative (97%) yield (Fig. 5A). CTP and ATP at 100 µM supported phosphorylation of 38% and 9% of the substrate, respectively (Fig. 5A,B). When 100 µM dNTPs were tested as phosphate donors, only dGTP was effective (Fig. 5A,C).
FIGURE 5.
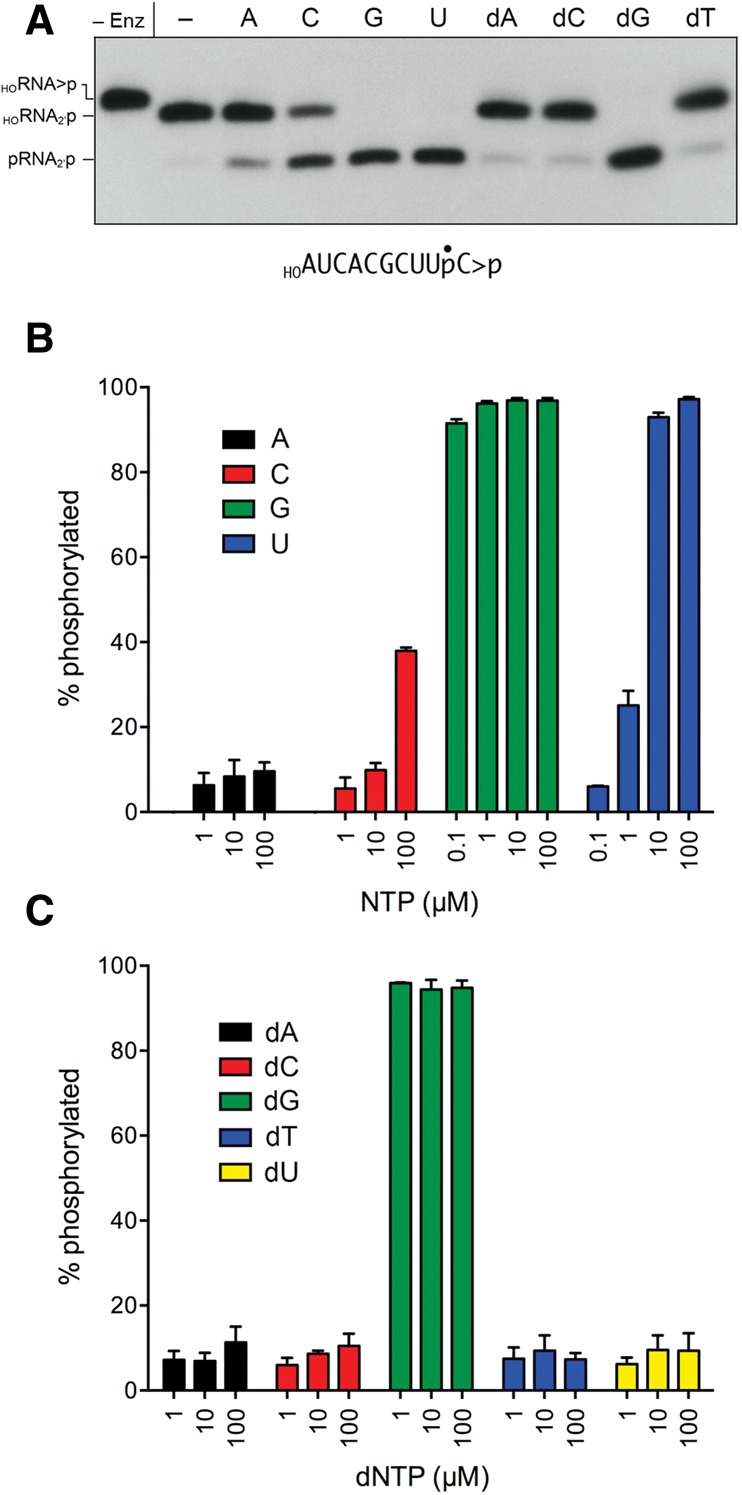
NTP donor specificity of the KlaTrl1 5′ kinase reaction. (A) Reaction mixtures (20 µL) containing 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 20 nM 3′ 32P-labeled 10-mer HORNA>p substrate (depicted at the bottom of the panel with the 32P-label denoted by •), 1 µM KlaTrl1-(385–813), and either no added NTP (lane –) or 0.1 µM of the indicated NTP were incubated at 22°C for 1 min, then quenched with an equal volume of 90% formamide, 30 mM EDTA. The products were analyzed by urea-PAGE and visualized by autoradiography. Enzyme was omitted from the control reaction in the leftmost lane. (B,C) Reaction mixtures (20 µL) containing 50 mM Tris-HCl (pH 8.0), 2 mM DTT, 10 mM MgCl2, 20 nM HOR10>p, 1 µM KlaTrl1-(385–813), and either 0.1, 1, 10, or 100 µM of the indicated NTP (either ATP, CTP, GTP, or UTP in panel B) or dNTP (dATP, dCTP, dGTP, dTTP, or dUTP in panel C) were incubated at 22°C for 1 min. The products were analyzed by urea-PAGE. The extents of 5′ phosphorylation were quantified by scanning the gel and are plotted in bar graph format as a function of NTP or dNTP concentration. Each datum is the average of three separate experiments ±SEM.
To better gauge the phosphate donor preferences of the K. lactis kinase, we back-titrated the nucleotide concentrations in 10-fold decrements and quantified the extents of phosphorylation, which are displayed in bar graph format in Figure 5, B and C. Lowering the GTP concentration by a factor of 100 or 1000, to 1 µM or 0.1 µM, had virtually no effect on the yields of phosphorylated RNA (96% and 92%, respectively) after a 1-min reaction in enzyme excess. In contrast, whereas UTP was an effective kinase substrate at the highest concentration tested (100 µM), back-titration progressively diminished the extent of phosphorylation, to 25% at 1 µM UTP and 6% at 0.1 µM UTP (Fig. 5B). The intermediate yield of product formation observed with 100 µM CTP (38%) fell off quickly at 10 µM and 1 µM CTP, to 10% and 5%, respectively (Fig. 5B). Thus, GTP is the strongly preferred phosphate donor among the four canonical rNTPs. Among the dNTPs titrated for kinase activity, dGTP supported 96% phosphorylation at 1 µM concentration (Fig. 5C), signifying that the K. lactis kinase was not stringently specific for the pentose sugar, so long as guanine was the nucleobase. However, dUTP was not an effective substrate, even at 100 µM, supporting only 9% phosphorylation (Fig. 5C); this result contrasts with the better performance of UTP as a kinase substrate, with its yield of 93% phosphorylation at 10 µM UTP (Fig. 5B). We surmise that the ribose sugar matters when the NTP nucleobase is suboptimal.
Purine NTP analogs illuminate nucleobase recognition by the K. lactis kinase
We gained insights into the nucleobase specificity of the yeast kinase by testing purine ribonucleoside triphosphate analogs as substrates for HORNA>p phosphorylation. The NTPs were included at 1, 10, and 100 µM concentration, and the extents of phosphorylation in a 1-min reaction under single-turnover conditions were quantified (Fig. 6). Inosine triphosphate was the most effective substrate, supporting 95% phosphorylation at 1 µM ITP (equivalent to the yield at 1 µM GTP), signifying that the exocyclic 2-NH2 moiety of GTP is not an important determinant of nucleobase recognition per se. In contrast, 2-aminopurine triphosphate was a much less effective substrate for the kinase reaction, with yields of 3%, 16%, and 52% phosphorylation at 1, 10, and 100 µM concentration, respectively. This result highlights the importance of the 6-oxo atom of guanine for phosphate donor function. This conclusion was fortified by the feeble donor activity of 6-chloropurine triphosphate, which supported 5% and 13% phosphorylation at 10 µM and 100 µM concentration, respectively. Because inosine and 6-chloropurine are isosteres differing only in the 6-oxo vs. 6-chloro atom, the several hundredfold difference in their efficacy as kinase substrates (on a concentration basis) provides a rough gauge of the contribution of the 6-oxo atom to nucleobase recognition, albeit specifically in the absence of a 2-NH2 moiety on the purine ring. In the same vein, the finding that 2-aminopurine, which lacks a 6-oxo atom, was an approximately fourfold better donor NTP than 6-chloropurine (at 100 µM concentration) roughly gauges the contribution of the 2-NH2 substituent to nucleobase recognition. Finally, we found that 6-O-methyl guanosine triphosphate was a very effective donor, sustaining 68% and 94% phosphorylation at 1 µM and 10 µM 6-OMeGTP, respectively. We infer that the 6-oxo atom is essential because it acts as a hydrogen-bond acceptor, presumably from an amino acid side chain hydrogen donor or a main chain amide donor on the kinase enzyme. This scenario would account for the unique ability of UTP to serve as an alternative phosphate donor, by virtue of its pyrimidine 4-oxo group accepting the putative hydrogen bond in lieu of the guanine 6-oxo.
FIGURE 6.
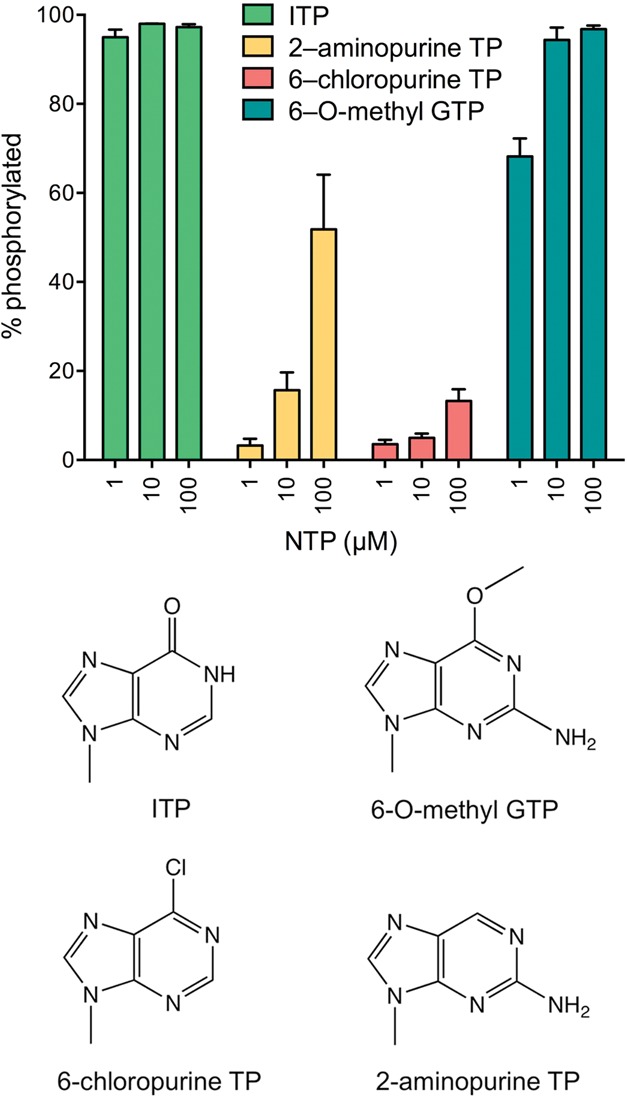
Purine NTP analogs as substrates for the KlaTrl1 kinase reaction. Reaction mixtures (20 µL) containing 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 20 nM HOR10>p, 1 µM KlaTrl1-(385–813), and 1, 10, or 100 µM of the indicated purine NTPs were incubated at 22°C for 1 min. The extents of 5′ phosphorylation are plotted in bar graph format as a function of NTP or dNTP concentration. Each datum is the average of three separate experiments ±SEM. The chemical structures of the nucleobases are shown below the graph.
Single-turnover kinetics of the K. lactis kinase reaction
The rate of 5′ phosphorylation of the 10-mer HORNA>p by the K. lactis healing domain under conditions of enzyme excess was too fast to measure manually. Therefore, we employed a rapid mix-quench apparatus to access the reaction on a millisecond time scale. Equal volumes of enzyme and substrate solutions were mixed at 22°C to achieve final concentrations of 20 nM HORNA>p, 1 µM kinase-CPDase, 10 mM Mg2+, and 100 µM GTP or UTP. The rapid mix reactions were quenched with EDTA, and the products were resolved by urea-PAGE. The kinetic profiles for the GTP and UTP reactions are shown in Figure 7, A and B, where each datum is the average of three experiments ±SEM. Nonlinear regression fitting of the data to a single exponential yielded rate constants of 14.3 ± 1.2 sec−1 with GTP as the phosphate donor and 0.7 ± 0.03 sec−1 with UTP. The salient points of this analysis are that (1) the chemistry of the single-turnover K. lactis kinase reaction was 20-fold faster with 100 µM GTP vs. 100 µM UTP as the phosphate donor, and (2) the rate of phosphorylation of the 10-mer HORNA>p substrate by K. lactis kinase was 300-fold faster than the rate of the plant AtRNL kinase (0.049 sec−1) (Fig. 1).
FIGURE 7.

Single-turnover kinetics of the KlaTrl1 kinase reaction. Rapid mix-quench assays of kinase activity under conditions of enzyme excess were performed as described in Materials and Methods, with either 100 µM GTP (panel A) or 100 µM UTP (panel B) as the phosphate donor. The extents of product formation are plotted as a function of reaction time. Each datum in the graphs is the average of three separate time-course experiments ±SEM. Nonlinear regression curve fits of the data to a single exponential are shown; the rate constants are indicated.
Kinetic profile of the composite KlaTrl1 HORNA>p splicing reaction
Recombinant full-length KlaTrl1 (Supplemental Fig. S1) was used to study the kinetics and outcome of the ligation reaction under single-turnover conditions (1 µM KlaTrl1 and 20 nM 3′ 32P-labeled 20-mer HORNA>p substrate) in the presence of 100 µM GTP and 100 µM ATP. The temporal progress of the reaction is shown in Figure 8A. Whereas the end-healing reactions were complete within 10 sec (the earliest time point sampled), the sealing of the pRNA>p strand proceeded slowly over 10 min to yield a mixture of monomer circle and multimeric products, formed by intra- and intermolecular end joining, respectively (Fig. 8A). We also detected the formation of an adenylylated RNA species, AppRNA2′p, migrating slower that the pRNA>p strand, that appeared in parallel with ligated products and decayed between 5 and 30 min as the circles and multimers accumulated. The fraction of the total labeled RNA that was ligated is plotted as a function of time in Figure 8B; 96% of the input RNA was sealed at the reaction endpoint. Nonlinear regression fitting of the data to a single exponential yielded an apparent rate constant of 0.0048 ± 0.00036 sec−1 for the KlaTrl1 splicing reaction. This value is 10-fold slower than the rate of single-turnover splicing of the same 20-mer HORNA>p substrate by AtRNL (0.048 sec−1) (Remus and Shuman 2013). It is also slower by a factor of 3000 than the rate of the K. lactis kinase reaction.
FIGURE 8.

Kinetics of healing and sealing a 20-mer HORNA>p substrate by KlaTrl1. A reaction mixture (240 µL) containing 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 100 µM GTP, 20 nM 3′ 32P-labeled 20-mer HORNA>p substrate (depicted at the bottom of panel A with the 32P-label denoted by •), and 1 µM KlaTrl1 was incubated at 22°C. Aliquots (20 µL) were withdrawn at the times specified and quenched immediately with an equal volume of 90% formamide, 30 mM EDTA. The time 0 sample was withdrawn and quenched prior to adding KlaTrl1. The products were analyzed by urea-PAGE. An autoradiogram of the gel is shown in panel A. The position and identities of the radiolabeled substrate and products are indicated on the left and right. The extent of ligation ([circle + multimers]/total RNA) is plotted as a function of time in panel B. Each datum is the average of three separate time-course experiments ±SEM. A nonlinear regression curve fit of the data to a single exponential is shown.
The temporal profile of the reaction of KlaTrl1 with the shorter 10-mer HORNA>p substrate is shown in Figure 9A. Again, the end-healing reactions were complete within 10 sec, but the ligation reaction with the pRNA>p strand occurred quite slowly, yielding a mixture of AppRNA2′p and sealed RNA circles that persisted up to 30 min. (No multimers were formed in this reaction.) The fraction ligated is plotted as a function of time in Figure 9B; 78% of the input RNA was circularized in 30 min. By fitting the data to a single exponential, we derived an apparent rate constant of 0.0027 ± 0.00015 sec−1 for the splicing reaction on the 10-mer.
FIGURE 9.

Reaction of KlaTrl1 with a 10-mer HORNA>p substrate. A reaction mixture (200 µL) containing 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM ATP, 100 µM GTP, 20 nM 3′ 32P-labeled 10-mer HORNA>p substrate (depicted at the bottom of panel A with the 32P-label denoted by •), and 1 µM KlaTrl1 was incubated at 22°C. Aliquots (20 µL) were withdrawn at the times specified and quenched immediately. The products were analyzed by urea-PAGE. An autoradiogram of the gel is shown in panel A. The position and identities of the radiolabeled substrate and products are indicated on the left and right. The extent of ligation (circle/total RNA) and the extent of RNA adenylylation ([AppRNAp + circle]/total RNA) are plotted as a function of time in panel B. Each datum is the average of three separate time-course experiments ±SEM. Nonlinear regression curve fits of the data to a single exponential are shown.
The experiments in Figures 8 and 9 show that the sealing phase of the K. lactis reaction is severely rate limiting, at least in part because the step of phosphodiester synthesis in the three-step ligation reaction is slowed or aborted. This is a point of distinction between AtRNL and KlaTrl1. During the course of 10-mer ligation by AtRNL, we observed no AppRNA2′p intermediate, signifying that the rate of phosphodiester synthesis (i.e., the attack of the 3′-OH on the AppRNA end) is much faster than the rate of adenylate transfer from ligase-AMP to the 5′-PO4 terminus to form AppRNA. In contrast, the adenylylated RNA species accumulated to a peak extent of 33% of total RNA during the KlaTrl1 reaction with the 10-mer, signifying that the step of phosphodiester synthesis was debilitated. The persistence of AppRNA at 30 min suggested that a percentage of the sealing reactions were aborted subsequent to RNA adenylylation, most likely because (1) the AppRNA2′p intermediate dissociates from the KlaTrl1 ligase domain, (2) the apo-ligase reacts with ATP to form ligase–AMP, and (3) ligase–AMP is unable to rebind and seal the released AppRNA2′p strand because the adenylate-binding pocket is occupied. This is the accepted explanation for trapping of adenylylated RNA and DNA species during abortive sealing by other polynucleotide ligases (Sriskanda and Shuman 1998; Ho and Shuman 2002). We derived a rate constant of 0.0043 ± 0.00015 sec−1 for the RNA adenylation step of the KlaTrl1 ligase reaction on the 10-mer substrate by plotting the sum of the AppRNA2′p and circular RNA species as a function of time and fitting the data to a single exponential (Fig. 9B).
DISCUSSION
The present study fills gaps in our understanding of the mechanism and specificity of plant tRNA ligase AtRNL and fungal tRNA ligase KlaTrl1. We exploited short HORNA>p substrates to determine, in a one-pot assay format, the order and rates of the CPDase, kinase, and ligase steps along the AtRNL splicing reaction pathway. Whereas the rate of the CPDase reaction was unaffected and the rate of the kinase reaction was affected only modestly by shortening the length of the RNA substrate, the rate of the sealing reaction was acutely sensitive, being slowed by a factor of 16 in the transition from 10-mer RNA to 8-mer and by eightfold in the transition from 8-mer RNA to 6-mer. The shift from 8-mer to 6-mer also changed the sealing mode from intramolecular to intermolecular end joining.
These results provide new insights into the minimal substrates and conformational constraints on the catalytic domains of AtRNL. The observed reaction sequence, independent of RNA length, is that the CPDase engages first, converting HORNA>p to HORNA2′p, which is then phosphorylated to pRNA2′p by the kinase. It is unclear whether the product 2′-PO4 terminus dissociates from the CPDase active site prior to engagement of the 5′-OH end by the kinase active site. (If the 2′-PO4 stays bound to the CPDase during the kinase reaction, it would imply, in the case of the 6-mer substrate, that the kinase phosphoacceptor site is located within ∼45 Å of the CPDase 2′-PO4 site, assuming an interval of 6.5 Å between the phosphates of an RNA chain.)
The ligase module of AtRNL is thought to have independent and specific binding sites for the 2′-PO4 and 5′-PO4 RNA ends. A key feature of the AtRNL ligation reaction is that transfer of AMP from the ligase–AMP adduct to the 5′-PO4 RNA end requires the 2′-PO4 end (Remus and Shuman 2013). The order of end capture by the ligase is not known, but there are several plausible scenarios, including one in which the half-healed HORNA2′p species dissociates from the CPDase and the 2′-PO4 terminus is quickly engaged by the ligase module, so that subsequent phosphorylation of the 5′-OH by the kinase occurs while the 2′-PO4 is bound to the ligase, after which the 5′-PO4 end dissociates from the kinase and is immediately captured by the ligase (thereby favoring intramolecular sealing). The fact that HORNA>p substrates as short as an 8-mer are sealed by AtRNL to yield exclusively circular products tells us that the active site of the ligase is sufficiently “open” to attain the proper orientation of the reactive RNA termini, either by having the 8-mer RNA adopt a minicircle conformation (with the ends lined up in series) or a fold-back hairpin conformation (with anti-parallel ends). The proper geometry for RNA 5′ adenylylation apparently cannot be achieved when the RNA is shortened to a 6-mer; consequently, the outcome shifts to intermolecular end joining.
We report that a single ribonucleoside-2′,3′-cyclic-PO4 moiety enables AtRNL to efficiently splice an otherwise all-DNA strand. Evidently, the kinase and ligase domains are not specific for RNA vs. DNA 5′ ends. We surmise that RNA specificity is enforced solely at the level of the 2′-PO4 requirement for adenylylation of the 5′-PO4 terminus by the ligase module. The DNA sealing ability of AtRNL expands its utility as a reagent for nucleic acid capture and sequencing. As conceived initially, Schutz et al. (2010) used AtRNL to ligate an RNA oligonucleotide tag to the population of cellular RNAs that naturally have 2′,3′-cyclic-PO4 termini. After treating the ligated products with Tpt1 to remove the 2′-PO4 at the splice junction, they used an oligonucleotide complementary to the tag to prime synthesis of a cDNA library, which was then amplified by PCR and sequenced. This method can be used to identify RNA species and processing intermediates that are not accessible by tagging with conventional RNA ligases such as T4 Rnl1 and T4 Rnl2 (Schutz et al. 2010).
Based on our findings here, we envision a new application for AtRNL in the tagging and characterization of DNAs with 2′,3′-cyclic-PO4 ends. DNA breaks with 2′,3′-cyclic-PO4 ends are generated by topoisomerase IB transesterification at sites where single ribonucleotides are embedded in the genome (Sekiguchi and Shuman 1997). Embedded ribonucleotides are among the most abundant natural lesions in DNA, and their cleavage by topoisomerase IB causes a sharp increase in deletion mutations at the incision site (Kim et al. 2011; Reijns et al. 2012). The metabolism of 2′,3′-cyclic-PO4 DNA breaks is entirely uncharted. A method to tag, quantify, and map the location of such breaks on a genome-wide scale would be a key advance—and AtRNL is an enabling reagent to accomplish this goal. Equally promising is the use of AtRNL to quantify and map the location of embedded ribonucleotides on a genome-wide scale. This can be accomplished by treating genomic DNA with alkali, which will yield a mixture of 2′-PO4 and 3′-PO4 ends at all sites where ribonucleotides are embedded. AtRNL can then selectively ligate an oligonucleotide tag to the DNA 2′-PO4 ends. The specificity of AtRNL ensures that no ligation will occur to a DNA 3′ end.
Our characterization of K. lactis tRNA ligase highlights important functional distinctions vis à vis the plant homolog. First, KlaTrl1 ligase domain is less effective than its AtRNL counterpart in sealing the short HORNA>p substrates used here for the kinetic analyses. This echoes earlier findings that, whereas AtRNL is as adept at splicing linear single-stranded RNAs as it is in joining the breaks in the anti-codon stem–loop of pre-tRNAs that are the physiological substrates for tRNA splicing, the yeast enzyme SceTrl1 exhibits a preference for broken stem–loop substrates and is less active in sealing short linear RNAs (Englert and Beier 2005; Nandakumar et al. 2008). Our observation that the KlaTrl1 ligation reaction on the 10-mer tends to abort after the formation of AppRNA2′p suggests that the fungal ligase active site is constrained in its ability to hold on to both ends of the 10-mer RNA.
The second, and more salient, difference between the fungal and plant tRNA ligases pertains to the kinase module. We find that (1) the KlaTrl1 kinase reaction is 300-fold faster than the AtRNL kinase reaction measured under single-turnover conditions with the same RNA substrate, and (2) the KlaTrl1 kinase is highly specific for GTP or dGTP as the phosphate donor. The GTP specificity of KlaTrl1 resonates with earlier findings for SceTrl1 (Westaway et al. 1993).
The strong GTP preference exhibited by the polynucleotide kinase domains of fungal tRNA ligases is unique among all polynucleotide kinase (Pnk) enzymes characterized to date. The best studied of these, with respect to structure, mechanism, and NTP substrate specificity, are bacteriophage T4 Pnk (Galburt et al. 2002; Wang et al. 2002) and Clostridium thermocellum Pnk (CthPnk) (Wang et al. 2012; Das et al. 2013b,c), both of which are end-healing components of a multienzyme RNA repair pathway and both of which can use any NTP as the phosphate donor. Crystal structures of CthPnk bound to ATP, GTP, CTP, UTP, and dATP revealed an extensive network of ionic and hydrogen-bonding contacts to the α, β, and γ phosphates. In contrast, there are no enzymic contacts to the ribose and none with the nucleobase other than a π-cation interaction with an arginine side chain. This arginine is conserved in T4 Pnk, where it makes the same π-cation stack on the nucleobase of the donor (Galburt et al. 2002), and it is also present in the kinase domains of AtRNL and the fungal Trl1 enzymes (Wang et al. 2006). Whereas the arginine contributes to donor NTP affinity in CthPnk (Das et al. 2013b), it does not explain the GTP specificity of the fungal Trl1 kinase domain. Our hypothesis, based on our nucleotide analog experiments, is that the fungal kinases make clade-specific hydrogen-bonding interactions with the guanine 6-oxo atom that enforce GTP specificity. This raises the prospect that the fungal kinase donor site can be targeted selectively by a small molecule inhibitor that interacts with the guanine specificity determinant(s) of Trl1 but does not bind to the donor sites of the many other cellular P-loop phosphotransferases, which either have no NTP donor preference or selectively use ATP as substrates.
MATERIALS AND METHODS
AtRNL purification
Recombinant wild-type AtRNL and mutants K152A, K700A-S701A, D726A, and T1001A were produced in Escherichia coli as His10Smt3 fusions and purified as described (Remus and Shuman 2013). The tagged proteins were isolated from soluble bacterial extracts by Ni-agarose chromatography, followed by treatment with Smt3-specific protease Ulp1 to cleave the tag. After depletion of the tag by adsorption to Ni-agarose, the AtRNL proteins were subjected to gel-filtration through a Superdex-200 column equilibrated in 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.05% Triton X-100, and 10% glycerol. Peak AtRNL fractions were pooled and stored at −80°C. Protein concentrations were determined by using the Biorad dye reagent with BSA as the standard.
KlaTrl1 production and purification
The full-length KlaTRL1 open reading frame (GenBank accession CAG98435) and the segment encoding the C-terminal kinase-CPDase module (aa 385–813) were amplified by PCR from K. lactis genomic DNA with primers that introduced a BamHI site flanking the start codon and an XhoI site 3′ from the stop codon. The PCR product was digested with BamHI and XhoI and inserted between the BamHI and XhoI sites of the bacterial expression vector pET28b-His10Smt3. The inserts of the resulting pET28b-His10Smt3-KlaTRL1 and pET28b-His10Smt3-KlaTRL1-(385–313) plasmids were sequenced to verify the in-frame junction between His10Smt3 and the Trl1 polypeptide and that no coding changes were introduced during amplification or cloning. The plasmids were transformed into E. coli BL21-CodonPlus(DE3). Cultures (4-L) derived from single transformants were grown at 37°C in Terrific Broth medium containing 50 µg/mL kanamycin until A600 reached 0.8. The cultures were chilled on ice for 30 min and then adjusted to 0.1 mM isopropyl β-D-thiogalactoside, 2% (v/v) ethanol. Incubation was continued at 17°C for 16 h with constant shaking. Cells were harvested by centrifugation and stored at −80°C. All subsequent procedures were performed at 4°C. The cell pellet was resuspended in 200 mL buffer A (50 mM Tris-HCl, pH 7.5, 1 M NaCl, 20 mM imidazole, 10% glycerol, 0.05% Triton X-100), and lysozyme was added to 0.2 mg/mL. After 30 min, the lysate was sonicated, and insoluble material was removed by centrifugation at 20,000g for 45 min. The supernatant was mixed for 1 h with 10 mL of His60 Ni Superflow resin (Qiagen) that had been equilibrated in buffer A. The resin was recovered by centrifugation and washed three times with 75 mL of buffer B (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol, 0.05% Triton X-100) containing 40 mM imidazole, before a final wash step with 75 mL of Tris-HCl, pH 7.5, 3 M KCl. The washed resin was then poured into a column. Bound proteins were eluted with 500 mM imidazole in buffer B. The elution profile was monitored by SDS-PAGE. Peak fractions containing His10Smt3-KlaTrl1 or His10Smt3-KlaTrl1-(383–813) were treated with the Smt3-specifc protease Ulp1 (at a His10Smt3-KlaTrl1:Ulp1 ratio of 500:1) during overnight dialysis against buffer B. The dialysate was mixed with 5 mL of His60 Ni Superflow resin that had been equilibrated in buffer B, and the mixture was poured into a column. The column was washed with buffer B, and bound material was eluted with 500 mM imidazole. The tag-free KlaTrl1 and KlaTrl1-(385–813) proteins were recovered in the flow-through and wash fractions; the cleaved His10Smt3 tag was bound to the resin and recovered in the imidazole eluate. The KlaTrl1 and KlaTrl1-(385–813) preparations were concentrated by centrifugal ultrafiltration to 10 mg/mL (in 4 mL) and then gel-filtered through a 120-mL 16/60 HiLoad Superdex 200 column equilibrated with buffer C (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol) at a flow rate of 1 mL/min while collecting 5-mL fractions. The peak KlaTrl1 and KlaTrl1-(385–813) fractions were pooled and concentrated by centrifugal ultrafiltration. Protein concentrations were determined by using the Biorad dye reagent with bovine serum albumin as the standard. The yields of KlaTrl1 and KlaTrl1-(385–813) were 25 mg and 40 mg, respectively.
Preparation of 3′ 32P-labeled RNA>p substrates
HORNA3′p oligonucleotides labeled with 32P at the penultimate phosphate were prepared by T4 Rnl1-mediated addition of [5′-32P]pCp to a 19-mer, 9-mer, 7-mer, or 5-mer synthetic oligoribonucleotides. The resulting 20-mer, 10-mer, 8-mer, and 6-mer HORNA3′p oligonucleotides were treated with E. coli RNA 3′-terminal phosphate cyclase (RtcA) and ATP to generate the 2′,3′-cyclic phosphate derivatives, HORNA>p, which were gel-purified, eluted from an excised gel slice, and recovered by ethanol precipitation.
Preparation of 3′ 32P-labeled DNA>p substrate
The 3′ 32P-labeled HOD17R3′p strand was prepared as follows. A 10-mer deoxyribonucleotide HOATTCCGATAGOH was 5′ 32P-labeled by using [γ32P]ATP and T4 polynucleotide kinase-phosphatase (Pnkp), then gel-purified. The labeled pD10 strand was mixed with an 18-mer CATATCCGTGTCGCCCT(rC)OH strand (D17R) and a complementary 36-mer DNA strand to form a nicked DNA duplex with a monoribonucleotide on the 3′-OH side of the nick and a 32P-labeled deoxynucleotide on the 5′-PO4 side of the nick. The nick was ligated by reaction with a 10-fold molar excess of Chlorella virus DNA ligase, 10 mM MgCl2, and 1 mM ATP for 1 h at 22°C to yield an internally 32P-labeled HOD17RpD10 strand. The ligation product was heated for 10 min at 95°C in the presence of a 10-fold molar excess of an unlabeled 36-mer DNA that competed with the radiolabeled ligation product for reannealing to the bridging template DNA strand. This mixture was digested with 50 µg RNase A for 10 min at 37°C, thereby converting the HOD17RpD10 species to a 3′ 32P-labeled HOD17Rp strand, which was gel-purified. HOD17Rp was converted to HOD17R>p by reaction with RNA 3′-terminal cyclase and ATP, then recovered by phenol:chloroform extraction and precipitation with ethanol.
Rapid kinetic assay of KlaTrl1 kinase activity
A Kintek RQF3 rapid chemical quench apparatus was used to assay the reaction of HORNA>p with a 50-fold molar excess of KlaTrl1-(385–813) at 22°C in the range of 0.05- to 1-sec reaction times. The reaction mixtures (40 µL) contained 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM GTP or UTP, 20 nM 32P-labeled HORNA>p substrate, and 1 µM AtRNL. The rapid kinetic measurements were initiated by mixing two buffer solutions (20 µL each). One solution contained 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM GTP or UTP, and 40 nM HORNA>p. The other solution contained 50 mM Tris-HCl, pH 8.0, 2 mM DTT, 10 mM MgCl2, 100 µM GTP or UTP, and 2 µM KlaTrl1-(385–813). The reactions were quenched by rapid mixing with 110 μL of 90% formamide, 50 mM EDTA. The samples were analyzed by urea-PAGE. The 32P-labeled RNAs were quantified by scanning the gel with a Fujix BAS2500 imager.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM42498. S.S. is an American Cancer Society Research Professor.
REFERENCES
- Apostol BL, Westaway SK, Abelson J, Greer CL 1991. Deletion analysis of a multifunctional yeast tRNA ligase polypeptide: Identification of essential and dispensable functional domains. J Biol Chem 266: 7445–7455 [PubMed] [Google Scholar]
- Chakravarty AK, Subbotin R, Chait BT, Shuman S 2012. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc Natl Acad Sci 109: 6072–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Shuman S 2013. 2′-Phosphate cyclase activity of RtcA: A potential rationale for the operon organization of RtcA with an RNA repair ligase RtcB in Escherichia coli and other bacterial taxa. RNA 19: 1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Chakravarty AK, Remus BS, Shuman S 2013a. Rewriting the rules for end joining via enzymatic splicing of DNA 3′-PO4 and 5′-OH ends. Proc Natl Acad Sci 110: 20437–20442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang LK, Smith P, Shuman S 2013b. Structural and biochemical analysis of the phosphate donor specificity of the polynucleotide kinase component of the bacterial Pnkp•Hen1 RNA repair system. Biochemistry 52: 4734–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang LK, Smith P, Jacewicz A, Shuman S 2013c. Structures of bacterial polynucleotide kinase in a Michaelis complex with GTP•Mg2+ and 5′-OH oligonucleotide and a product complex with GDP•Mg2+ and 5′-PO4 oligonucleotide reveal a mechanism of general acid-base catalysis and the determinants of phosphoacceptor recognition. Nucleic Acids Res 42: 1152–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KK, Bingman CA, Phillips GN, Raines RT 2013. Structure of the noncanonical RNA ligase RtcB reveal the mechanism of histidine guanylylation. Biochemistry 52: 2518–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Beier H 2005. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res 33: 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Xia S, Okada C, Nakamura A, Tanavde V, Yao M, Eom SH, Koningsberg WH, Söll D, Wang J 2012. Structural and mechanistic insights into guanylylation of RNA-splicing ligase RtcB joining RNA between 3′-terminal phosphate and 5′-OH. Proc Natl Acad Sci 109: 15235–15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galburt EA, Pelletier J, Wilson G, Stoddard BL 2002. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure 10: 1249–1260 [DOI] [PubMed] [Google Scholar]
- Greer CL, Peebles CL, Gegenheimer P, Abelson J 1983. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell 32: 537–546 [DOI] [PubMed] [Google Scholar]
- Ho CK, Shuman S 2002. Bacteriophage T4 RNA ligase 2 (gp24.1) exemplifies a family of RNA ligases found in all phylogenetic domains. Proc Natl Acad Sci 99: 12709–12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Shuman S 2011. Active site mapping and substrate specificity of bacterial Hen1, a manganese-dependent 3′ terminal RNA ribose 2′O-methyltransferase. RNA 17: 429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA,Pommier Y, Jinks-Robertson S 2011. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332: 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska M, Filipowicz W, Gross HJ 1982. RNA ligation via 2′-phosphomonoester, 3′,5′-phosphodiester linkage: Requirement of 2′,3′-cyclic phosphate termini and involvement of a 5′-hydroxyl polynucleotide kinase. Proc Natl Acad Sci 79: 1474–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar J, Shuman S 2004. How an RNA ligase discriminates RNA damage versus DNA damage. Mol Cell 16: 211–221 [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Schwer B, Schaffrath R, Shuman S 2008. RNA repair: An antidote to cytotoxic eukaryal RNA damage. Mol Cell 31: 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick L, Hurwitz J 1986. Purification of wheat germ RNA ligase: Characterization of a ligase-associated 5′-hydroxyl polynucleotide kinase activity. J Biol Chem 261: 6684–6693 [PubMed] [Google Scholar]
- Pick L, Furneaux H, Hurwitz J 1986. Purification of wheat germ RNA ligase: Mechanism of action of wheat germ RNA ligase. J Biol Chem 261: 6694–6704 [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lürhmann R, Söll D, et al. 2011. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331: 760–764 [DOI] [PubMed] [Google Scholar]
- Reijns MAM, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian development. Cell 149: 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S 2013. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43298–43398 [DOI] [PubMed] [Google Scholar]
- Schutz K, Hesselberth JR, Fields S 2010. Capture and sequence analysis of RNAs with terminal 2′,3′-cyclic phosphates. RNA 16: 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RC, Greer CL, Gegenheimer P, Abelson J 1983. Enzymatic mechanism of an RNA ligase from wheat germ. J Biol Chem 258: 8374–8383 [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S 2004. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci 101: 2788–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi J, Shuman S 1997. Site-specific ribonuclease activity of eukaryotic topoisomerase I. Mol Cell 1: 89–97 [DOI] [PubMed] [Google Scholar]
- Sriskanda V, Shuman S 1998. Mutational analysis of Chlorella virus DNA ligase: Catalytic roles of domain I and motif VI. Nucleic Acids Res 26: 4618–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Shuman S 2011. RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem 286: 7727–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Chakravarty AK, Maughan B, Shuman S 2011. A novel mechanism of RNA repair by RtcB via sequential 2′,3′-cyclic phosphodiesterase and 3′-phosphate/5′-hydroxyl ligation reactions. J Biol Chem 286: 43134–43143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S 2005. Structure–function analysis of yeast tRNA ligase. RNA 11: 966–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Lima CD, Shuman S 2002. Structure and mechanism of T4 polynucleotide kinase: An RNA repair enzyme. EMBO J 21: 3873–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Englert M, Beier H, Shuman S 2006. Structure–function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res 34: 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Das U, Smith P, Shuman S 2012. Structure and mechanism of the polynucleotide kinase component of the bacterial Pnkp-Hen1 RNA repair system. RNA 18: 2277–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway SK, Belford HG, Apostol BL, Abelson J, Greer CL 1993. Novel activity of a yeast ligase deletion polypeptide: Evidence for GTP-dependent tRNA splicing. J Biol Chem 268: 2435–2443 [PubMed] [Google Scholar]