

This paper describes the effects of modulating kinase activity on alternative splicing of Rac1 in tumor cells. Specifically, knockdown of SRPK1, a kinase that phosphorylates SR proteins, reduced translocation of SRSF1 into the nucleus and thereby reduced the ability of this factor to promote inclusion of a tumor-specific exon in the Rac1 pre-mRNA.
Keywords: alternative splicing, SRPK1, SRSF1, signal transduction pathways, colorectal cancer
Abstract
The premessenger RNA of the majority of human genes can generate various transcripts through alternative splicing, and different tissues or disease states show specific patterns of splicing variants. These patterns depend on the relative concentrations of the splicing factors present in the cell nucleus, either as a consequence of their expression levels or of post-translational modifications, such as protein phosphorylation, which are determined by signal transduction pathways. Here, we analyzed the contribution of protein kinases to the regulation of alternative splicing variant Rac1b that is overexpressed in certain tumor types. In colorectal cells, we found that depletion of AKT2, AKT3, GSK3β, and SRPK1 significantly decreased endogenous Rac1b levels. Although knockdown of AKT2 and AKT3 affected only Rac1b protein levels suggesting a post-splicing effect, the depletion of GSK3β or SRPK1 decreased Rac1b alternative splicing, an effect mediated through changes in splicing factor SRSF1. In particular, the knockdown of SRPK1 or inhibition of its catalytic activity reduced phosphorylation and subsequent translocation of SRSF1 to the nucleus, limiting its availability to promote the inclusion of alternative exon 3b into the Rac1 pre-mRNA. Altogether, the data identify SRSF1 as a prime regulator of Rac1b expression in colorectal cells and provide further mechanistic insight into how the regulation of alternative splicing events by protein kinases can contribute to sustain tumor cell survival.
INTRODUCTION
Gene expression is initiated by transcription into a premessenger RNA, which undergoes a series of processing steps including pre-mRNA splicing. Through alternative splicing, more than one mRNA can be produced from the same gene and generate transcript isoforms that differ in stability or translation efficiency or that encode functional protein variants, thus increasing the diversity of products expressed by the genome. Current estimates based on high-throughput sequencing approaches identified alternative splicing events in >90% of human protein-coding genes (Pan et al. 2008; Wang et al. 2008; Blencowe 2012).
The regulation of such alternative splice-site choices is mediated by the binding of splicing factors (SFs) to gene-specific cis-acting sequence elements so that protein–protein interactions required for productive spliceosome assembly are either promoted or disrupted. These gene-specific elements can either enhance (splice enhancer [SE]) or suppress (splice silencer [SS]) inclusion of a given exon and be located in the exon itself (ESE, ESS) or in the surrounding introns (ISE, ISS) (Pagani and Baralle 2004; Wang and Burge 2008).
Individual tissues differ in their alternative splicing patterns due to different expression levels of key SFs, which determine such patterns through a combinatorial mode of action (Singh and Valcárcel 2005; Wang and Burge 2008; Calarco et al. 2011). For example, members of the arginine-serine rich (SR) proteins and heterogenous nuclear ribonucleoproteins (hnRNPs) compete for splice-site regulatory elements (Eperon et al. 2000; Smith and Valcárcel 2000).
Changes in SF expression were also reported in a variety of tumor types (Ghigna et al. 1998; Stickeler et al. 1999; Venables 2006), and overexpression of SRSF1 (previously called ASF or SF2) has been shown to exert oncogenic effects in cells (Karni et al. 2007). Recent data support a view that the alternative spliced exons in a given tissue form functional networks, which rewire protein–protein interactions and establish tissue-specific proteome signatures (Buljan et al. 2012; Ellis et al. 2012). This is an important notion because changes in genome-wide splicing patterns can also be induced following either physiological or pathological stimuli (Shin and Manley 2004; Blaustein et al. 2007; Stamm 2008; Heyd and Lynch 2010) with concomitant remodeling of protein interaction networks or signaling pathways. In tumors, SF expression levels or activity were found to be altered due to SF gene mutations or deregulated signal transduction pathways that affect, for example, changes in nuclear-cytoplasmic SF localization ratios and accordingly lead to changes in the cell's splicing pattern (Srebrow and Kornblihtt 2006; Venables 2006; David and Manley 2010). One particular example is the tumor-related splicing variant Rac1b, an isoform of the signaling GTPase Rac1 (Matos et al. 2003), in which a usually skipped exon 3b is retained. The resulting protein isoform Rac1b is overexpressed in a specific subtype of colorectal tumors and required to sustain tumor cell survival (Jordan et al. 1999; Matos and Jordan 2008; Matos et al. 2008) but was also reported in breast, lung, and thyroid tumors (Schnelzer et al. 2000; Radisky et al. 2005; Liu et al. 2012; Stallings-Mann et al. 2012; Silva et al. 2013; Zhou et al. 2013). Interestingly, Rac1b was found to be predominantly in the signaling-competent GTP-bound conformation (Schnelzer et al. 2000; Matos et al. 2003; Fiegen et al. 2004; Radisky et al. 2005), so that small changes in its expression level yield significant cellular responses.
Although much remains to be learned about the mechanisms by which deregulated cancer cell signaling connects to the splicing machinery, a couple of instructive examples have been elucidated in recent years. For example, activation of the p38-MAP kinase pathway led to phosphorylation of hnRNPA1, resulting in its cytoplasmic sequestration and altered splicing of an adenovirus reporter gene (Van der Houven van Oordt et al. 2000). Inclusion of exon v5 into the cell surface tumor marker CD44 depended on activation of the Ras-ERK pathway (Weg-Remers et al. 2001; Matter et al. 2002). Also, phosphorylation of SRp40 by AKT2 has been shown to regulate alternative splicing of protein kinase C βII (Patel et al. 2005; Jiang et al. 2009). Nucleo-cytoplasmic transport of the splicing regulator polypyrimidine tract-binding protein (PTB) was found to be modulated by the cAMP-dependent protein kinase PKA (Xie et al. 2003). The phosphatidylinositol 3 (PI3)-kinase pathway mediates alternative splicing of fibronectin in response to growth factor stimulation (Blaustein et al. 2004).
One identified phosphorylation target is the SR protein SRSF1 (Xiao and Manley 1997), and its nuclear versus cytoplasmic localization is modulated by phosphorylation in HeLa cells (Sanford et al. 2005). Responsible protein kinases are AKT1, which phosphorylates SRSF1 in vitro (Blaustein et al. 2005), and SRPK1, which binds SRSF1 with high affinity and progressively phosphorylates 10–12 serines in the N-terminal region of the RS domain (RS1). This modification is required for nuclear import of SRSF1 and its typical localization in speckles (Ngo et al. 2005; Ghosh and Adams 2011). In one particular example, the IGF receptor activates SRPK1 to switch between two VEGF splicing isoforms with opposite angiogenic properties (Nowak et al. 2010; Amin et al. 2011).
Here, we searched for protein kinases involved in alternative splicing of Rac1b and identified a crucial role for SRPK1-mediated nuclear import of SRSF1 in colorectal cells.
RESULTS
Identification of regulatory protein kinases involved in the skipping of RAC1 exon 3b
Previously, we described that the splicing factors SRSF1 and SRSF3 (former designated as ASF/SF2 and SRp20, respectively) regulate alternative splicing of Rac1b in colorectal cancer cells in an antagonistic manner (Gonçalves et al. 2009). In HT29 cells, for instance, high levels of SRSF1 promoted the inclusion of alternative exon 3b, whereas in SW480 cells, high levels of SRSF3 favored exon 3b skipping.
To screen for signaling proteins that influence the levels and activity of these splicing factors, leading to Rac1b overexpression, we chose, however, to use the nontransformed NCM460 colonocytes (Moyer et al. 1996). In contrast to the cancer cells lines used in our previous studies (Gonçalves et al. 2009), NCM460 cells do not carry BRAF (HT29) nor K-Ras (SW480, DLD-1) mutations, thus avoiding potential confounding effects from these oncogenic mutations on putative upstream regulators of Rac1b alternative splicing. To ascertain that NCM460 cells maintained the antagonistic regulation of Rac1 splicing via SRSF1 and SRSF3, they were first tested by cotransfection of a splicing-reporter RAC1 minigene and either SRSF1 or SRSF3, as previously described (Gonçalves et al. 2009). As shown in Figure 1A, expression of SRSF1 also increased inclusion of alternative exon 3b in NCM460 cells, whereas that of SRSF3 promoted exon skipping. We then tested regulation of the endogenous Rac1b levels in NCM460 cells using depletion of either SRSF1 or SRSF3 by RNA interference and also observed the described antagonistic regulation (Fig. 1B).
FIGURE 1.
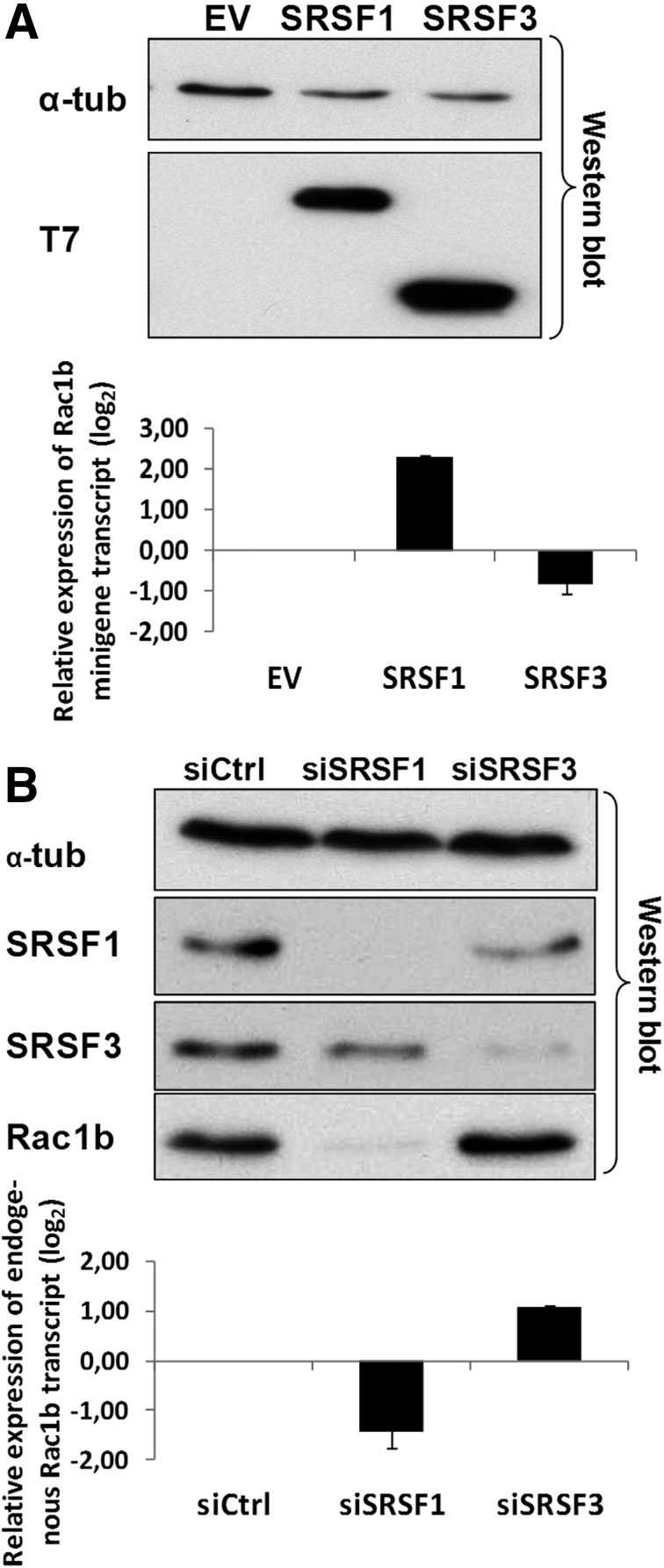
Effect of SRSF1 and SRSF3 on alternative splicing of Rac1b in NCM460 cells. (A) The normal colonocyte cell line NCM460 was cotransfected with a RAC1 minigene and the indicated T7-tagged splicing factors. The Western blot detection of T7-SRSF1 and T7-SRSF3 is shown 24 h after transfection in NCM460 with endogenous α-tubulin as loading control. qPCR quantification of the corresponding minigene-derived transcripts is shown in the lower panel. (B) NCM460 cells were transfected with the indicated siRNAs and analyzed after 48 h. A Western blot detection of endogenous levels of Rac1b is shown, together with those of SRSF1 and SRSF3, to document successful knockdown and of α-tubulin as loading control. The lower panel shows quantification of the corresponding splicing changes in endogenous Rac1 and Rac1b transcript levels by qPCR.
Once validated as a working model, these cells were transfected with a selection of shRNA plasmids from a previously described library (Moura-Alves et al. 2011; Moita et al. 2012), directly targeting a set of 20 kinases and phosphatases (see Table 1) earlier implicated in the regulation of other alternative splicing events (Blaustein et al. 2005; Ngo et al. 2005; Heyd and Lynch 2010; Liu et al. 2011; Qian et al. 2011).
TABLE 1.
List of kinases and phosphatases targeted by RNA interference in this study
As a crude primary screening method, protein extracts from these cells were analyzed by Western blot for changes in endogenous Rac1b levels. Results indicated that in the NCM460 colonocytes, none of the shRNAs produced an obvious increase in isoform Rac1b; however, the targeting of AKT1, AKT2, and AKT3, as well as of GSK3β and SRPK1, led to a clear decrease in Rac1b protein (Fig. 2).
FIGURE 2.

Effect of shRNA-mediated depletion of selected protein kinases on endogenous Rac1b protein levels. NCM460 cells were transiently transfected with shRNAs against the indicated kinases, lysed after 48 h, and analyzed by Western blot for endogenous Rac1b protein and α-tubulin as loading control. Band intensities from the primary screen blot were determined by densitometry and plotted in the graph below to display fold changes in Rac1b protein expression compared to control cells and identify candidate kinases for subsequent analysis.
The role of these five positive kinases in regulating Rac1b levels was then confirmed and explored in more detail by using commercially available, validated siRNA oligonucleotides. The successful knockdown of all the targeted kinases was confirmed by Western blot analysis, which also included the other candidate protein kinases to exclude possible indirect effects on Rac1b due to cross-talk between the kinases’ expression levels. All siRNAs robustly reproduced the effects of their respective shRNA on Rac1b protein levels, with the exception of siAKT1 (Fig. 3A). Although siAKT was isoform specific, we found that the AKT1 shRNA depleted all AKT isoforms (data not shown), explaining the observed discrepancy. Remarkably, when Rac1b transcript levels were assessed following normalization to total Rac1 transcript levels, only the depletion of GSK3β and SRPK1 decreased endogenous Rac1b expression (Fig. 3B). This indicated that only these two kinases were actually regulating the alternative splicing event that generates the Rac1b mRNA; AKT2 and AKT3 depletion appeared to act post-splicing in these cells, affecting both Rac1 and Rac1b protein levels.
FIGURE 3.

siRNA-mediated depletion of selected candidate protein kinases to validate their effect on endogenous Rac1b levels. NCM460 cells were transfected with the indicated siRNA oligonucleotide and analyzed 48 h after. (A) Western blots showing the degree of depletion of the targeted kinases and the corresponding effect on endogenous Rac1b protein (α-tubulin as loading control). Also shown are the protein levels of the remaining identified candidate protein kinases. (B) Quantitative PCR analysis of endogenous Rac1b transcript levels. The ratio of Rac1b over total Rac1 transcripts was calculated and fold changes in relative Rac1b expression were graphically displayed. Note that only depletion of GSK3β and SRPK1 led to decreased Rac1b transcript levels.
Inhibition of GSK3β decreases Rac1b alternative splicing through SRSF1
GSK3β is a component of the APC-axin complex (Ikeda et al. 1998) regulating the phosphorylation and subsequent proteolytic degradation of β-catenin (Aberle et al. 1997). Interestingly, we previously described that in HT29 colorectal cancer cells, the β-catenin-activated transcription factor TCF4 promoted expression of the SRSF3 gene (Gonçalves et al. 2008) and reported later that SRSF3 acts as an inhibitor of Rac1b splicing (Gonçalves et al. 2009). We therefore used HT29 as a Rac1b overexpressing cell line to further investigate the role of GSK3β and SRPK1 in sustaining increased Rac1b splicing.
We then determined whether siRNA-mediated GSK3β knockdown would lead to nuclear accumulation of β-catenin and a subsequent increase in SRSF3 expression in these cells. As shown in Figure 4A, a cell fractionation protocol previously described for the analysis of nuclear factors (Barros et al. 2009) revealed no considerable increment in nuclear β-catenin under GSK3β knockdown conditions. Similarly, siGSK3β-treated cells showed no increase in SRSF3 expression (Fig. 4B). This indicated that another mechanism was decreasing Rac1b levels following GSK3β depletion.
FIGURE 4.
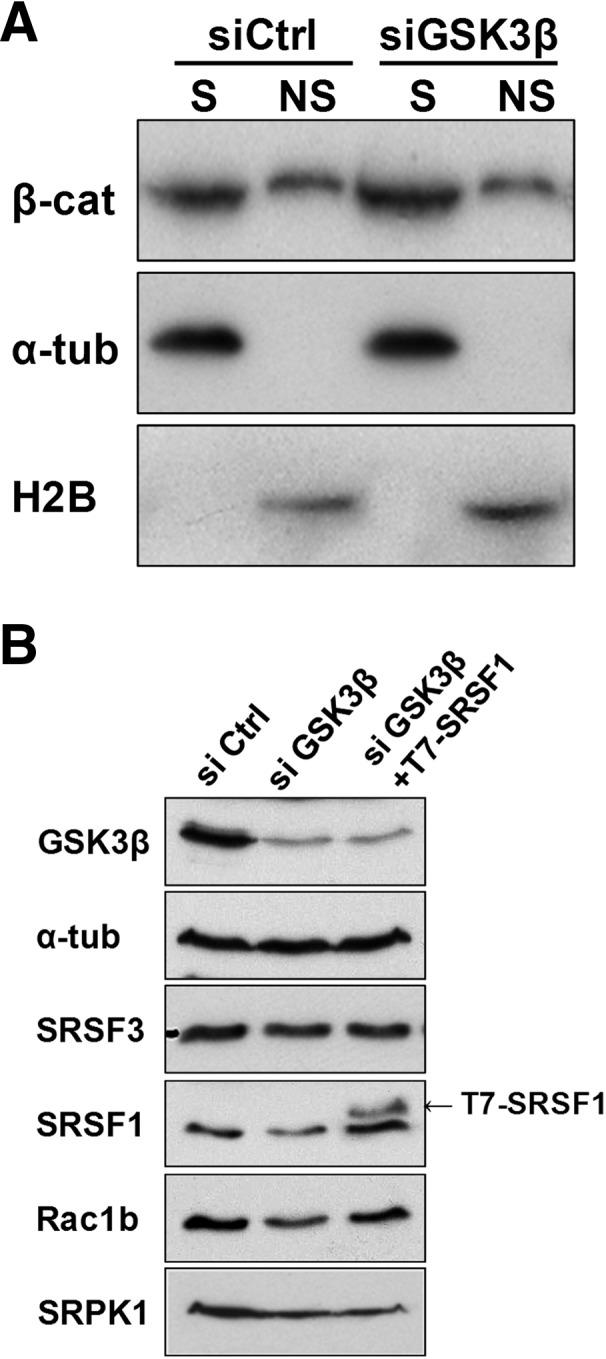
Analysis of the mechanism mediating the effect of GSK3β on Rac1b. (A) HT29 colorectal cells were transfected with the indicated siRNA oligonucleotide and nuclear chromatin-associated β-catenin was detected 48 h after transfection by Western blot. A detergent-based cell fractionation methodology separated β-catenin into a soluble (S) and a nonsoluble, chromatin-bound (NS) pool. Histone2B and α-tubulin protein levels were probed as controls for NS and S fractions, respectively. (B) Effect of GSK3β depletion on endogenous SRSF3, SRSF1, Rac1b, and SRPK1 protein levels in HT29 cells. Note that the depletion led to decreased levels of SRPK1, SRSF1, and Rac1b but had no effect on SRSF3. The third lane corresponds to cotransfection with a T7-SRSF1 expression vector 24 h after GSK3β depletion. Detection of GSK3β and α-tubulin served as controls.
Previous reports have shown that GSK3β can modulate alternative splicing events by directly interfering with SR proteins, namely SRSF1 (Hernández et al. 2004; Zhou et al. 2012), which is a positive modulator of Rac1b splicing (Gonçalves et al. 2009). We therefore analyzed the effects of GSK3β depletion on endogenous SRSF1 and observed a clear decrease of the SR protein levels (Fig. 4B). Cotransfection of ectopic, T7-tagged SRSF1 with siGSK3β clearly countered the effect of GSK3β knockdown on Rac1b levels, confirming that GSK3β affects Rac1b splicing via SRSF1 (Fig. 4B). Interestingly, we also observed that GSK3β depletion led to a reduction in SRPK1 protein levels (Fig. 4B), whereas SRPK1 transcript levels did not suffer a corresponding decrease (data not shown). The effect of GSK3β depletion was specific as the reciprocal depletion of SRPK1 did not affect GSK3β or AKT protein levels (data not shown). Together, these data indicate that GSK3β can act upstream of SRPK1 in the regulation of Rac1b alternative splicing.
Inhibition of SRPK1 decreases SRSF1-dependent alternative splicing of Rac1b
The N-terminal region of the RS domain in SRSF1 is a substrate of SRPK1, and its phosphorylation was reported to be involved in the assembly of SRSF1 into nuclear speckles (Ngo et al. 2005). However, it remained to be determined whether SRPK1 depletion affected SRSF1 expression levels in colorectal cells. We found that in HT29 cells the knockdown of SRPK1 or GSK3β by siRNA notably decreased the levels of SRSF1 protein (Fig. 4B or 5A, respectively) but not of the SRSF1 transcript (Fig. 5B). This indicated that SRPK1 was acting post-transcriptionally on the SRSF1, possibly through phosphorylation of its RS domain. Indeed, the probing of these lysates with an anti-phospho-SR protein antibody confirmed a stronger decrease in phopho-SRSF1 levels upon SRPK1 knockdown in HT29 colorectal cells (Fig. 5A). Consistently, when we treated these cells with 25 μM (24 h) of the SRPK-selective kinase inhibitor SRPIN340 (Fukuhara et al. 2006), we saw a clear decrease in SRSF1 phosphorylation and total protein levels with a comparable decrease in Rac1b expression (Fig. 5C). An identical treatment with the control compound SRPIN349 (that has no inhibitory effect on SRPKs) showed no effect in either SRSF1 or Rac1b levels.
FIGURE 5.

Effect of SRPK1 depletion or inhibition of its kinase activity on endogenous Rac1b and SRSF1 levels. (A) Western blot analysis of SRPK1, Rac1b, and SRSF1 protein levels in HT29 cells following transfection with SRPK1-specific siRNAs for 48 h. Levels of phosphorylated SRSF1 were also detected. (B) Quantitative PCR analysis of endogenous Rac1b and SRSF1 transcript levels in HT29 cells following transfection with either SRPK1-specific or, for comparison, GSK3β-specific siRNAs. (C) Inhibition of SRPK1 activity in HT29 cells by incubation with 25 μM SRPIN340 or control components during 24 h. Shown is a Western blot analysis of the endogenous levels of Rac1b and SRSF1 protein as well as of SRSF1 phosphorylation.
Thus, we concluded that interfering with SRPK1, either by knockdown or by inhibiting its activity, leads to an SRSF1-dependent decrease in alternative splicing of Rac1b. To investigate whether this effect would entail changes in SRSF1 subcellular localization, a T7-tagged SRSF1 was transfected into HT29 cells prior to SRPK1 knockdown and cells analyzed by confocal fluorescence microscopy. A T7-tagged factor was used because the SRSF1 antibody employed in Western blots failed to recognize the protein in its native conformation. Figure 6A shows that T7-SRSF1 was exclusively detected in the nucleus of control HT29 cells, whereas depletion of SRPK1 led to a clear shift of the factor's localization to the cytoplasm.
FIGURE 6.
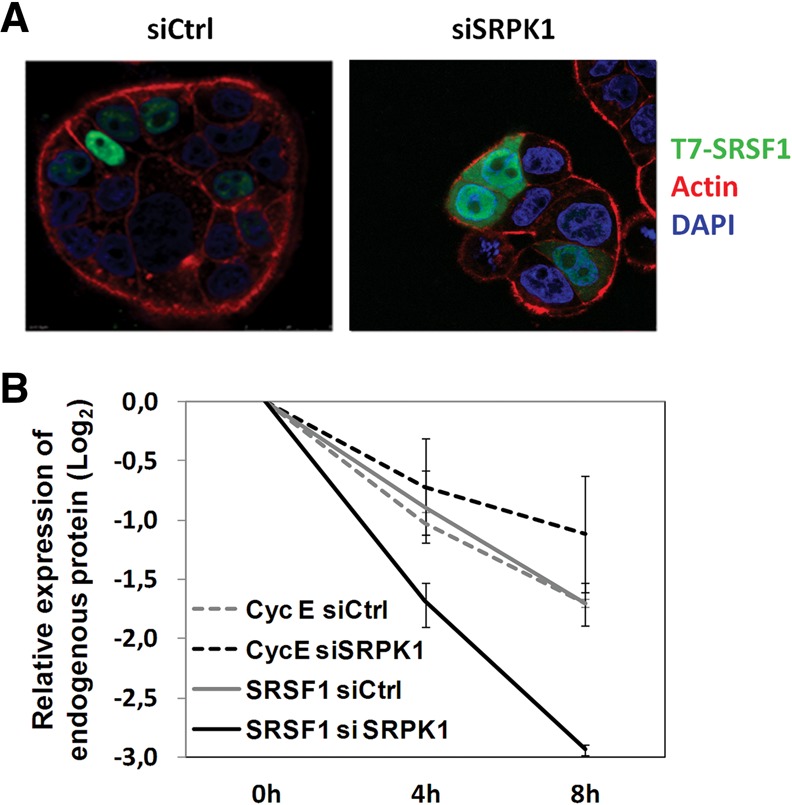
Effect of SRPK1 depletion on SRSF1 subcellular localization and proteolytic degradation. (A) HT29 cells were transfected with the indicated siRNAs, cotransfected with T7-tagged SRSF1 24 h later, and analyzed by confocal immunofluorescence microscopy 20 h later. Shown are overlay images from cell staining for SRSF1 (green), actin (red), and DAPI (blue). Note the increased cytoplasmic staining of SRSF1 after depletion of SRPK1. (B) HT29 cells were transfected with the indicated siRNAs for 48 h and then treated with 500 μg/mL cycloheximide for the indicated periods. A graphical display of endogenous SRSF1 and CyclinE protein levels obtained by densitometry of Western blots bands is shown. Note the faster decay in SRSF1 protein levels in SRPK1-depleted (solid black line) compared to control cells (solid gray line), whereas Cyclin E levels (dashed lines) did not differ under the same conditions.
We then asked whether this cytosol-localized SRSF1 had an accelerated rate of turnover, which would explain the observed decrease in SRSF1 steady-state levels following SRPK1 depletion and its inability to re-enter the nucleus. To test this, we inhibited de novo protein synthesis with cycloheximide (CHX) for several hours and analyzed endogenous SRSF1 protein decay in the presence of control or SRPK1-specific siRNAs. Cyclin E was chosen as control because it was found in pilot experiments to have a decay rate similar to that of SRSF1. Figure 6B shows that, in contrast to Cyclin E, endogenous SRSF1 protein levels decreased faster after SRPK1 depletion, indicating an acceleration of SRSF1 decay rate when it localizes to the cytosol.
DISCUSSION
The splicing factor SRSF1 has been previously identified to promote inclusion of exon 3b into the RAC1 pre-mRNA in colorectal cells, increasing the expression of alternative spliced Rac1b (Gonçalves et al. 2009). In this paper, we report that the two protein kinases SRPK1 and GSK3β act upstream of SRSF1 and are required to sustain Rac1b levels in colorectal cells.
SRPK1 is a known activator of SRSF1. It phosphorylates a stretch of serine residues in the RS1 domain, which is required for proper nuclear localization of SRSF1 (Ngo et al. 2005; Ghosh and Adams 2011). We confirmed in colorectal cells that depletion of SRPK1 by RNAi also led to decreased nuclear localization of SRSF1 (Fig. 6A), thus explaining the concomitant reduction in Rac1b levels. A similar role of SRPK1 has been observed in other alternative splicing events, for example, in regulating the balance of pro- and anti-angiogenic VEGF isoforms (Nowak et al. 2010; Amin et al. 2011), or E1A minigene-derived alternative transcripts (Zhong et al. 2009; Zhou et al. 2012).
Besides validating the role of SRPK1 in colorectal cells, our data revealed a further interesting regulatory property, namely, that in SRPK1-depleted cells, the expression levels of the SRSF1 protein but not of the corresponding mRNA decreased (Fig. 5A,B). Using cycloheximide to transiently block de novo protein synthesis, we could indeed demonstrate that SRSF1 suffered from an increased decay rate, presumably due to proteolytic degradation when accumulating in the cytoplasm (Fig. 6B). This effect could also be observed when the catalytic activity of SRPK1 was repressed by incubating cells with the inhibitor SRPIN340 (Fig. 5C). Indeed, we observed that SRSF1 protein levels can be regulated by proteasomal degradation because they remained stable in SRPK1-depleted or SRPIN340-treated cells when incubated with proteasome inhibitors MG132 or Lactacystin (data not shown). Whether these inhibitors reversed the effects of depletion or inhibition of SRPK1 on Rac1b levels could, however, not be determined because they also stabilized β-catenin and this led, as we previously reported (Gonçalves et al. 2008), to increased transcriptional activation of SRSF3, a negative regulator of RAC1 exon 3b inclusion and thus of Rac1b levels (Gonçalves et al. 2009).
Overexpression of SRPK1 has been described in several types of solid tumors, such as colon and breast carcinomas (Plasencia et al. 2006; Hayes et al. 2007), and may further increase nuclear activity of SRSF1 and promote target alternative splicing events. It will be interesting to determine whether the overexpression of Rac1b observed in colon, breast, thyroid, and lung tumors correlates with increased SRPK1 expression.
A second kinase identified in our screen and required to sustain Rac1b levels was GSK3β. Apparently, this kinase did not act through its well-known role in the Wnt pathway because in the cell lines used, we did not detect the expected nuclear accumulation of β-catenin and consequently no changes in the expression levels of SRSF3, which is a transcriptional target of the β-catenin/TCF4 complex (Gonçalves et al. 2008). Although previous data have reported that GSK3β can regulate splicing through direct phosphorylation of SR proteins such as SRSF2 (former SC35) (Hernández et al. 2004), we observed in colorectal cells that its effect on Rac1b was dependent on SRSF1 (Fig. 4B). Because GSK3β depletion led to a reduction in both SRSF1 and SRPK1 protein levels, we suggest an indirect effect of GSK3β on SRSF1 and Rac1b via SRPK1. The mechanism by which depletion of GSK3β leads to the observed decrease in SRPK1 levels still remains to be determined, i.e., whether GSK3β acts directly on SRPK1 through phosphorylation or whether additional downstream effector proteins or protein complex formation are involved. For example, a genome-wide kinome screen revealed previously unpredicted networks regulated by GSK3β (Lu et al. 2011).
Taken together, our data reinforce previous results indicating a crucial role for SRSF1 in the regulation of Rac1b splicing in colorectal cells. Various other tumor-related splicing events were identified as targets for SRSF1 and its overexpression has oncogenic effect on cells (Karni et al. 2007). Interestingly, in mouse mammary epithelial cells, Rac1b expression was reported to be primarily regulated through repression by hnRNP A1 (Pelisch et al. 2012), which apparently fails to modulate Rac1b splicing in colorectal cells (Gonçalves et al. 2009). It remains to be determined whether these differences reflect tissue-specific regulation patterns or differences in the nucleotide sequences between both species. Nonetheless, other SR proteins besides SRSF1 can be expected to participate in the regulation of Rac1b splicing.
Another protein kinase reported to act as a regulator of SR proteins in alternative splicing events is AKT. Although AKT2 was reported to phosphorylate SRSF5 (SRp40) in myoblasts (Patel et al. 2005), AKT1 phosphorylated SRSF1 and SRSF7 (9G8) proteins directly in HeLa cells (Blaustein et al. 2005). A further study in EGF-stimulated HeLa cells identified that AKT1 promoted SRPK1 and SRPK2 autophosphorylation and translocation to the nucleus, with corresponding changes in alternative splicing of an E1A minigene (Zhou et al. 2012), raising the possibility that the described abilities of immunopurified AKT to phosphorylate SR proteins may be caused by associated SRPKs.
We thus expected to observe a role for AKT1, AKT2, or AKT3 in SRPK- or SRSF1-mediated changes in Rac1b alternative splicing. Surprisingly, our data in colorectal cells revealed that AKT2 and AKT3 did not affect Rac1b at the transcript but rather at the protein level (Fig. 3A,B). It remains to be investigated whether this effect is due to changes in translation efficiency or proteolytic degradation of Rac1b, in which case conflicting results on Rac1b ubiquitylation were reported (Esufali et al. 2007; Visvikis et al. 2008). Although AKT can phosphorylate and inhibit GSK3β, this mechanism is apparently not used for regulating Rac1b in colorectal cells because only GSK3β but not AKT depletion affected Rac1b transcript levels.
Altogether, our data identify regulatory protein kinases involved in the regulation of alternative splicing of tumor-related Rac1b in colorectal cells. Although the effect of SRPK1 fits well with published data, the role observed experimentally for GSK3β or AKT in these cells differs in part from results reported for the same kinases in other cellular systems or splicing events. This underlines that basic principles can be learned from the study of individual splicing variants; however, generally applicable rules to the regulation of alternative splicing are not easily derived. This is due to tissue-specific signaling networks that direct protein kinase activity toward selected target proteins. Nonetheless, if individual cancer-driving splicing events can be linked to specific signaling cascades, the growing list of small molecule protein kinase inhibitors may represent a therapeutic resource to correct aberrant splicing events.
MATERIALS AND METHODS
Cell culture and transfection
HT29 cells were maintained in RPMI, whereas NCM460 colon cells were grown in M3:Base (INCELL), all supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen). Cell lines were regularly checked for absence of mycoplasm infection. Cells were grown in 24-well plates or 35-mm dishes to 60%–80% confluence, transfected using LipofectAMINE 2000 (Invitrogen), according to the manufacturer's instructions, and analyzed 20–24 h later (48 h for the shRNAs and siRNAs). Total amounts of transfected DNA were 0.75 μg per well of 24-well plates, or 4 μg DNA for 35-mm dishes. If required, the amount of DNA was adjusted with empty vector. Plasmid transfection efficiencies were judged microscopically by expression of GFP and reached 40%–60% in NCM460 and HT29 cells. For RNA interference experiments, NCM460 and HT29 cells at 30%–40% confluence were transfected in 24-well plates or 35-mm dishes with 75 pmol or 400 pmol, respectively, of the indicated siRNAs using LipofectAMINE 2000 and analyzed after 48 h. Some siRNA oligos were ordered from MWG Biotech with the following sequence: control siLuc: 5′-CGUACGCGGAAUACUUCGATT; siSRPK1: 5′-UUAUUCAGCAAGUGUUACATT; siSRp20: 5′-GAGCUAGAUGGAAGAACAC; siSF2: 5′-AGAAGAUAUGACCUAUGCA. The other siRNA oligos were ordered from Santa Cruz Biotechnology with the following catalog numbers: siAKT1: sc-29195; siAKT2: sc-29197; siAKT3: sc-38912; and siGSK3β: sc-35527. To inhibit de novo protein synthesis, HT29 cells were treated with 500 μg/mL cycloheximide for the indicated periods 40 h after siRNA transfection. For SRPK inhibition, HT29 cells at 60%–80% confluence were treated with 25 μM SRPIN340 during 24 h, using 25 μM of SRPIN349 (has no inhibitory effect on SRPKs) and DMSO as controls. Both SRPIN340 and 349 were kindly provided by Dr. Masatoshi Hagiwara. For proteasomal inhibition, cells were treated for 8 h with either 10 μM MG132 or 20 μM Lactacystin (Merck Millipore).
DNA plasmids
The following previously published constructs were used: T7-tagged constructs of SRSF1 and SRSF3 (Cáceres et al. 1997); splicing reporter minigene RAC1 (Gonçalves et al. 2009). Previously validated shRNA encoding plasmids targeting protein kinase transcripts were part of the LKO.1 shRNA constructs obtained from the RNAi Consortium (TRC). All constructs were confirmed by automated DNA sequencing.
Analysis of transcript expression
Total RNA was extracted from cell lysates with the RNAeasy kit (Qiagen) and 1 μg reverse transcribed using random primers (Invitrogen) and Ready-to-Go You-Prime Beads (GE Healthcare). Relative changes in Rac1b expression were calculated from quantitative PCR reactions (qPCR) as the ratio between the specific amplicons for Rac1b (78 bp) and for total Rac1 (75 bp) transcripts (i.e., Rac1 + Rac1b) as previously described (Gonçalves et al. 2009). Transcript expression was analyzed with each amplification performed in duplicate reactions and repeated in at least three independent experiments. No amplification was obtained when RNA was mock reverse transcribed without adding reverse transcriptase.
SDS-PAGE and Western blotting
Samples were prepared and detected as described (Matos and Jordan 2006). The antibodies used for Western blots were: mouse anti-α-tubulin (clone B-5-1-2) from Sigma; T7-Tag Antibody from Novagen/VWR; rabbit anti-Rac1b from Millipore; mouse anti-SR proteins (clone 1H4), mouse anti-ASF/SF2 (SRSF1) and mouse anti-SRp20 (SRSF3) from Invitrogen; mouse anti-SRPK1 and mouse anti-β-catenin from BD Transduction Laboratories; rabbit anti-AKT and rabbit anti-GSK3β (27C10) from Cell Signalling; mouse anti-cyclin E (ab3927) from Abcam; and anti-Histone H2b (sc-10808) from Santa Cruz Biotechnology. Shown are Western blot images that are representative of at least three independent experiments.
Cell fractionation
Nuclear proteins were separated into a soluble pool not retained in the nucleus and into a chromatin-bound insoluble pool as previously described (Barros et al. 2009). Briefly, cells were scraped and lysed on ice for 10 min in 200 μL fractionation buffer [50 mM Tris at pH 7.9, 0.1% (v/v) NP40; 1.5 mM MgCl2, 10 mM KCl and a protease inhibitor cocktail (Sigma)]. The soluble fraction was collected by centrifuging the lysate and adding the supernatant to 50 μL 5× Laemmli SDS sample buffer. The pellet containing the insoluble nuclear fraction was washed once in fractionation buffer and then resuspended in 250 μL 1× Laemmli sample buffer. Equal volumes of both fractions were analyzed side by side on Western Blots. Results were confirmed in at least three independent experiments.
Confocal immunofluorescence microscopy
Cells were grown on 10 × 10-mm glass coverslips, transfected, and incubated as indicated above, then washed twice in PBS, immediately fixed with 4% (v/v) formaldehyde in PBS for 60 min at room temperature, and subsequently permeabilized with 0.5% (v/v) Triton X-100 in PBS for 30 min at room temperature. Cells were then labeled for 60 min with a 1:200 dilution of T7-Tag antibody (Novagen), washed 3× in PBS for 5 min with gentle shaking, followed by 30 min incubation with a 1:250 dilution of Alexa Fluor 488 (Invitrogen) and phalloidin-TRITC (Sigma). Coverslips were washed 3× in PBS, briefly stained with 0.5 ng/mL DAPI (Sigma), washed again, and then mounted in VectaShield (Vector Laboratories) and sealed with nail polish. Images were recorded with the 405 nm, 488 nm, and 532 nm laser lines of a Leica TCS-SPE confocal microscope and processed with Adobe Photoshop software.
ACKNOWLEDGMENTS
We thank J. Cáceres for providing plasmid vectors and M. Hagiwara for the SRPK inhibitor used in this study. This work was supported by the Fundação para a Ciência e Tecnologia, Portugal (Grants PEst-OE/BIA/UI4046/2011 to the BioFig research unit, and PTDC/SAU-MII/100780/2008 and PTDC/SAU-IMU/110303/2009 to L.F.M, contract Ciência2007 to P.M., and fellowship BPD 63395/2009 to V.G). L.F.M. received support from Fundação Luso-Americana para o Desenvolvimento.
REFERENCES
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R 1997. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16: 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin EM, Oltean S, Hua J, Gammons MVR, Hamdollah-Zadeh M, Welsh GI, Cheung M-K, Ni L, Kase S, Rennel ES, et al. 2011. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20: 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros P, Jordan P, Matos P 2009. Rac1 signaling modulates BCL-6-mediated repression of gene transcription. Mol Cell Biol 29: 4156–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Coso OA, Bissell MJ, Kornblihtt AR, Srebrow A 2004. Mammary epithelial-mesenchymal interaction regulates fibronectin alternative splicing via phosphatidylinositol 3-kinase. J Biol Chem 279: 21029–21037 [DOI] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Tanos T, Muñoz MJ, Wengier D, Quadrana L, Sanford JR, Muschietti JP, Kornblihtt AR, Cáceres JF, et al. 2005. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 12: 1037–1044 [DOI] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Srebrow A 2007. Signals, pathways and splicing regulation. Int J Biochem Cell Biol 39: 2031–2048 [DOI] [PubMed] [Google Scholar]
- Blencowe BJ 2012. An exon-centric perspective. Biochem Cell Biol 90: 603–612 [DOI] [PubMed] [Google Scholar]
- Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM 2012. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell 46: 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Misteli T, Screaton GR, Spector DL, Krainer AR 1997. Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J Cell Biol 138: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco JA, Zhen M, Blencowe BJ 2011. Networking in a global world: Establishing functional connections between neural splicing regulators and their target transcripts. RNA 17: 775–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL 2010. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev 24: 2343–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JD, Barrios-Rodiles M, Colak R, Irimia M, Kim T, Calarco JA, Wang X, Pan Q, O'Hanlon D, Kim PM, et al. 2012. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell 46: 884–892 [DOI] [PubMed] [Google Scholar]
- Eperon IC, Makarova OV, Mayeda A, Munroe SH, Cáceres JF, Hayward DG, Krainer AR 2000. Selection of alternative 5′ splice sites: Role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol Cell Biol 20: 8303–8318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esufali S, Charames GS, Bapat B 2007. Suppression of nuclear Wnt signaling leads to stabilization of Rac1 isoforms. FEBS Lett 581: 4850–4856 [DOI] [PubMed] [Google Scholar]
- Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, Vetter IR, Ahmadian MR 2004. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem 279: 4743–4749 [DOI] [PubMed] [Google Scholar]
- Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Herzenberg LA, et al. 2006. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci 103: 11329–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigna C, Moroni M, Porta C, Riva S, Biamonti G 1998. Altered expression of heterogenous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res 58: 5818–5824 [PubMed] [Google Scholar]
- Ghosh G, Adams JA 2011. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J 278: 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves V, Matos P, Jordan P 2008. The β-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA 14: 2538–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves V, Matos P, Jordan P 2009. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum Mol Genet 18: 3696–3707 [DOI] [PubMed] [Google Scholar]
- Hayes GM, Carrigan PE, Miller LJ 2007. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res 67: 2072–2080 [DOI] [PubMed] [Google Scholar]
- Hernández F, Pérez M, Lucas JJ, Mata AM, Bhat R, Avila J 2004. Glycogen synthase kinase-3 plays a crucial role in τ exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer's disease. J Biol Chem 279: 3801–3806 [DOI] [PubMed] [Google Scholar]
- Heyd F, Lynch KW 2010. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell 40: 126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A 1998. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J 17: 1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Patel NA, Watson JE, Apostolatos H, Kleiman E, Hanson O, Hagiwara M, Cooper DR 2009. Akt2 regulation of Cdc2-like kinases (Clk/Sty), serine/arginine-rich (SR) protein phosphorylation, and insulin-induced alternative splicing of PKCβII messenger ribonucleic acid. Endocrinology 150: 2087–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P, Brazåo R, Boavida MG, Gespach C, Chastre E 1999. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 18: 6835–6839 [DOI] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR 2007. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14: 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Xie N, Rennie P, Challis JRG, Gleave M, Lye SJ, Dong X 2011. Consensus PP1 binding motifs regulate transcriptional corepression and alternative RNA splicing activities of the steroid receptor coregulators, p54nrb and PSF. Mol Endocrinol 25: 1197–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee W, Jiang Z, Chen Z, Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J, et al. 2012. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 22: 2315–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, Ruths D, Tseng JT, Yu S, Yu Q, et al. 2011. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 30: 4567–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos P, Jordan P 2006. Rac1, but not Rac1B, stimulates RelB-mediated gene transcription in colorectal cancer cells. J Biol Chem 281: 13724–13732 [DOI] [PubMed] [Google Scholar]
- Matos P, Jordan P 2008. Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res 6: 1178–1184 [DOI] [PubMed] [Google Scholar]
- Matos P, Collard JG, Jordan P 2003. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem 278: 50442–50448 [DOI] [PubMed] [Google Scholar]
- Matos P, Oliveira C, Velho S, Gonçalves V, da Costa LT, Moyer MP, Seruca R, Jordan P 2008. B-RafV600E cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology 135: 899–906 [DOI] [PubMed] [Google Scholar]
- Matter N, Herrlich P, König H 2002. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420: 691–695 [DOI] [PubMed] [Google Scholar]
- Moita CF, Chora Â, Hacohen N, Moita LF 2012. RNAi screen for kinases and phosphatases that play a role in antigen presentation by dendritic cells. Eur J Immunol 42: 1843–1849 [DOI] [PubMed] [Google Scholar]
- Moura-Alves P, Neves-Costa A, Raquel H, Pacheco TR, D'Almeida B, Rodrigues R, Cadima-Couto I, Chora Â, Oliveira M, Gama-Carvalho M, et al. 2011. An shRNA-based screen of splicing regulators identifies SFRS3 as a negative regulator of IL-1β secretion. PLoS One 6: e19829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer MP, Manzano LA, Merriman RL, Stauffer JS, Tanzer LR 1996. NCM460, a normal human colon mucosal epithelial cell line. In Vitro Cell Dev Biol Anim 32: 315–317 [DOI] [PubMed] [Google Scholar]
- Ngo JCK, Chakrabarti S, Ding J-H, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu X-D, Ghosh G 2005. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell 20: 77–89 [DOI] [PubMed] [Google Scholar]
- Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G, Hagiwara M, Harper SJ, Woolard J, Ladomery MR, et al. 2010. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J Biol Chem 285: 5532–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani F, Baralle FE 2004. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat Rev Genet 5: 389–396 [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Patel NA, Kaneko S, Apostolatos HS, Bae SS, Watson JE, Davidowitz K, Chappell DS, Birnbaum MJ, Cheng JQ, Cooper DR 2005. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CβII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem 280: 14302–14309 [DOI] [PubMed] [Google Scholar]
- Pelisch F, Khauv D, Risso G, Stallings-Mann M, Blaustein M, Quadrana L, Radisky DC, Srebrow A 2012. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem 113: 2319–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasencia C, Martínez-Balibrea E, Martinez-Cardús A, Quinn DI, Abad A, Neamati N 2006. Expression analysis of genes involved in oxaliplatin response and development of oxaliplatin-resistant HT29 colon cancer cells. Int J Oncol 29: 225–235 [DOI] [PubMed] [Google Scholar]
- Qian W, Liang H, Shi J, Jin N, Grundke-Iqbal I, Iqbal K, Gong C-X, Liu F 2011. Regulation of the alternative splicing of τ exon 10 by SC35 and Dyrk1A. Nucleic Acids Res 39: 6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. 2005. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436: 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Ellis JD, Cazalla D, Cáceres JF 2005. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc Natl Acad Sci 102: 15042–15047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E 2000. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 19: 3013–3020 [DOI] [PubMed] [Google Scholar]
- Shin C, Manley JL 2004. Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol 5: 727–738 [DOI] [PubMed] [Google Scholar]
- Silva AL, Carmo F, Bugalho MJ 2013. RAC1b overexpression in papillary thyroid carcinoma: A role to unravel. Eur J Endocrinol 168: 795–804 [DOI] [PubMed] [Google Scholar]
- Singh R, Valcárcel J 2005. Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 12: 645–653 [DOI] [PubMed] [Google Scholar]
- Smith CW, Valcárcel J 2000. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem Sci 25: 381–388 [DOI] [PubMed] [Google Scholar]
- Srebrow A, Kornblihtt AR 2006. The connection between splicing and cancer. J Cell Sci 119: 2635–2641 [DOI] [PubMed] [Google Scholar]
- Stallings-Mann ML, Waldmann J, Zhang Y, Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields AP, Radisky DC 2012. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci Transl Med 4: 142ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S 2008. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 283: 1223–1227 [DOI] [PubMed] [Google Scholar]
- Stickeler E, Kittrell F, Medina D, Berget SM 1999. Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene 18: 3574–3582 [DOI] [PubMed] [Google Scholar]
- Van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Cáceres JF 2000. The MKK3/6-p38–signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol 149: 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP 2006. Unbalanced alternative splicing and its significance in cancer. Bioessays 28: 378–386 [DOI] [PubMed] [Google Scholar]
- Visvikis O, Lorès P, Boyer L, Chardin P, Lemichez E, Gacon G 2008. Activated Rac1, but not the tumorigenic variant Rac1b, is ubiquitinated on Lys 147 through a JNK-regulated process. FEBS J 275: 386–396 [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge CB 2008. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 14: 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weg-Remers S, Ponta H, Herrlich P, König H 2001. Regulation of alternative pre-mRNA splicing by the ERK MAP-kinase pathway. EMBO J 20: 4194–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SH, Manley JL 1997. Phosphorylation of the ASF/SF2 RS domain affects both protein–protein and protein–RNA interactions and is necessary for splicing. Genes Dev 11: 334–344 [DOI] [PubMed] [Google Scholar]
- Xie J, Lee J-A, Kress TL, Mowry KL, Black DL 2003. Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci 100: 8776–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X-Y, Ding J-H, Adams JA, Ghosh G, Fu X-D 2009. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev 23: 482–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Licciulli S, Avila JL, Cho M, Troutman S, Jiang P, Kossenkov AV, Showe LC, Liu Q, Vachani A, et al. 2013. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 32: 903–909 [DOI] [PMC free article] [PubMed] [Google Scholar]