

Haploid male germ cells contain a unique, unusually large cytoplasmic RNP granule, the chromatoid body (CB), which emerges during the switch between the meiotic and post-meiotic phases of spermatogenesis. This paper reveals the RNA and protein composition of CBs isolated from mouse testes, and therefore provides an important basis for the functional characterization of RNP granules. The results of this study suggest a central role for the CB in the control of the highly complex transcriptome of round spermatids.
Keywords: chromatoid body, male germ cells, piRNA, post-transcriptional, ribonucleoprotein granule
Abstract
The genome of male germ cells is actively transcribed during spermatogenesis to produce phase-specific protein-coding mRNAs and a considerable amount of different noncoding RNAs. Ribonucleoprotein (RNP) granule-mediated RNA regulation provides a powerful means to secure the quality and correct expression of the requisite transcripts. Haploid spermatids are characterized by a unique, unusually large cytoplasmic granule, the chromatoid body (CB), which emerges during the switch between the meiotic and post-meiotic phases of spermatogenesis. To better understand the role of the CB in male germ cell differentiation, we isolated CBs from mouse testes and revealed its full RNA and protein composition. We showed that the CB is mainly composed of RNA-binding proteins and other proteins involved RNA regulation. The CB was loaded with RNA, including pachytene piRNAs, a diverse set of mRNAs, and a number of uncharacterized long noncoding transcripts. The CB was demonstrated to accumulate nascent RNA during all the steps of round spermatid differentiation. Our results revealed the CB as a large germ cell-specific RNP platform that is involved in the control of the highly complex transcriptome of haploid male germ cells.
INTRODUCTION
Gametes are unique in their capability to give rise to a new organism and to thus ensure the perpetuation of the species. In addition to the genetic information, the epigenetic status of the cells can be inherited, which signifies the importance of securing both the genetic integrity and the correct epigenetic marks in the germline (Daxinger and Whitelaw 2012). Male germ cell differentiation is an exceptional developmental process that is characterized by the coordinated progression of cell differentiation through the mitotic proliferation phase, meiosis, and post-meiotic reconstruction of cell morphology to assemble the sperm-specific structures. Transitions between the different phases are tightly coordinated and controlled by orchestrated gene expression patterns (Chalmel et al. 2007; Laiho et al. 2013). Dramatic chromosomal modifications during meiotic events and during haploid chromatin compaction set additional requirements for the genome control due to the transient changes in the chromatin environment (Meikar et al. 2012).
All cell types have to selectively display precise genetic information at a given time and space during development. The complexity of the transcriptome has exceedingly expanded with the discovery of noncoding RNAs (ncRNAs) that are produced from the nonprotein-coding regions of the genome. A diverse network of post-transcriptional processes that interconnect the mRNAs, regulatory ncRNAs, and RNA-binding proteins in ribonucleoprotein particles (RNPs) provides an important additional regulatory level to gene expression (Keene 2010). RNP particles can form larger RNP composites, or RNP granules, that have a central role in determining cytoplasmic RNA localization and in compartmentalizing specific RNA regulation pathways. For example, processing bodies (P-bodies or GW/P bodies) and stress granules in mammalian cells are known to coordinate mRNA decay, storage, and transport (Buchan and Parker 2009; Kulkarni et al. 2010).
Germ cells of various organisms are characterized by special cytoplasmic RNP granules that are commonly called germ granules. The germ granules seem to be central structures in the processes involving PIWI proteins and PIWI-interacting RNAs (piRNAs), which are the male germ cell-predominant small RNAs with diverse, largely uncharacterized, functions (Meikar et al. 2011; Siomi et al. 2011). Mammalian germ granules exist in the cytoplasm of male germ cells during the whole course of spermatogenesis (Chuma et al. 2009). In prenatal prospermatogonia, two types of granules exist: germ granule-resembling pi-bodies and piP-bodies that have additional proteins that are also found in the somatic processing bodies (Aravin et al. 2009). These two types of granules are closely connected and participate in piRNA-mediated transposon silencing. After puberty, the most prominent germ granules are the intermitochondrial cement (IMC) in spermatocytes, which forms between mitochondrial clusters and the chromatoid body (CB) in haploid spermatids.
The CB appears in late pachytene spermatocytes and condenses into one single finely filamentous, lobulated, perinuclear granule in post-meiotic round spermatids (Fawcett et al. 1970; Meikar et al. 2011). The CB is a very large RNP granule (0.5–1 µm), and it has several intriguing features, such as its nonrandom movements and frequent contacts with the nuclear envelope and Golgi-acrosomal system. The important role of the CB in male reproduction is highlighted by the infertile phenotype of many knockout mice lacking various CB components (Kotaja and Sassone-Corsi 2007).
The known molecular features and its strategic emergence suggest that the CB is involved in the regulation of the transition from the meiotic to the post-meiotic phase and the subsequent haploid differentiation. The CB was described more than a hundred years ago, yet the reason for its existence and the nature of the CB-mediated RNA regulation have remained obscure. To gain a comprehensive view of the molecular composition of the CB, we applied the CB isolation protocol previously developed in our laboratory (Meikar et al. 2010) and identified the full protein and RNA content of the CB. This is the first time the composition of any mammalian germ granule has been revealed in detail. This study provides a fundamental basis for the future functional analysis of both the somatic RNP granules and the germline-specific germ granules.
RESULTS
CBs accumulate RNA
To study whether newly synthesized RNA in round spermatids is targeted to the CB, stage-specific pieces of the seminiferous tubules were cultured in the presence of the nucleotide analog 5-ethynyl uridine (EU), which is incorporated into the RNA upon synthesis. After 12 h of incubation, the nuclei of the pachytene spermatocytes and the round spermatids were intensively marked by EU (Supplemental Fig. S1A). Interestingly, a clear signal was also found in the CBs of the round spermatids (Fig. 1A). The CB localization was confirmed by phase contrast microscopy, as well as by costaining with anti-MVH (DDX4) antibody (Fig. 1A). We repeated the EU culture experiment with the seminiferous tubules containing rounds spermatids at defined stages of the epithelial cycle (XII, I–II, and IV–V), and the CB was always clearly labeled (Supplemental Fig. S1B). This finding indicates that the translocation of RNA into the CB was not restricted to a specific developmental stage. A chase experiment showed that the EU-labeled RNA signal was retained in the CB for at least 12 h (Fig. 1B). We also localized the poly(A)-containing RNAs in the CB throughout all steps of round spermatid differentiation by in situ hybridization using a polyT probe (Supplemental Fig. S2).
FIGURE 1.

CBs accumulate RNA. (A) Pieces of the seminiferous tubules (stage II–V) were incubated in the absence (Control) or presence of 5-ethynyl uridine (EU) to detect the newly synthesized, EU-labeled RNA (green). RNA accumulated in the nuclei and in the CBs (arrows). The CB localization was confirmed by immunostaining with anti-MVH antibody (red) and phase contrast microscopy. The nuclei were stained blue with DAPI. Scale bar, 10 µm. (B) EU-containing RNA retained inside the CB for at least 12 h after the removal of EU from the culture media. (C) The general CB RNA profile. CB RNA was separated by agarose and polyacrylamide gel electrophoresis and stained by SYBR Gold. The CB RNA can be grouped by size into ∼30 nt (small), ∼50–300 nt (medium), and >300 nt (large) RNAs. (D) Small RNAs in the CB mainly correspond to pachytene piRNAs. piRNAs derived from the pachytene piRNA clusters were enriched in the CB compared with the round spermatids (RS).
We previously developed and validated a CB isolation protocol (Meikar et al. 2010). We applied this protocol to isolate the CBs from total mouse testes (Supplemental Fig. S3A–E). Immunostaining with anti-MVH antibody was used to track the CBs during the isolation procedure (Supplemental Fig. S3A). It revealed that the CBs were enriched in the pellet fraction of the cell lysate after low-speed centrifugation and immonoprecipitated by the anti-MVH-bead complexes. This was confirmed by a parallel Western blot analysis with antibodies detecting different CB components (Supplemental Figs. S3B,C, S4A; Meikar et al. 2010). The electron microscopy of the isolated CBs revealed that intact CBs, with similar size and appearance as native CBs in round spermatids were bound in the anti-MVH-bead complexes (Supplemental Fig. S3D), while the negative control beads with rabbit IgG were depleted of CBs. Furthermore, we did not observe any other evident structures attached to the antibody-bead complexes at the electron microscopy level.
On the basis of a size separation profile in gel electrophoresis, the CB RNA can be roughly divided into three parts: small RNAs with a predominant ∼30-nt band, a medium-size smear between 50 and 300 nt and larger RNA molecules (Fig. 1C). To find out whether the general RNA profile changed during round spermatid development, we extracted the RNA from the CBs that were isolated from juvenile testes at 22 and 26 d post-partum (dpp) (Supplemental Fig. S3F). Phase contrast microscopy confirmed that the 22-dpp testis contained step 1–2 spermatids, while the 26-dpp testis contained step 7–8 spermatids (data not shown). No considerable changes in the characteristic ∼30-nt band were observed between the CB profiles of the early and late round spermatids (Supplemental Fig. S3G).
CBs accumulate pachytene piRNAs
Previously, we detected piRNAs in CBs using Northern blot analysis and in situ hybridization (Meikar et al. 2010). The characteristic ∼30-nt band in the total testis RNA that contains pachytene piRNAs, coimmunoprecipitated with the CBs from the pellet fraction during the CB-isolation procedure (Fig. 1C; Supplemental Fig. S3E). To study the composition of the ∼30-nt band that is characteristic of the CB RNA, we obtained small RNA libraries from all of the steps involved in the CB purification process (LYS, SUP, PEL, PF, CB-IP, and Ctrl-IP, see Materials and Methods) and the enriched population of round spermatids (RS). The CBs are only present in RSs, and the RS RNA includes the CB-associated RNAs. The small RNAs sequenced from the RS were divided in six main categories: pachytene piRNAs (59%), genic piRNAs (15%), miRNAs (7%), structural RNAs (7%), transposon piRNAs (9%), and simple repeats (2%). Reads that mapped uniquely to pachytene piRNA cluster coordinates (Girard et al. 2006) were annotated as pachytene piRNAs. Reads that mapped outside piRNA clusters to transposable elements and genes were considered as transposon piRNAs and genic piRNAs, respectively. piRNAs derived from pachytene piRNA clusters and transposable elements were more represented in the CB (79% and 17%, respectively) when compared with the RS sample (Fig. 1D). On the other hand, microRNAs (miRNAs) were generally under-represented in the CB when compared with the RS (Fig. 1D). Also, piRNAs from genes were not specifically enriched in the CB (Fig. 1D). These results show that from all of the RS small RNAs, pachytene piRNAs and transposon piRNAs are preferably accumulated in the CB. Overall, the CB piRNA content was similar to MIWI-bound piRNAs (data not shown), which is in agreement with the observation that MIWI is one of the main protein components of the CB (Meikar et al. 2010).
The CB proteome supports its involvement in RNA regulation and piRNA pathways
To analyze the presence of the piRNA pathway components in the CB at the protein level, we subjected the CB protein extract to mass spectrometric analysis. Analysis was performed from five independent experiments. After extracting the unspecific protein hits that were obtained from the negative control IPs, the results were combined to reveal a reproducible list of around 100 CB-associated proteins. We analyzed the relative abundance of the individual proteins in the CB by dividing their emPAI values by the sum of the whole emPAI value of the CB proteins. Two PIWI family members, PIWIL1/MIWI and PIWIL2/MILI, which bind piRNAs during this specific spermatogenic phase were among the most abundant proteins in the CB (Fig. 2A). In addition to MIWI and MILI, the nine most abundant proteins included the DEAD box helicases DDX4/MVH, DDX25/GRTH, and DDX3L, the Tudor domain-containing proteins TDRD6 and TDRD7, the poly(A)-binding protein PABP, and the heat-shock protein HSP72/HSPA2 (Fig. 2A). Together, these proteins made up approximately two-thirds of all of the CB proteins. MVH and MIWI appear to be the two CB core components that together contribute to ∼40 mole percent of the CB proteome. The high abundance of these proteins in the CB has also been shown by silver staining of the proteins extracted from the isolated CBs (Meikar et al. 2010). We isolated CBs from 22- and 26-dpp testes to study the dynamics of the CB protein composition and showed that the general profile of the most abundant CB proteins remained the same during round spermatid development (data not shown). We identified an RNA-binding protein SAM68 in the CB sample isolated from 22- but not from 26-dpp mice (data not shown). This demonstrates the sensitivity and specificity of the CB purification protocol as SAM68 is previously reported to localize in the CB only in the very early round spermatids (Messina et al. 2012).
FIGURE 2.

Proteomic analysis of the CB. (A) The main CB components MVH/DDX4, MIWI/ PIWIL1, TDRD6, TDRD7, GRTH/ DDX25, PABP, HSP72/HSPA2, DDX3L, and MILI/PIWIL2 are presented as the mole percentage of the total CB protein content. (B) Pie chart of the most represented domains in the 88 CB proteins. (C) Selection of the main Gene Ontology (GO) terms of the molecular functions, biological processes, and cellular components of the 88 CB proteins. The numbers on the bars represent the CB-protein hits in the corresponding GO term. (FDR) False discovery rate of a given GO term.
A list of 88 CB proteins that were identified in at least three separate experiments (Supplemental Table S1) was generated to perform functional annotation and association analysis using DAVID Bioinformatics Resources 6.7 (Huang da et al. 2009). The most common domains in the CB proteome were nucleotide and nucleic acid binding domains, the DEAD-box helicase domain, and the K-homology domain; all of these domains are related to RNA recognition and binding (Fig. 2B). Another well-represented protein domain in the CB proteome was the Tudor domain, which is related to the structural organization of RNPs.
Gene Ontology (GO) term analysis identified 81 Biological Process terms (GOTERM_BP_FAT), 34 Cellular Component terms (GOTERM_CC_FAT), and 32 Molecular Function terms (GOTERM_MF_FAT) among the CB proteins. The most highly represented biological processes in the CB-specific proteome were mRNA metabolic processes (RNA splicing, mRNA processing, etc.), reproductive cellular process, regulation of translation, and gamete generation (sexual reproduction, germ cell development, spermatogenesis, etc.) (Fig. 2C). The most represented cellular component terms were the ribonucleoprotein complex (chromatoid body, P granule, germplasm), non-membrane-bounded organelle, and spliceosome. The most represented molecular functions in the CB proteome were RNA binding, helicase activity, and purine ribonucleotide binding. Altogether, the proteome analysis clearly demonstrated that the CB has a role in RNA-related processes, and the proteins associated with the piRNA pathway comprise the most prominent functionally associated group of proteins in the CB.
CBs contain transcripts that are derived from pachytene piRNA clusters
To analyze the detailed long RNA content of the CB, we prepared several transcriptome libraries from all of the steps of the CB purification process and an enriched population of round spermatids using the NSR protocol, which detects both poly(A) and non-poly(A) transcripts excluding rRNA (Armour et al. 2009). We obtained three independent replicates of anti-MVH IPs (isolated CBs) or anti-acrosin IPs (control). While ∼79% of the reads sequenced from the round spermatid libraries or the libraries prepared from the lysate (LYS), supernatant (SUP), pellet (PEL), and filtrated pellet (PF) steps of the protocol (∼5.6 million reads) were successfully mapped to genome, only ∼63% of the reads (∼2.5 million reads) from the anti-MVH IP mapped to the genome and ∼30% of the reads from the nonspecific anti-acrosin IP mapped to genome (Supplemental Table S2).
We compared the RNA content of the CB vs. the RNA prepared from either total testis lysate (LYS) or round spermatids (RS) (Fig. 3A). We mapped reads to five major categories: exons, introns, structural RNAs, transposable elements (TE), and other intergenic regions. Structural RNAs included rRNA, tRNA, and snoRNA (from UCSC genome browser). Overall, we did not observe any enrichment for specific categories of RNA in the CB compared with the round spermatids transcriptome. On closer observation, some individual TEs were depleted in the CB when compared with the round spermatids (Fig. 3B). The ratio between the exonic and intronic reads was different between the total testis lysate and the CB and round spermatids samples, with more intronic reads in the testis lysate (Fig. 3A). The anti-acrosin and rabbit IgG negative control IP samples yielded only low amount of reads.
FIGURE 3.

CB long RNA analysis. (A) CB RNA reads mapped to five main categories: exons, introns, structural RNA, transposable elements (TE), and other intergenic regions. The majority of the reads were from the exonic regions of the genes. (CB-IP) Isolated CBs; (LYS) total testis lysate; (RS) round spermatid lysate. (B) Transcript abundances in the CB vs. the RS. (Gene) mRNAs; (TE) transposable elements; (rRNA) ribosomal RNAs; (snoRNA) small nucleolar RNAs; (lincRNAs) annotated long intergenic RNAs. (C) Transcripts from pachytene piRNA clusters were enriched during the CB isolation procedure. (LYS) Whole testis lysate after sonication, (SUP) supernatant fraction after centrifugation, (PEL) CB-containing pellet fraction after centrifugation, (PF) pellet fraction after additional filtration, (CB) immunoprecipitation of the PF fraction using anti-MVH antibody. (D) Precursors of the pachytene piRNA clusters were specifically enriched in the CB compared with the round spermatid transcriptome (RS). (E) Correlation between the abundance of long RNA transcripts and small RNAs in the CB.
Approximately 7% of the long RNA reads of the CB mapped to noncoding transcripts other than structural RNAs and transposable elements (Fig. 3A). Among them, almost 20% represented transcripts that derived from pachytene piRNA clusters, which were specifically enriched after all steps of the CB purification process (Fig. 3C). The cluster transcripts were also enriched in the CB when compared with the round spermatid transcriptome (Fig. 3D; Supplemental Table S3). This suggests that the CB could participate in the processing of piRNAs from longer precursors. However, the absence of the core primary piRNA processing components in the CB (Supplemental Table S1), as well as the unaffected pachytene piRNA production in knockout mice with disrupted CB structure (Tanaka et al. 2011) contradicts this hypothesis. Finally, we did not observe any correlation between the abundance of long RNA transcripts and small RNA production in the CB (Fig. 3E). Although we cannot exclude the possibility that the CB is involved in processing a minor fraction of pachytene piRNAs or other small RNAs, these observations suggest that piRNA biogenesis does not take place in the CB.
Novel RNA transcripts are enriched in the CB
In addition to the long piRNA cluster transcripts and some annotated long noncoding transcripts (Fig. 3B), the CB transcriptome also included 4992 nonannotated transcripts that originated from regions of the genome with no reported activities (Supplemental Table S4). Reads deriving from the novel nonannotated transcripts were almost twofold enriched in the CB compared with the round spermatid sample (data not shown). The novel CB-enriched transcripts mapped evenly to all chromosomes, except for the Y-chromosome, which was virtually devoid of any hits (Fig. 4A). We analyzed the expression of some randomly selected transcripts using RT-qPCR. The majority of the analyzed transcripts were predominantly expressed in the testis. Cuff1617 was most highly expressed in the brain but was found also in the testis; lower levels of Cuff1617 were also found in the liver, kidneys, ovaries, and adrenal glands (Fig. 4B). We also analyzed the expression of four transcripts in the enriched populations of pachytene spermatocytes, round spermatids, and elongating spermatids. We showed that two transcripts had relatively higher expression in the round spermatids, and two transcripts were relatively higher in the spermatocytes (Fig. 4C). All four transcripts were down-regulated in the elongating spermatids.
FIGURE 4.
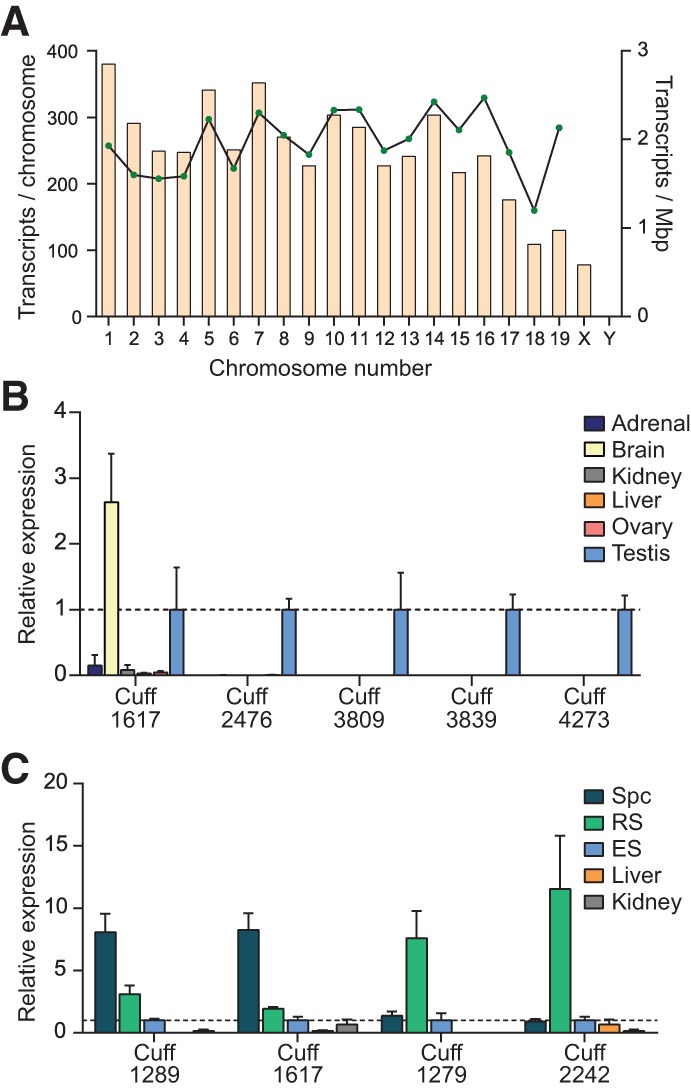
Novel long noncoding RNAs were enriched in the CB. (A) Distribution of the CB-enriched noncoding transcript genes on mouse chromosomes. (B) Relative expression levels of five novel transcripts in different mouse tissues as detected by RT-qPCR. (C) Expression levels of four transcripts were compared in the spermatocytes (Spc), round spermatids (RS), and elongating spermatids (ES). Note that two of the transcripts were mainly expressed in the round spermatids, whereas the other two transcripts have higher expression in the spermatocytes.
CBs accumulate mRNAs and proteins involved in mRNA processing
The CB was originally hypothesized to be involved in the translational control of mRNAs (Parvinen 2005). Our deep sequencing revealed that, in addition to its piRNA-related functions, the CB may play a substantial role in mRNA regulation. In the transcriptome analysis, the most abundant transcripts in the CB were from the exonic regions of genes (Fig. 3A). In total, 8059 genic transcripts were detected in the CB (Supplemental Table S5). Only 915 of these genes were two- to 15-fold enriched in the CB vs. the round spermatids (Supplemental Table S6). Intronic reads were also found in the CB transcriptome, but we did not observe any enrichment for intron retention for the protein-coding genes (data not shown).
The high number of mRNAs in the CB was corroborated by the CB proteome that includes proteins that bind RNA during nuclear RNA maturation steps and remain bound in the cytoplasmic mature mRNAs (Fig. 5A). A large portion of these proteins is composed of the shuttling heterogeneous nuclear ribonucleoproteins (hnRNPs) that coat the newly synthesized RNA and participate in mRNA splicing, export, and translation (Han et al. 2010). Poly(C) and poly(A)-binding proteins that bind to newly synthesized RNA and can shuttle between the nucleus and the cytoplasm (Gorgoni and Gray 2004) were also found in the CB. The CB contains several proteins involved in the regulation of splicing, such as hnRNPA2/B1, DDX5, hnRNPA3, PABP1, hnRNPM, TRA2B, PCBP1, hnRNPH, and hnRNPF.
FIGURE 5.
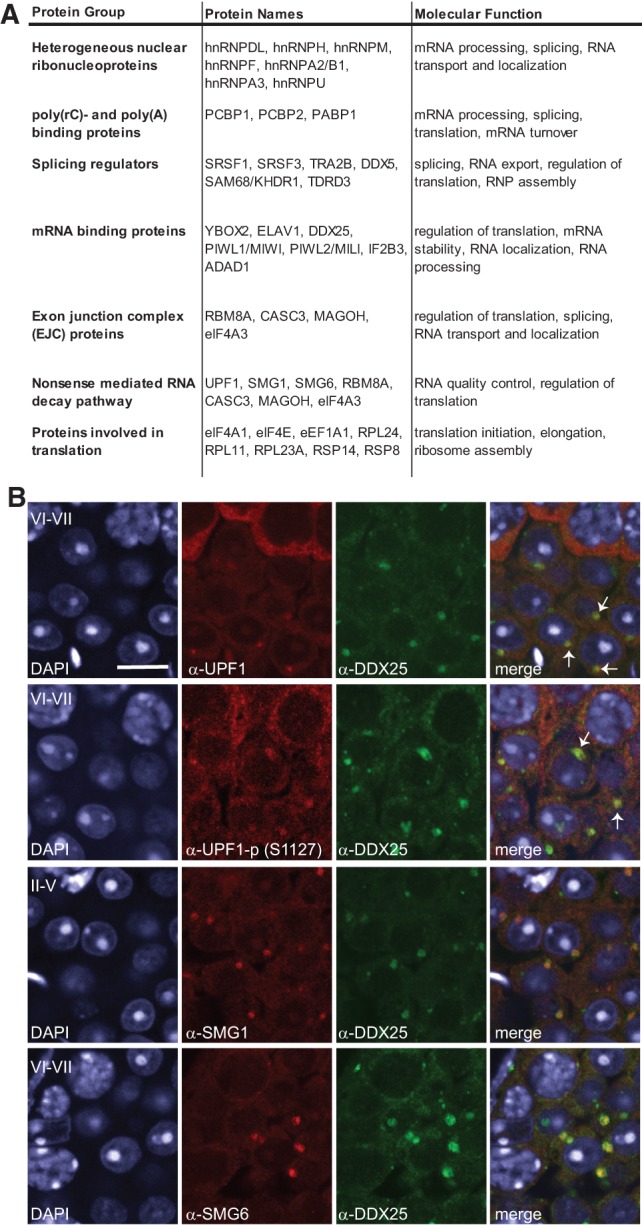
CBs accumulate proteins involved in mRNA maturation and regulation. (A) The CB proteins that have known functions in mRNA maturation, processing, splicing, and mRNA binding are listed in the table. (B) Confocal fluorescence microscopy to show the localization of the NMD components UPF, phosphorylated UPF1, SMG1, and SMG6 (red) in round spermatids. Costaining with DDX25 (green) confirmed their localization in the CB. DDX25 has earlier been reported as a CB component (Sato et al. 2010) and we further validated its CB localization by coimmunostaining with anti-MVH antibody (Supplemental Fig. S5B). Nuclei are stained with DAPI (blue). White arrows point to some selected CBs. Scale bar, 10 µm.
Furthermore, we demonstrated the enrichment of the exon–junction complex (EJC) proteins in the CB. The EJC is assembled onto the exon borders of the mRNA during splicing and travels with the mRNP to the cytoplasm, where it acts as an effector for downstream events in mRNA metabolism. The EJC is composed of four conserved core proteins: the MAGOH-RBM8A/Y14 heterodimer, eIF4A3, and CASC3 (Bono and Gehring 2011). All of these proteins were identified as CB components (Fig. 5A). The CB localization of eIF4A3 was confirmed in the isolated CB extracts by immunoblot analysis (Supplemental Fig. S4A). eIF4A3 and RBM8A were also localized to the CB using immunofluorescence staining (Supplemental Fig. S4B). eIF4A3 was found to be enriched in the CBs during all steps of round spermatid differentiation (Supplemental Fig. S4C).
The components of the NMD pathway in the CB
Interestingly, many proteins that are involved in the nonsense-mediated RNA decay (NMD) pathway were identified as CB components in the mass spectrometric analysis (Fig. 5A). The NMD machinery has a well-known role in a translation-coupled quality control as it can recognize and degrade aberrant mRNAs with premature termination codons (PTC) (Kervestin and Jacobson 2012). In addition to the EJC that enables the NMD machinery to localize PTCs, the CB contains an important NMD mediator UPF1 (Fig. 5B) that triggers downstream activities in the pathway upon the recognition of PTCs. UPF1 is activated by the SMG1 kinase that can phosphorylate UPF1 at several serine/threonine–glutamine (S/TQ) motifs (Yamashita et al. 2001; Okada-Katsuhata et al. 2012). Phosphorylation of UPF1 by SMG1 has been demonstrated to create binding platforms for other NMD factors (the SMG5:SMG7 complex and for SMG6) (Okada-Katsuhata et al. 2012). We were able to localize phosphorylated UPF1 in the CB by using an antibody recognizing phosphorylated serine at the C terminus of UPF1. This indicates that the CB may act as a platform for UPF1-triggered downstream processes. SMG1 was also accumulated in the CB (Fig. 5B).
Aberrant mRNAs recognized by the NMD machinery are targeted to different decay pathways involving deadenylation, decapping, and exonucleolysis (Kervestin and Jacobson 2012). The CB proteome does not include enzymes acting in these processes (Supplemental Table 1). However, we identified an endonuclease SMG6 as a CB component (Fig. 5B). SMG6 is an NMD factor that has endonuclease activity and that provides an alternative decay pathway for NMD substrates (Huntzinger et al. 2008; Eberle et al. 2009). SMG6 signal was highly enriched in the CB (Fig. 5B) and it localized in the CB throughout the round spermatid differentiation (data not shown).
A diverse set of cellular mRNAs is targeted to the CB
To better understand the nature of the CB-accumulated mRNAs, we first integrated a transcriptomic data set from microarray data including three types of enriched mouse germ cell populations (spermatogonia, pachytene spermatocytes, and round spermatids) and one population of testicular somatic cells (Sertoli cells) (Chalmel et al. 2007). Using this data set, we filtered out a set of 14,788 loci that had detectable expression (above the overall median intensity) in the round spermatid samples, termed rSpt+. We then further analyzed the transcripts assembled from the CB RNA sequencing that matched with the annotated transcripts from RefSeq. The set of CB transcripts corresponding to 7367 annotated loci (EntrezGene IDs) was termed CB+; of these transcripts, 3704 correspond to the highly abundant transcripts above the median of the FPKM (fragments per kilobase of transcript per million mapped reads) values and were termed CB-High+. By the direct comparison of both the spermatid and CB transcriptome data sets, we found that (1) approximately half (45.2%, 6686/14,788) of the mRNAs expressed in the round spermatids (rSpt+) were detected in our CB+ mRNA list; and (2) 90.7% (6686/7367) of the CB+ genes were expressed in the round spermatids.
To analyze the cellular origins of the transcripts detected in the mouse CBs, we overlapped the list of CB-High+ loci with genes detected in round spermatids and those associated with somatic (SO), mitotic (MI), meiotic (ME), or post-meiotic (PM) expression patterns (Chalmel et al. 2007). The analysis revealed that the CB-High+ genes were significantly over-represented in the clusters showing a meiotic (21.4%, 769/3592, P = 2.72 × 10−79) or a post-meiotic (18.9%, 679/3592, P = 2.64 × 10−68) induction of expression during spermatogenesis (Fig. 6A,B). On the other hand, round spermatid genes without differential expression during spermatogenesis (Not-DET) were unambiguously under-represented in the set of CB-High+ genes (P = 3.2 × 10−62) and were enriched in the set of genes undetectable in CBs (CB−) (P = 7.04 × 10−43). Taken together, these results indicate a clear tendency of the CBs to accumulate a significant proportion of germline-expressed genes that show differential expression and are likely to be involved in the mouse spermatogenic process, especially during the meiotic and post-meiotic phases (Fig. 6; Supplemental Fig. S5).
FIGURE 6.
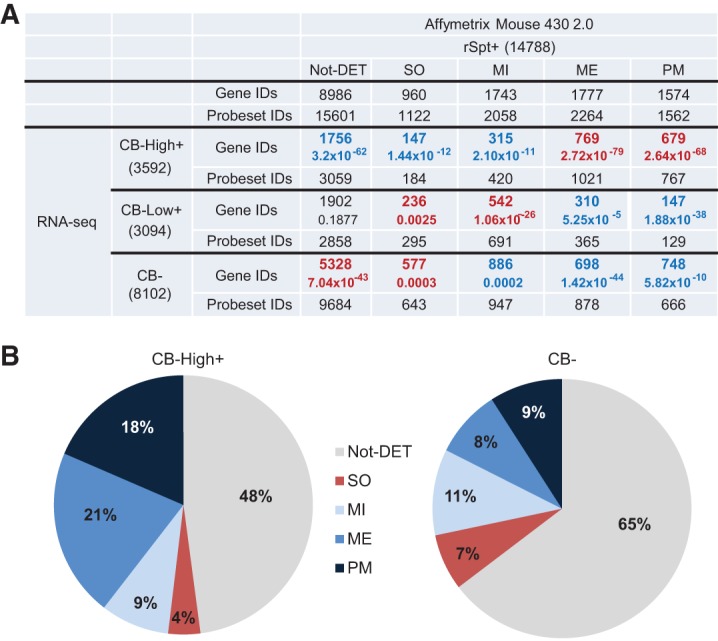
Expression profiling of the mRNAs localized in the CBs. (A) The round spermatid (rSpt+) mRNAs that were found in the CB at high levels (CB-High+) or at low levels (CB-Low+), or were not found in the CB (CB−) were compared with the list of genes shown to be differentially expressed in the somatic cells of the testis (SO), mitotic spermatogonia (MI), meiotic spermatocytes (ME), or post-meiotic spermatids (PM) (Chalmel et al. 2007). Not-DET represents the group of genes that are expressed in round spermatids but are not differentially expressed in the testis. The table summarizes the intersections with the corresponding P-values (hypergeometric). The number of total nonredundant gene IDs for each category is provided in parentheses. (B) Pie charts representing the distribution of the CB-High+ and CB− genes from the previous table.
We performed a GO term enrichment analysis using the AMEN suite (Chalmel and Primig 2008). Briefly, 42 of the GO terms that were either enriched or depleted in the CB are presented in Figure 7. The CB-High+ genes that showed peak expression in the meiotic (ME) or post-meiotic (PM) cells were often found to be enriched in biological process GO terms connected to germ cell-specific processes, such as spermatid differentiation, flagellar construction (centrosome organization and cilium morphogenesis), sperm motility, fertilization, or meiosis and cell cycle. In contrast, the CB− genes that showed peak expression in the ME or PM cells appeared to be more enriched in GO terms related to chromatin organization and gene silencing. Distinct GO terms also appeared to be enriched among the CB-High+ and CB− genes that were not differentially expressed during spermatogenesis (Not-DET).
FIGURE 7.

GO term enrichment analysis. Forty-two significantly enriched GO terms are listed, followed by the total number of genes associated with the term. The genes expressed in the round spermatids (rSpt+) were divided into those detectable in high abundance in the CB (CB-High+) or undetectable in the CB (CB−). These classes were further divided into the clusters of differentially expressed genes during spermatogenesis with peak expression during the meiosis (ME) or post-meiotic phases (PM), or not differentially expressed during spermatogenesis (Not-DET). For each cluster of genes, the numbers of genes observed vs. expected by chance for each GO term are indicated. The total number of genes associated with a biological process term and the genes in the classes are shown at the top of the column. A color scale of P-values for enriched (red) and depleted (blue) terms is shown at the bottom. To allow a direct comparison of the CB-High+ and CB− classes, the enrichment calculations were computed by considering only the genes expressed in rSpt, instead of all the genes in the genome.
To identify whether any specific motif was required for CB targeting, we performed de novo motif predictions followed by enrichment analysis. Somewhat surprisingly, no motif enrichment was over-represented in the transcripts that were highly abundant in the CB (CB-High+) compared with the cytoplasmic transcripts (CB−) (Supplemental Fig. S6). Instead, we observed the opposite; 43 significantly enriched 3′ UTR nucleotide motifs were more abundant in the CB− transcripts than in the CB-High+ transcripts. Most of these enrichments were observed for both the meiotic (ME) and post-meiotic (PM) transcripts but not for the Not-DET transcripts.
DISCUSSION
Postnatal male germ cells evidently have a need for accurate and effective post-transcriptional control mechanisms. This is highlighted by a recent study demonstrating that the transcription of the genome was substantially more widespread in the testis than in other organs (Soumillon et al. 2013). In particular, post-meiotic spermatids have a remarkably diverse transcriptome, which encompasses protein-coding and lncRNA genes as well as poorly conserved nongenic elements (Soumillon et al. 2013). By revealing the molecular composition of the CB, we demonstrated the strong association of this cytoplasmic germ granule with RNA-related processes and its possible specialization in the control of the complex transcriptome of the round spermatids. Differentiating male germ cells undergo unique processes, including meiosis and the haploid differentiation program, as well as the preparation of the gamete for genetic and epigenetic inheritance upon fertilization. It is clear that unique mechanisms are required to cope with these processes. The CB is composed of several ubiquitously expressed proteins that are involved in pre-mRNA binding and processing. However, a substantial number of the CB components are germ-cell specific, supporting its specialized function in the regulation of germline development.
The purity and integrity of the isolated CBs is of principal importance for this study, and the success of the CB isolation was validated by several methods (Supplemental Fig. S3). The proteomic analysis was highly reproducible. In addition to the abundant CB components such as MVH, MIWI, DDX25, TDRD6, and TDRD7, the CB component list included many other less-abundant proteins. The CB localization of at least one-fourth of these proteins has been validated by immunostainings (this study; Kotaja and Sassone-Corsi 2007; Meikar et al. 2011; O Meikar, K Sõstar, M Da Ros, N Kotaja, unpubl.). The CB proteome included many proteins with reported nuclear localization/function, but the closer examination revealed that most of these proteins are shuttling and can be translocated to the cytoplasm. The EJC proteins provide an example of such proteins, and their CB localization was confirmed by immunofluorescence (Supplemental Fig. S5). Living cell microscopy has revealed that the CB is actively and nonrandomly moving in the cytoplasm, and it makes frequent contacts with other cellular structures, such as the nuclear envelope, Golgi complex, and vesicular transport system (Parvinen et al. 1997). This interactive and dynamic nature of the CB in cellular environment hampers the analysis of the CB as a completely isolated structure, and the cross-linking may cause unspecific copurification of some closely associated molecules. However, all of our results suggest that the current protocol is sufficiently sensitive and specific for the downstream analysis.
By far, the most pronounced molecular pathway in the CB is the piRNA pathway. The piRNA-binding PIWI proteins MIWI and MILI and the Tudor-domain containing proteins that associate with the PIWI proteins were among the most prominent CB components according to our analysis. Several Tudor proteins were identified in the CB, including TDRD1, TDRD3, TDRD5, TDRD6, TDRD7, RNF17, and STK31. Considering the well-characterized role of the Tudor proteins as molecular scaffolds (Pek et al. 2012) and given the disrupted CB architecture in the TDRD6 and TDRD7 knockout animals (Vasileva et al. 2009; Tanaka et al. 2011), these proteins most likely serve as structural components by creating the CB protein mesh and recruiting other proteins through dimethylarginine binding (Pek et al. 2012) or through other protein–protein interactions. Several other proteins that have been associated with the piRNA pathways on the basis of their molecular interactions or the phenotypic characteristics of the knockout mice models were also found in the CB. These include, for example, the MAEL and MOV10L proteins (Aravin et al. 2009; Frost et al. 2010; Zheng et al. 2010; Zheng and Wang 2012).
Germ granules have been implicated in piRNA pathways in both mammals and lower organisms (Siomi et al. 2011). However, the exact role of the germ granules in piRNA biogenesis or functions is still unknown. Small RNA analysis confirmed the enrichment of a wide range of pachytene piRNAs in the CBs, and we were also able to identify long transcripts derived from piRNA clusters. However, the available data argue against the idea that the primary processing of pachytene piRNAs takes place in the CB. The processing of pachytene piRNAs has been demonstrated to begin in meiotic cells (Aravin et al. 2006; Li et al. 2013), prior to the formation of the CB. Genetic mouse studies have also revealed that the intact CB is not required for piRNA production (Tanaka et al. 2011). Moreover, the initial steps of primary piRNA biogenesis are compartmentalized on the surface of the mitochondria that contain the conserved primary piRNA components, including GAZS, GPAT2, and endoribonuclease Zucchini/PLD6 (Ma et al. 2009; Huang et al. 2011; Watanabe et al. 2011; Ipsaro et al. 2012; Nishimasu et al. 2012; Czech et al. 2013; Handler et al. 2013; Shiromoto et al. 2013; Vagin et al. 2013). These proteins were not detected in the CB pulldowns. These results conjecture that pachytene piRNAs are synthesized in meiotic spermatocytes, most likely in conjunction with mitochondria-associated germ granules such as the intermitochondrial cement (IMC), and are later loaded into the CB along with the PIWI proteins. This places the CB downstream from the initial steps of piRNA processing. CBs could, for example, function by facilitating mature piRNA–PIWI complex formation. Similar function has been suggested for Drosophila somatic Yb bodies; the Yb body component Armitage is required for Piwi localization into Yb bodies, and piRNAs are freed from Piwi proteins in the absence of Armitage or Yb (Saito et al. 2010). CB could also function by gathering PIWI–piRNA-target RNA complexes in order to store them or to keep them dormant. This role has been demonstrated for somatic GW/P bodies that can sequester and stabilize translationally repressed miRISC-bound target mRNAs (Castilla-Llorente et al. 2012). Alternatively, the general RNA flux into the CB could provide material for a potential CB-associated scanning mechanism to identify piRNA targets.
The function of the CB is clearly not restricted to piRNA-related processes. The CB retains a considerable portion of the round spermatid mRNA transcriptome. In particular, transcripts that are specifically induced in spermatocytes or round spermatids are enriched in the CB. We found a significant enrichment of GO terms related to gamete generation among these CB-targeted transcripts with meiotic or post-meiotic expression patterns; however, in general, the CB transcriptome included a diverse set of mRNAs involved in different biological processes. De novo motif prediction failed to reveal any specific sequence motifs that would explain their targeting to the CB. Instead, our data suggest a presence of possible CB-exclusion signals rather than CB-targeting signals. Although the molecular mechanism by which mRNAs are targeted to the CB remains elusive, it is clear that some selectivity exists and that the CB transcripts represent a specific set of round spermatid transcripts.
Among the CB long RNAs, we identified thousands of previously uncharacterized intergenic noncoding transcripts that are potentially spliced and processed in the same way as mRNAs and might follow the same targeting routes. The accumulation of different RNA species in the CB suggests a more general role in the RNA-related processes instead of a functional specialization in a clearly defined class of RNAs. Importantly, we demonstrated that the flow of RNA into the CB was not restricted to a certain developmental phase and the CB receives newly synthesize RNA during all of the steps (1–8) of round spermatid differentiation. It is evident that further functional characterization and specific genetically modified mouse models will be required to address the exact nature of the CB-targeted RNAs and the reason for their CB targeting.
A very interesting functional clue was provided by the presence of the nonsense-mediated decay (NMD) machinery in the CB. In addition to the exon–junction complex (EJC) components that provide the recognition platform for NMD, the CB accumulated UPF1/RENT, which is a key mediator of NMD, SMG1 kinase, which activates UPF1 through phosphorylation, and SMG6, which is an endonuclease involved in the NDM pathway (Huntzinger et al. 2008; Eberle et al. 2009; Kervestin and Jacobson 2012). SMG1-mediated UPF1 phosphorylation (and the subsequent dephosphorylation) was recently shown to be the only critical requirement for both, EJC-enhanced and the alternative, EJC-independent NMDs (Metze et al. 2013). It is important to note that according to our immunofluorescence results, the CB-localized UPF1 is at least partially phosphorylated. NMD acts mainly by promoting the degradation of mRNAs containing premature termination codons. Interestingly, NMD can also regulate the stability of various types of other substrates, including alternatively spliced mRNAs, mRNAs containing upstream open reading frames (ORFs) and transcripts that are derived from transposons, pseudogenes, or out-of-frame gene rearrangements (Mendell et al. 2004; Rehwinkel et al. 2005; Kervestin and Jacobson 2012). The presence of the NMD core components in the CB and the general diversity of NMD targets conjecture a potentially powerful mechanism in the CB to regulate a high number and a broad range of transcripts.
In this study, we have generated an atlas of CB components, which will be exceedingly applicable in the functional characterization of the germ granules and analogous somatic RNP granules. While the exact mechanisms of the CB-mediated post-transcriptional regulatory functions require further investigation, it is clear that this tremendous germline-specific RNP granule has an integral role in the production of fertile spermatozoa.
MATERIALS AND METHODS
Seminiferous tubule cultures
Mouse testes were decapsulated in Dulbecco's Modified Eagle's Medium/Nutrient Mixture F-12 Ham (D8437, Sigma). Segments of the seminiferous epithelial cycle representing stages X–XI, XII–I, II–V, and VII–VIII of spermatogenesis were cut as previously described (Kotaja et al. 2004). The isolated pieces of tubules were incubated on glass slides in 30 μL of medium supplemented with 1 mM ethynyl uridine (EU) at 34°C O/N in a highly humidified atmosphere containing 5% CO2. Spermatogenic cells were spread out of the cultured seminiferous tubules to monolayers using the squash technique (Kotaja et al. 2004), snap-frozen in liquid nitrogen, and fixed in 96% EtOH for 3 min. The nascent RNA was visualized using the Click-iT RNA Alexa Fluor 488 Imaging Kit (Molecular Probes, Invitrogen, C10329). For the chase experiment, EU was removed from the media after 8 h of incubation, and the tubule culture was continued for 12 h.
Isolation of the chromatoid bodies
CBs were isolated according to Meikar et al. (2010) with some modifications. Germ cells were released from four testes of adult C57BL/6NHsd mice with 0.05% (w/v) collagenase (Worthington). The cells were filtered through a 100-µm cell strainer (BD Falcon), washed with Phosphate buffered saline (PBS), and fixed in 0.1% PFA solution (Electron Microscopy Sciences, USA) for 20 min at RT. The fixed cells were lysed by sonication (UCD-200, Diagenode) in 1.5 mL of RIPA buffer (50 mM Tris-HCl at pH 7.5, 1% NP-40, 0.5% w/v sodium deoxycholate, 0.05% w/v sodium dodecyl sulfate, 1 mM EDTA, 150 mM NaCl, 1x complete mini mix (Roche), 0.2 mM PMSF, and 1 mM DTT) (sample name: LYS). The CB-enriched pellet fraction was separated by centrifugation at 300g for 10 min, resuspended in RIPA buffer, sonicated for an additional 2 × 30 sec (sample name: PEL; leftover supernatant sample name: SUP), and filtered through 5.00-µm filter units (SLSV 025LS, Millipore) (sample name: PF). The CBs were immunoprecipitated using Dynabead Protein G (Invitrogen) coupled to either an in-house (Kotaja et al. 2006) or a commercial (Abcam, ab13840) anti-MVH/DDX4 antibody O/N at 4°C (sample name: CB). For the negative controls, a non-CB specific antibody, such as anti-acrosin (Santa Cruz, sc-67151) or rabbit IgG (Neomarkers, NC-100-P), was used (sample name: Ctrl). The crosslinks of the isolated CBs were reversed by incubation at 70°C for 45 min.
RNA extraction and gel electrophoresis
RNA was extracted from the isolated CBs with the Trisure reagent (Bioline) using the standard protocol. Extra chloroform and ethanol purification steps were added to remove the traces of phenol. Isolated RNA was analyzed using a NanoDrop (Thermo Scientific) and Bioanalyzer (Agilent). For the CB RNA profile analysis, the isolated RNA from the CBs of adult and juvenile mice was [γ32P]ATP-labeled through an exchange reaction using the T4 Polynucleotide Kinase kit (EK0032, Fermentas). The RNA was separated in 10%–15% denaturing urea-polyacrylamide or 1%–2% agarose gels, post-stained with SYBR Gold (Invitrogen), and visualized using a Fastgene Blue LED Illuminator (Nippon Genetics). The radioactivity was detected using a FujuFilm BAS-5000 PhosphorImager system.
Small RNA cloning and analysis
RNA prepared from the CB and control samples was size selected for species that were 19–33 nt in length, and small RNA cloning was performed according to (Malone et al. 2012). The samples were sequenced using an Illumina Genome Analyzer II for 36 cycles in a single end sequencing run. After clipping the 3′ end adaptor sequence (CTGTAGGCACCATCAATCGTATGCCGTCTTCTGCTTG), only reads that were longer than 15 nt in length were selected, collapsed, and mapped to the mouse genome (2007 NCBI37/mm9) without mismatches. The pachytene cluster coordinates used for the piRNA analysis were previously described (Girard et al. 2006).
Transcriptome libraries and analysis
Transcriptome libraries were prepared according to Armour et al. (2009) using the not-so-random priming (NSR) method. The samples were sequenced on Illumina Genome Analyzer II for 50 to 75 cycles in a pair-ends sequencing run. First, 8 nt of the reads were trimmed and mapped to the mouse genome (2007 NCBI37/mm9). UCSC RefSeq July 2011 and the piRNA cluster coordinates previously described in Girard et al. (2006) were used to map the reads to genes, piRNA clusters, and known noncoding transcripts. Tophat v1.3.3 was used with a default parameter set that includes reads from both strands. For the identification of novel transcripts, we used Cufflinks. Any novel transcripts that overlapped with the exons of known RefSeq transcripts or the piRNA clusters were removed from the analysis. Subsequently, all predicted novel transcripts from all replicates were merged and redundant calls were removed. Transcripts shorter than 500 nt were removed from the analysis.
Mass spectrometric analysis
Reversed CB samples were separated in mini-protean TGX pre-cast gradient gels (BioRad) and stained with SimplyBlue SafeStain (Life Technologies). Gel pieces were excised from the sample lanes, followed by in-gel digestion with trypsin (Promega) and extraction of the peptides. The peptides were analyzed using LC-MS/MS with an Agilent 1200 series nanoflow system (Agilent Technologies) connected to a LTQ Orbitrap mass-spectrometer (Thermo Electron) equipped with a nanoelectrospray ion source (Proxeon, Odense). The data were analyzed using the Mascot search engine and the NCBI MOUSE database. For the approximate quantitation, the exponentially modified Protein Abundance Index (emPAI) was used.
Immunofluorescence
Immunofluorescence staining was performed on drying-down preparations, stage-specific squash preparations (Kotaja et al. 2004), or paraffin-embedded testis sections. For drying-down preparations, samples were spread on slides predipped in 1% paraformaldehyde, 0.15% Triton X-100, and incubated overnight in a humid chamber. Then the slides were dried. Paraffin-embedded testes (fixed in 4% PFA at RT for O/N) were cut and mounted onto slides. After rehydration, antigen retrieval was performed in boiling 10-mM sodium citrate buffer (pH 6.0). Before the incubations with antibodies, all of the prepared slides were post-fixed in 4% paraformaldehyde for 5 min, permeabilized with 0.2% Triton X-100 for 2 min, and blocked in 10% BSA for 1 h. The primary antibodies against MVH (rabbit, in-house and Abcam, ab13840), RBM8A (rabbit, Proteintech, 14958-1-AP), DDX25 (goat, SantaCruz, sc-51271), EIF4A3 (rabbit, Proteintech, 17504-1-AP), UPF1 (rabbit, Cell Signaling Technologies), D15G6, phosphorylated UPF1 (Ser1127) (rabbit, Millipore, 07-1016), SMG1 (rabbit, Abcam, ab30916), and SMG6 (rabbit, Abcam, ab87539) were used in dilution 1:100–1:500. AlexaFluor 488/594 conjugated secondary antibodies (Life Technologies) were used in dilution 1:500. Sections were mounted in a medium with DAPI (Santa Cruz). Photomicrographs were taken with Olympus DP72 digital color camera mounted onto a Leica DMRB microscope (Leica Microsystems) using PL FLUOTAR 40×/0.70 and N PLAN 40×/0.65 PH 2 objectives and cellSens Entry 1.5 (Olympus) digital imaging software. All images were processed using Photoshop (Adobe).
RT-qPCR analysis
RNA was extracted from whole-mouse tissues and from the enriched populations of spermatocytes and round spermatids isolated by centrifugal elutriation (Barchi et al. 2009) using TRIsure (Bioline). DNase treatment was performed using DNase I, Amplification Grade (Invitrogen). cDNA synthesis and qPCR quantification were performed using the DyNAmo cDNA Synthesis Kit (Finnzymes) and the DyNAmo Flash SYBR Green qPCR Kit (Finnzymes), respectively. Expression of RPL13A and YWHAZ was used to normalize the expression of the noncoding RNA targets. The assay was performed in three independent technical and biological replicates. Primer sequences are provided in the Supplemental Experimental procedures.
DATA DEPOSITION
Sequencing data are available for download from the Gene Expression Omnibus (GEO) under accession number GSE54288.
SUPPLEMENTAL MATERIAL
Supplemental material is available for the article.
ACKNOWLEDGMENTS
This work was supported by the Academy of Finland; the Emil Aaltonen Foundation; the Sigrid Jusélius Foundation; the Novo Nordisk Foundation (to N.K.); the Turku Doctoral Programme of Biomedical Sciences (to M.D.R.); the National Institutes of Health (5R01GM062534 to G.J.H.); a kind gift from Kathryn W. Davis (to G.J.H.). G.J.H. is an investigator at the Howard Hughes Medical Institute. K.A.W. is a George A. and Marjorie H. Anderson Fellow at the Watson School of Biological Sciences. We thank the Tartu University Proteomics Unit for the MS analysis, the Turku Centre for Biotechnology, Sanja Vanhatalo, and the whole staff of the Department of Physiology for their contributions and technical support. We thank Ralph Burgess for sorting the round spermatids and Michelle Rooks, Dick McCombie, Elena Ghiban, Danea Rebbolini, and Laura Cardone for help with the Illumina sequencing. We also thank Assaf Gordon for help with the data analysis.
REFERENCES
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. 2006. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207 [DOI] [PubMed] [Google Scholar]
- Aravin AA, van der Heijden GW, Castaneda J, Vagin VV, Hannon GJ, Bortvin A 2009. Cytoplasmic compartmentalization of the fetal piRNA pathway in mice. PLoS Genet 5: e1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour CD, Castle JC, Chen R, Babak T, Loerch P, Jackson S, Shah JK, Dey J, Rohl CA, Johnson JM, et al. 2009. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods 6: 647–649 [DOI] [PubMed] [Google Scholar]
- Barchi M, Geremia R, Magliozzi R, Bianchi E 2009. Isolation and analyses of enriched populations of male mouse germ cells by sedimentation velocity: The centrifugal elutriation. Methods Mol Biol 558: 299–321 [DOI] [PubMed] [Google Scholar]
- Bono F, Gehring NH 2011. Assembly, disassembly and recycling: The dynamics of exon junction complexes. RNA Biol 8: 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Parker R 2009. Eukaryotic stress granules: The ins and outs of translation. Mol Cell 36: 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla-Llorente V, Spraggon L, Okamura M, Naseeruddin S, Adamow M, Qamar S, Liu J 2012. Mammalian GW220/TNGW1 is essential for the formation of GW/P bodies containing miRISC. J Cell Biol 198: 529–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmel F, Primig M 2008. The Annotation, Mapping, Expression and Network (AMEN) suite of tools for molecular systems biology. BMC Bioinformatics 9: 86 10.1186/1471-2105-9-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, Chung SS, Demougin P, Gattiker A, Moore J, Patard JJ, Wolgemuth DJ, Jegou B, et al. 2007. The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci 104: 8346–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuma S, Hosokawa M, Tanaka T, Nakatsuji N 2009. Ultrastructural characterization of spermatogenesis and its evolutionary conservation in the germline: Germinal granules in mammals. Mol Cell Endocrinol 306: 17–23 [DOI] [PubMed] [Google Scholar]
- Czech B, Preall JB, McGinn J, Hannon GJ 2013. A transcriptome-wide RNAi screen in the Drosophila ovary reveals factors of the germline piRNA pathway. Mol Cell 50: 749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E 2012. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet 13: 153–162 [DOI] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55 [DOI] [PubMed] [Google Scholar]
- Fawcett DW, Eddy EM, Phillips DM 1970. Observations on the fine structure and relationships of the chromatoid body in mammalian spermatogenesis. Biol Reprod 2: 129–153 [DOI] [PubMed] [Google Scholar]
- Frost RJ, Hamra FK, Richardson JA, Qi X, Bassel-Duby R, Olson EN 2010. MOV10L1 is necessary for protection of spermatocytes against retrotransposons by Piwi-interacting RNAs. Proc Natl Acad Sci 107: 11847–11852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA 2006. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202 [DOI] [PubMed] [Google Scholar]
- Gorgoni B, Gray NK 2004. The roles of cytoplasmic poly(A)-binding proteins in regulating gene expression: A developmental perspective. Brief Funct Genomic Proteomic 3: 125–141 [DOI] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R 2010. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem J 430: 379–392 [DOI] [PubMed] [Google Scholar]
- Handler D, Meixner K, Pizka M, Lauss K, Schmied C, Gruber FS, Brennecke J 2013. The genetic makeup of the Drosophila piRNA pathway. Mol Cell 50: 762–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA 2011. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell 20: 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsaro JJ, Haase AD, Knott SR, Joshua-Tor L, Hannon GJ 2012. The structural biochemistry of Zucchini implicates it as a nuclease in piRNA biogenesis. Nature 491: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD 2010. Minireview: Global regulation and dynamics of ribonucleic acid. Endocrinology 151: 1391–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A 2012. NMD: A multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaja N, Sassone-Corsi P 2007. The chromatoid body: A germ-cell-specific RNA-processing centre. Nat Rev Mol Cell Biol 8: 85–90 [DOI] [PubMed] [Google Scholar]
- Kotaja N, Kimmins S, Brancorsini S, Hentsch D, Vonesch JL, Davidson I, Parvinen M, Sassone-Corsi P 2004. Preparation, isolation and characterization of stage-specific spermatogenic cells for cellular and molecular analysis. Nat Methods 1: 249–254 [DOI] [PubMed] [Google Scholar]
- Kotaja N, Bhattacharyya SN, Jaskiewicz L, Kimmins S, Parvinen M, Filipowicz W, Sassone-Corsi P 2006. The chromatoid body of male germ cells: Similarity with processing bodies and presence of Dicer and microRNA pathway components. Proc Natl Acad Sci 103: 2647–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni M, Ozgur S, Stoecklin G 2010. On track with P-bodies. Biochem Soc Trans 38: 242–251 [DOI] [PubMed] [Google Scholar]
- Laiho A, Kotaja N, Gyenesei A, Sironen A 2013. Transcriptome profiling of the murine testis during the first wave of spermatogenesis. PLoS One 8: e61558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, et al. 2013. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell 50: 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Buchold GM, Greenbaum MP, Roy A, Burns KH, Zhu H, Han DY, Harris RA, Coarfa C, Gunaratne PH, et al. 2009. GASZ is essential for male meiosis and suppression of retrotransposon expression in the male germline. PLoS Genet 5: e1000635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone C, Brennecke J, Czech B, Aravin A, Hannon GJ 2012. Preparation of small RNA libraries for high-throughput sequencing. Cold Spring Harb Protoc 2012: 1067–1077 [DOI] [PubMed] [Google Scholar]
- Meikar O, Da Ros M, Liljenback H, Toppari J, Kotaja N 2010. Accumulation of piRNAs in the chromatoid bodies purified by a novel isolation protocol. Exp Cell Res 316: 1567–1575 [DOI] [PubMed] [Google Scholar]
- Meikar O, Da Ros M, Korhonen H, Kotaja N 2011. Chromatoid body and small RNAs in male germ cells. Reproduction 142: 195–209 [DOI] [PubMed] [Google Scholar]
- Meikar O, Da Ros M, Kotaja N 2012. Epigenetic regulation of male germ cell differentiation. Subcell Biochem 61: 119–138 [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073–1078 [DOI] [PubMed] [Google Scholar]
- Messina V, Meikar O, Paronetto MP, Calabretta S, Geremia R, Kotaja N, Sette C 2012. The RNA binding protein SAM68 transiently localizes in the chromatoid body of male germ cells and influences expression of select microRNAs. PLoS One 7: e39729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metze S, Herzog VA, Ruepp MD, Muhlemann O 2013. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA 19: 1432–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ishizu H, Saito K, Fukuhara S, Kamatani MK, Bonnefond L, Matsumoto N, Nishizawa T, Nakanaga K, Aoki J, et al. 2012. Structure and function of Zucchini endoribonuclease in piRNA biogenesis. Nature 491: 284–287 [DOI] [PubMed] [Google Scholar]
- Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S 2012. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40: 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvinen M 2005. The chromatoid body in spermatogenesis. Int J Androl 28: 189–201 [DOI] [PubMed] [Google Scholar]
- Parvinen M, Salo J, Toivonen M, Nevalainen O, Soini E, Pelliniemi LJ 1997. Computer analysis of living cells: Movements of the chromatoid body in early spermatids compared with its ultrastructure in snap-frozen preparations. Histochem Cell Biol 108: 77–81 [DOI] [PubMed] [Google Scholar]
- Pek JW, Anand A, Kai T 2012. Tudor domain proteins in development. Development 139: 2255–2266 [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Letunic I, Raes J, Bork P, Izaurralde E 2005. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11: 1530–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Ishizu H, Komai M, Kotani H, Kawamura Y, Nishida KM, Siomi H, Siomi MC 2010. Roles for the Yb body components Armitage and Yb in primary piRNA biogenesis in Drosophila. Genes Dev 24: 2493–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Tsai-Morris CH, Dufau ML 2010. Relevance of gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) in the structural integrity of the chromatoid body during spermatogenesis. Biochim Biophys Acta 1803: 534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiromoto Y, Kuramochi-Miyagawa S, Daiba A, Chuma S, Katanaya A, Katsumata A, Nishimura K, Ohtaka M, Nakanishi M, Nakamura T, et al. 2013. GPAT2, a mitochondrial outer membrane protein, in piRNA biogenesis in germline stem cells. RNA 19: 803–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA 2011. PIWI-interacting small RNAs: The vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258 [DOI] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the Mammalian testis. Cell Rep 3: 2179–2190 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Hosokawa M, Vagin VV, Reuter M, Hayashi E, Mochizuki AL, Kitamura K, Yamanaka H, Kondoh G, Okawa K, et al. 2011. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc Natl Acad Sci 108: 10579–10584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV, Yu Y, Jankowska A, Luo Y, Wasik KA, Malone CD, Harrison E, Rosebrock A, Wakimoto BT, Fagegaltier D, et al. 2013. Minotaur is critical for primary piRNA biogenesis. RNA 19: 1064–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileva A, Tiedau D, Firooznia A, Muller-Reichert T, Jessberger R 2009. Tdrd6 is required for spermiogenesis, chromatoid body architecture, and regulation of miRNA expression. Curr Biol 19: 630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Chuma S, Yamamoto Y, Kuramochi-Miyagawa S, Totoki Y, Toyoda A, Hoki Y, Fujiyama A, Shibata T, Sado T, et al. 2011. MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev Cell 20: 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S 2001. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev 15: 2215–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Wang PJ 2012. Blockade of pachytene piRNA biogenesis reveals a novel requirement for maintaining post-meiotic germline genome integrity. PLoS Genet 8: e1003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Xiol J, Reuter M, Eckardt S, Leu NA, McLaughlin KJ, Stark A, Sachidanandam R, Pillai RS, Wang PJ 2010. Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc Natl Acad Sci 107: 11841–11846 [DOI] [PMC free article] [PubMed] [Google Scholar]