

The endoribonuclease RNase E in a range of bacteria has a conserved N-terminal catalytic domain and a less well-conserved C-terminal tail. While the role of the C-terminal tail in the formation of the degradosome, including the interaction with the polynucleotide phosphorylase PNPase, has been studied extensively in Escherichia coli, less is known about RNase E in other bacteria. In this study, Zhang and colleagues characterize RNase E in Anabaena and Synechocystis and find that, despite having C-termini that are very different from E. coli and contain an unusual RRRRRRSSA motif, the cyanobacterial enzymes also bind PNPase.
Keywords: RNase E, PNPase recognition site, RNA degradosome, cyanobacteria
Abstract
RNase E, a central component involved in bacterial RNA metabolism, usually has a highly conserved N-terminal catalytic domain but an extremely divergent C-terminal domain. While the C-terminal domain of RNase E in Escherichia coli recruits other components to form an RNA degradation complex, it is unknown if a similar function can be found for RNase E in other organisms due to the divergent feature of this domain. Here, we provide evidence showing that RNase E forms a complex with another essential ribonuclease—the polynucleotide phosphorylase (PNPase)—in cyanobacteria, a group of ecologically important and phylogenetically ancient organisms. Sequence alignment for all cyanobacterial RNase E proteins revealed several conserved and variable subregions in their noncatalytic domains. One such subregion, an extremely conserved nonapeptide (RRRRRRSSA) located near the very end of RNase E, serves as the PNPase recognition site in both the filamentous cyanobacterium Anabaena PCC7120 and the unicellular cyanobacterium Synechocystis PCC6803. These results indicate that RNase E and PNPase form a ribonuclease complex via a common mechanism in cyanobacteria. The PNPase-recognition motif in cyanobacterial RNase E is distinct from those previously identified in Proteobacteria, implying a mechanism of coevolution for PNPase and RNase E in different organisms.
INTRODUCTION
In eukaryotes such as mammals and plants, many mRNAs species are relatively stable, with half-lives of more than several hours (Ross 1995; Narsai et al. 2007). In contrast, the average half-life of bacterial mRNA lasts only about a few minutes (Hambraeus et al. 2003; Selinger et al. 2003; Redon et al. 2005; Steglich et al. 2010), reflecting rapid responses of bacterial cells to constantly changing environments. The degradation of bacterial mRNA is a highly dynamic and tightly regulated process, involving many RNA processing enzymes and assisting proteins (Silva et al. 2011). Using the model organism Escherichia coli, bacterial RNA degradation mechanisms have been extensively studied. E. coli cells possess more than twenty RNA processing/degradation-related proteins required for the metabolism of various intracellular RNA species (Arraiano et al. 2010).
Among all known E. coli ribonucleases, RNase E plays a central role in mRNA turnover. This protein is a 1061-aa endoribonuclease that can be divided into two structurally distinct parts: the N-terminal catalytic domain and the C-terminal noncatalytic domain. RNase E cleaves single-strand RNA substrates through the activity of the catalytic domain and recruits interacting proteins through the noncatalytic domain, forming an RNA degradation complex known as the RNA degradosome (Carpousis 2007). In addition to RNase E, the major components of the E. coli RNA degradosome also include the polynucleotide phosphorylase (PNPase), the DEAD box RNA helicase RhlB, and the glycolytic enzyme enolase (Carpousis et al. 1994; Vanzo et al. 1998). RNA degradation mediated by the RNA degradosome is a highly cooperative and efficient process: RNase E cuts single-stranded RNA substrates preferably at AU-rich sites, PNPase further converts the generated fragments into diphosphate mononucleotides through its 3′-5′ phosphorolytic activity, and the RNA helicase RhlB unwinds structured RNAs to facilitate their turnover by RNase E and PNPase (Liou et al. 2002). Enolase in the RNA degradosome does not seem to act on RNA directly. However, it may modulate the activity and the substrate specificity of the RNA degradosome (Morita et al. 2004).
RNase E homologs have been identified in a large variety of bacterial species, as well as in chloroplasts (Schein et al. 2008; Kaberdin et al. 2011). One striking feature of the RNase E family is that the sequence of their catalytic domains is highly conserved, in contrast to that of the noncatalytic domain which is extremely divergent, even among those from closely related species. Therefore, although the recognition motifs for RNA degradosome components in the noncatalytic domain of E. coli RNase E (hereafter EcRne) have been well defined, it is still very difficult to predict if similar RNA degradation complexes are also present in other species by sequence comparison. Due to such difficulty, RNase E-based RNA degradation complexes have been experimentally identified only in a few bacteria, which mostly belong to Proteobacteria (Kaberdin et al. 2011). For example, the RNA degradosome in Caulobacter crescentus was shown to contain RNase E, PNPase, a DEAD-box RNA helicase, and the Krebs cycle enzyme aconitase (Hardwick et al. 2011), while the RNA degradosome in the psychrotrophic bacterium Pseudomonas syringae Lz4W was found to be composed of RNase E, the exoribonuclease RNase R, and the DEAD-box helicase RhlE (Purusharth et al. 2005).
Although the E. coli-type RNA degradosome seems to be commonly present in a certain group of bacteria (i.e., Proteobacteria), many other bacteria, such as the Gram-positive bacterium Bacillus subtilis, do not harbor genes encoding RNase E homologs. Apparently, in those species lacking RNase E, RNA degradation is conducted somewhat differently. Recent studies revealed that B. subtilis possesses RNase J1/J2 and RNase Y, two novel endoribonucleases that are absent in E. coli (Even et al. 2005; Shahbabian et al. 2009). These proteins, in conjunction with the RNA helicase CshA, enolase, and phosphofructokinase, were suggested to form an RNA degradation complex that is compositionally different from but functionally equivalent to the E. coli RNA degradosome (Lehnik-Habrink et al. 2012).
Our current understanding of bacterial RNA degradation machineries is derived from the analysis of a limited number of species in Proteobacteria, Actinobacteria, and Fimicutes (Kaberdin et al. 2011). In the majority of other bacteria, how ribonucleases cooperate in RNA degradation remains largely unknown. Cyanobacteria represent a large and unique group of oxygen-evolving photosynthetic prokaryotes that are physiologically and phylogenetically distinct from other bacterial phyla. Unlike B. subtilis, cyanobacteria do not harbor RNase Y homologs; they, instead, possess proteins homologous to E. coli RNase E. The first cyanobacterial RNase E protein was indentified and characterized 15 yr ago in Synechocystis PCC6803 (Kaberdin et al. 1998), one unicellular and nondiazotrophic cyanobacterial strain extensively used as a model in photosynthesis and bioenergy research. Like E. coli RNase E, Synechocystis RNase E (SynRne) also consists of an N-terminal catalytic domain and a C-terminal noncatalytic domain (Kaberdin et al. 1998). SynRne was able to confer viability to an E. coli rne mutant (Horie et al. 2007). Furthermore, SynRne also preferentially cleaves RNA substrates within AU-rich regions. This property was shown to contribute to dark-induced mRNA instability in vivo (Horie et al. 2007). The noncatalytic domain of SynRne displays no sequence similarity to its counterpart region in E. coli RNase E and does not associate with any components of the E. coli RNA degradosome (Kaberdin et al. 1998). To date, its role remains unknown.
In this study, we explored the role of the noncatalytic region cyanobacterial RNase E in the filamentous and heterocyst-forming cyanobacterium Anabaena PCC7120, as well as in the unicellular cyanobacterium Synechocystis PCC6803. We demonstrate a mechanism of interaction between PNPase and RNase E that is conserved in cyanobacteria but distinct from those so far reported in other bacteria.
RESULTS
RNase E homologs are universally present in cyanobacteria
Cyanobacteria is a bacterial phylum that comprises numerous species with great morphological, ecological, and genetic diversity. Although RNase E homologs were found in some cyanobacterial strains (Shahbabian et al. 2009), it is still unclear whether the enzyme is universally present in this phylum. Currently, the GenBank database has the entries of the genomic data for as many as 60 cyanobacterial strains, which represent a diverse group of cyanobacteria. We performed a BLAST analysis using the query of Synechocystis PCC6803 RNase E, the first cyanobacterial RNase E protein experimentally investigated (Kaberdin et al. 1998), and determined that a single copy of an RNase E homolog exists in each available cyanobacterial genome (data not shown). Like EcRne, all cyanobacterial RNase E homologs consist of an N-terminal catalytic domain and a C-terminal noncatalytic domain. Our sequence analysis also revealed that the catalytic domains of cyanobacterial RNase E shared high sequence similarities to those of other bacteria, whereas the noncatalytic domains show no detectable conservation, differing greatly from those of other bacteria. The domain structures of the RNase E proteins from the filamentous cyanobacterium Anabaena PCC7120 and the unicellular cyanobacterium Synechocystis PCC6803 were compared to those of E. coli and C. crescentus. As shown in Figure 1A, the catalytic domains of these proteins have similar sizes. In contrast, the noncatalytic domains of the two cyanobacterial RNase Es are much shorter than and show no similarity to those of E. coli and C. crescentus.
FIGURE 1.

(A) The domain structures of the RNase E proteins from Caulobacter crescentus, Escherichia coli, Anabaena PCC7120, and Synechocystis PCC6803. The catalytic domain of each RNase E is composed of a S1 domain and an RNase_E_G domain. The C-terminal noncatalytic domains of the two cyanobacterial strains show no similarity to those of other bacteria including the two proteobacterial strains listed here. (B) Alignment of RNase E proteins from 17 representative cyanobacterial strains. The alignment was performed using MEGA5 (Tamura et al. 2011) and refined manually. Four short conserved subregions (C1–C4) and three variable subregions (V1–V3) were revealed in the noncatalytic domains. The regions not shown are depicted with triple dots (…). Gaps in the alignment are depicted with short horizontal lines (-). The residues with high conservation are in bold. (C) Schematic structure of the Anabaena RNase E based on the alignment shown above. The variable and conserved regions of AnaRne revealed by the sequence alignment are depicted as lines and filled boxes, respectively.
AnaRne cleaves 9S RNA in a similar way as EcRne
Almost all known RNase E proteins exhibit catalytic activities similar to those of EcRne (Kaberdin et al. 1998; Lee and Cohen 2003; Kaberdin and Bizebard 2005; Zeller et al. 2007; Schein et al. 2008; Hardwick et al. 2011; Lee et al. 2011). In this study, we also checked whether AnaRne possesses a catalytic activity associated with RNase E proteins. The recombinant proteins consisting of Anabaena RNase E (AnaRne, 1–687 aa) or only its catalytic domain (AnaRneN, 1–412 aa), as well the catalytic domain of E. coli RNase E (EcRneN, 1–420 aa), were overexpressed as N-terminally His-tagged fusions in E. coli cells and purified using Ni-NTA columns. The activities of the purified enzymes on E. coli 9S RNA were tested at 30°C and 37°C, respectively. As shown in Figure 2, the degradation patterns of 9S RNA for the three proteins were similar, implying that AnaRne and AnaRneN cleave the RNA substrate like EcRne does. The result for SynRne is the same as reported (Kaberdin et al. 1998). We noticed that AnaRne, the full-length protein, appeared to be more active than its catalytic domain AnaRneN alone at either 30°C or 37°C, a result similar to that reported for E. coli RNase E (Cormack et al. 1993). The enzymatic assay also showed that EcRneN acted on the substrate less efficiently than AnaRneN at 30°C, but more efficiently at 37°C, probably reflecting the difference in the optimum growth temperatures for E. coli and Anabaena PCC7120.
FIGURE 2.

Comparison of the catalytic properties of EcRneN (the catalytic domain of E. coli RNase E), AnaRne (the full length of Anabaena RNase E), and AnaRneN (the catalytic domain of Anabaena RNase E) on the 9S RNA as substrate. (CK-) 9S RNA incubated for 60 min in the reaction buffer alone. In each of the other reactions, an equal amount of substrate and enzyme were used. The electrophoresis was performed in a 6% PAGE gel containing 7 M urea, and the gel was stained with ethidium bromide. The bands of 9S RNA and its end product p5S RNA are indicated by arrows.
Interaction of AnaRneC and AnaPnp in vivo
The noncatalytic domains in the RNase E proteins from E. coli, C. crescentus, and several other organisms have been shown to recruit other proteins to form an RNA degradosome (Vanzo et al. 1998; Aït-Bara and Carpousis 2010; Erce et al. 2010; Hardwick et al. 2011). In order to determine if similar RNA degradation complexes exist in cyanobacteria, we carried out a copurification experiment to see if any proteins associate with the noncatalytic domain of Anabaena RNase E (AnaRneC). To facilitate the experiment, we constructed AnaRneC and GFP fusion proteins with either an N-terminus or a C-terminus 8X His-tag (named hGFP-AnaRneC and AnaRneC-GFPh, respectively) (Fig. 3) and expressed them in Anabaena cells under a nickel-inducible promoter. As a negative control, a His-tagged GFP (hGFP) was also expressed. After induction in Ni2+-containing medium, the Anabaena cells expressing hGFP-AnaRneC, AnaRneC-GFPh, or hGFP were respectively lysed, and the total soluble proteins were applied to the Ni-NTA purification columns. The proteins bound to the columns were eluted and analyzed with SDS-PAGE (Fig. 3). As expected, there was only one band corresponding to the size of hGFP in the control sample. In contrast, two major bands appeared in the protein sample isolated from the cells expressing either hGFP-AnaRneC or AnaRneC-GFPh. The lower band was the size corresponding to the molecule weight of the fusion protein of hGFP-AnaRneC or AnaRneC-GFPh (61.5 kDa). We noticed that the upper band matched the size of the Anabaena PNPase (AnaPnp, 77.8 kDa). To test if the upper band was, indeed, AnaPnp, a duplicate SDS-PAGE gel was subjected to immunoblotting analysis with specific polyclonal antibodies against AnaPnp. As shown in Figure 3B, a clear immunoblotting signal appeared, indicating that the protein copurified with hGFP-AnaRneC or AnaRneC-GFPh was AnaPnp.
FIGURE 3.

Copurification of AnaPnp with the His-tagged GFP-AnaRneC fusions expressed in Anabaena cells using Ni-NTA beads. (A) Schematic presentation of the His-tagged GFP and GFP-AnaRneC fusions expressed in Anabaena cells. (B) Total proteins from Anabaena cells expressing the His-tagged recombinant proteins were applied to the Ni-NTA columns for purification. Proteins eluted were analyzed by SDS-PAGE gels, followed by silver staining or immunodetection using polyclonal antibodies against AnaPnp. Lanes 1–3, samples from Anabaena cells expressing hGFP, hGFP-EC, and EC-GFPh, respectively.
AnaRneC directly interacts with AnaPnp
Because E. coli RNase E is an RNA-binding protein, some proteins thought to be not directly associated with RNase E could be coisolated with RNase E in an RNA-mediated manner (Miczak et al. 1996). Thus, it was necessary to check a direct interaction between AnaRneC and AnaPnp. We purified the recombinant proteins of AnaRneC and AnaPnp from E. coli cells and investigated their interaction by a Far-Western dot-blot assay. Micrococcal nuclease was included in the assay to exclude the possible interference of RNA. As shown in Figure 4A, a strong interaction signal was revealed with purified AnaRneC and AnaPnp, and the interaction was not affected by treatment of Micrococcal nuclease. We additionally detected the complex of AnaRneC and AnaPnp by native gel electrophoresis after incubating the purified proteins in presence of micrococcal nuclease (Fig. 4B). These data indicate direct association between AnaRneC and AnaPnp.
FIGURE 4.

(A) Far-Western dot-blot assay investigating the interaction between the recombinant AnaRneC and AnaPnp purified from E. coli cells with or without the treatment with micrococcal nuclease (MNase). AnaPnp and BSA were dotted onto the membranes as a positive control and a negative control, respectively. (B) Determining the complex of AnaRneC and AnaPnp by gel electrophoresis. AnaRneC and AnaPnp were incubated under native conditions for 30 min, then the mixture was separated on the native-PAGE gel (the middle lane in the left panel). The band of protein complex (indicated by an arrow) in the native gel was excised and subsequently subjected to SDS-PAGE electrophoresis (the middle lane in the right panel). Purified AnaRneC and AnaPnp were included as size markers in both the native-PAGE gel and the SDS-PAGE gel. Note that AnaRneC did not migrate as a sharp band in the native-PAGE gel. (C) Bacterial two-hybrid assay investigating the interaction between AnaRneC and AnaPnp. The E. coli XL1-Blue MRF′ Kan cells cotransformed by the indicated two-hybrid plasmid pairs were spotted on the selective and the nonselective plates, respectively. Cells cotransformed with pBT-LGF2 and pTRG-Gal11P (supplied in the kit) were used as the positive control (CK+), and cells cotransformed with the empty vectors of pBT and pTRG were used as the negative control. Note that, due to unclear reasons, cells containing pTRG-AnaPnp/pBT or pTRG-AnaPnp/pBT showed weak growth.
We further investigated the interaction between AnaPnp and AnaRneC with the BacterioMatch II Two-Hybrid System (Agilent). The coding sequences of AnaPnp and AnaRneC were cloned into the bait vector pBT and the target vector pTRG, respectively. The resulting plasmids as well as the empty vectors were cotransformed into the host strain E. coli XL1-Blue MRF′ Kan in various combinations (Fig. 4C). The transformed cells were then grown on the selective and the nonselective plates, respectively. Cell growth on the selective plates was indicative of the interaction between the bait and the target proteins. As shown in Figure 4C, AnaPnp and AnaRneC exhibited strong and reciprocal interactions, in agreement with the copurification results. Moreover, both AnaRneC and AnaPnp exhibited strong self-interaction, suggesting that they form dimers or oligomers in vivo. Note that cells harboring pTRG-AnaPnp/pBT or pTRG-AnaRneC/pBT also showed some extent of growth on selective plates, which could be the result of nonspecific activation of the reporter gene.
Conserved and variable subregions exist in the noncatalytic domains of cyanobacterial RNase E
In E. coli, it is well known that PNPase binds to a microdomain at the C-terminus of RNase E (Callaghan et al. 2004). The noncatalytic domain of EcRne has several additional microdomains that contribute to membrane association, substrate binding, and interaction with other components of the RNA degradosome (Callaghan et al. 2004; Khemici et al. 2008). However, these microdomains are hardly detected in the RNase E sequences from the species that do not belong to γ-proteobacteria. Due to the lack of sequence similarity, we could not determine the PNPase binding site and other functional motifs in the noncatalytic domains of Anabaena RNase E. However, based on a careful alignment for the RNase E proteins from all sequenced cyanobacterial strains, we were able to identify four conserved (C1–4) and three variable subregions (V1–3) within their noncatalytic domains (Fig. 1B). The motif C1, consisting of ∼20 residues, is relatively less conserved in the RNase E proteins from two evolutionarily ancient strains (i.e., Thermosynechococcus BP-1 and Gloeobacter PCC 7421) and is missing in unicellular marine strains. C2, a highly positively charged region of ∼9 residues, is adjacent to C1 and not found in RNase E proteins from some Prochlorococcus strains. Among the four conserved subregions, only C3 and C4 are present in all cyanobacterial RNase Es, with the former being ∼50 aa in length and the latter a segment of nine residues with the consensus sequence RRRRRRSSA. The occurrence of these conserved motifs in the RNase E proteins from diverse cyanobacterial species suggests that they may have important functions. The three variable regions display great diversity in different species and may thus have more species-specific roles.
C4 region in AnaRneC is responsible for the interaction with AnaPnp
As revealed by sequence alignment, the noncatalytic domain of AnaRne contains four conserved and three variable regions (Fig. 1). To define the specific region(s) responsible for AnaPnp binding, a series of deletion mutants of AneRneC were constructed, and their interaction with AnaPnp was investigated by using the Far-Western dot-blot assay. In this assay, nitrocellulose membranes in duplicate were first dotted with AneRneC variants along with AnaPnp and BSA (the positive control and negative control, respectively). Subsequently, one membrane was incubated with AnaPnp and the other was not. Finally, both membranes were immunodetected with the antibodies against AnaPnp. As expected, except for the positive control, no immunoblotting signal appeared on the membrane without AnaPnp incubation (Fig. 5). On the membrane with AnaPnp incubation, the interaction levels between AneRneC variants and AnaPnp were estimated by the intensity of the immunoblotting signals. The result obtained showed that the AneRneC variant lacking C4 lost completely the ability to interact with AnaPnp. In contrast, deletion of other subregions in AneRneC did not disrupt the interaction between AnaRneC and AnaPnp, although the absence of C3 and V3 appeared to weaken the interaction slightly. These data indicate that C4 is the key element for AnaPnp recognition by AnaRneC.
FIGURE 5.

Far-Western dot-blot assay studying the role of each subregion of AnaRneC in AnaPnp–AnaRneC interaction. AnaRneC and its subregion deletion variants were dotted onto duplicate nitrocellulose membranes. Subsequently, one of the membranes was incubated with 2 µM AnaPnp and the other one was not. Both membranes were finally subjected to immunodetection with the antibody against AnaPnp (see Materials and Methods for details). The deleted regions in AnaRneC variants are illustrated with boxes with gray stripes. BSA and AnaPnp were used as the negative control and the positive control, respectively.
We further tested whether the motif C4, when fused to other proteins, could confer on the fusion proteins the ability to interact with AnaPnp. The green fluorescent protein GFP was chosen for the test as GFP alone was found unable to interact with AnaPnp in pilot experiments (data not shown). We fused the GFP protein at its C-terminus to four versions of C4, named C4a (STANRRRRRRSSASDSD, C4 with four flanking residues at both ends), C4b (RRRRRRSSA, the C4 subregion defined in Fig. 1B), C4c (RRRRRR, C4 without the last three neutral residues: SSA), and C4d (RRRRR, C4 without the last three neutral residues plus one arginine residue), resulting in the fusion proteins of GFP-C4a, GFP-C4b, GFP-C4c, and GFP-C4d, respectively (Fig. 6). The interactions between these proteins and AnaPnp were then investigated by a Far-Western blot assay. As shown in Figure 6, GFP-C4a and GFP-C4b bound to AnaPnp equally well. In contrast, the interaction of AnaPnp to GFP-C4c or GFP-C4d was greatly reduced, implying a critical role of the last three neutral residues in C4. These results indicate that the C4 peptide of RRRRRRSSA within a given protein is sufficient to serve as the AnaPnp recognition site.
FIGURE 6.

Investigation of the interaction between AnaPnp and GFP-C4 variants. (A) Schematic illustration of GFP-C4 variants. (B) Far-Western blot assay demonstrating the interaction between AnaPnp and GFP-C4 variants. All proteins were separated on a 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membrane was then incubated with AnaPnp, followed by immunodetection with the antibody against AnaPnp (see Materials and Methods for details). AnaPnp and AnaRneC were included as the positive controls; EC-DC4 and GFP were included as the negative controls. The amount of protein in each lane was 10 µg, except that 100 ng was loaded for AnaPnp.
The synthetic nonapeptide of C4 was also tested to see if it could act as a competitor to interfere with the formation of the AnaRne–AnaPnp complex using the Far-Western dot-blot assay. First, AnaRneC, AnaPnp, and BSA were dotted onto multiple nitrocellulose membranes; then, each membrane was incubated with a fixed concentration of AnaPnp (2 µM) and a varying amount of the synthetic peptide of C4 (0–20 µM). After the incubation, the membranes were immunodetected with the antibody against AnaPnp. The data in Figure 7A showed that the interaction between AnaRneC and AnaPnp was weakened by the addition of the peptide C4 in a concentration-dependent manner. Nearly 50% of the interaction signal was lost when the C4 peptide concentration was at 4 µM, while almost no interaction signal could be observed when the concentration was increased to 20 µM. These data demonstrated that the synthetic C4 nonapeptide could efficiently compete with AnaRneC for binding to AnaPnp.
FIGURE 7.

(A) Competition assay of the synthetic peptide C4 on the interaction between AnaRneC and AnaPnp by Far-Western dot-blot. AnaRneC, AnaPnp (the positive control), and BSA (the negative control) were dotted onto nitrocellulose membranes. After incubation with the indicated amount of AnaPnp and C4, the membranes were immunodetected with the antibody against AnaPnp (see Materials and Methods for details). (B) Effect of the C4 peptide on the interaction between SynRneC and SynPnp. The experiment was performed as described in A. (C) Interaction between SynPnp and GFP-C4b. The experiment was also performed as described in A. SynPnp and GFP proteins were dotted on the membranes as the positive control and the negative control, respectively.
RNase E–PNPase interaction is conserved in cyanobacteria
In the alignment for cyanobacterial RNase E sequences, C4 displays extremely high conservation among all cyanobacterial species (Fig. 1B). The C4 motif in AnaRne serves as the binding site for AnaPnp, making it tempting to hypothesize that RNase E associates with PNPase by the same mechanism in other cyanobacteria. We tested this hypothesis with the RNase E and PNPase proteins from the cyanobacterium Synechocystis PCC6803, a species that is distantly related to Anabaena PCC7120. The interaction between the recombinant noncatalytic domain of Synechocystis RNase E (SynRneC) and Synechocystis PNPase (SynPnp), as well as the effect of the synthetic nonapeptide of C4 on such interaction, were explored in a Far-Western dot-blot assay. As demonstrated in Figure 7B, a strong interaction signal was observed between SynRneC and SynPnp. Additionally, the interaction signal of the two Synechocystis proteins was gradually weakened when the concentration of the C4 peptide was increased in the incubation solution of SynPnp, showing that the effect of the C4 peptide on the SynRneC–SynPnp interaction is similar to that for AnaRneC and AnaPnp. Moreover, the chimeric protein of GFP-C4 was also able to specifically bind to SynPnp, as revealed by Far-Western dot-blot assay (Fig. 7C). It should be noted that the C4 nanopeptide in AnaRne is slightly different from its cognate motif in SynRne (GRRRRRSSA), with the first arginine residue in the former being substituted by a glycine residue in the latter (Fig. 1B). However, the difference did not seem to affect PNPase recognition. In summary, these data imply that C4-mediated interaction between RNase E and PNPase occurs not only in Anabaena PCC7120 but also in Synechocystis PCC6803.
DISCUSSION
To date, our understanding of ribonucleases and RNA metabolism in cyanobacteria remains very limited. RNase E is one of the most important ribonucleases identified in E. coli and many other bacteria, and its homologs are also present in cyanobacteria. Both E. coli RNase E and cyanobacterial RNase E feature a highly conserved N-terminal catalytic domain and a highly divergent C-terminal noncatalytic domain (Fig. 1B). Their catalytic domains share high similarities in structure, enzymatic activity, and membrane-binding behavior (Kaberdin et al. 1998; Murashko et al. 2012). In contrast, their C-terminal domain displays no obvious sequence homology at all. The noncatalytic domain of E. coli RNase E acts as the scaffold for the assembly of the RNA degradosome. However, it is hard to predict if a degradosome-like RNA degradation complex also exists in cyanobacteria. In this study, we coisolated AnaPnp with the noncatalytic domain of AnaRne from the cells of Anabaena PCC7120, indicating their interaction occurring in vivo. The interaction was further confirmed by Far-Western dot-blotting, Native-PAGE, and bacterial two-hybrid assays. Sequence alignment for all cyanobacterial RNase E proteins revealed four conserved and three variable subregions within AnaRneC. Using region-deletion variants of AnaRneC, one highly conserved subregion, C4, was identified as the AnaPnp recognition site. The RNase E and PNPase from the cyanobacterium Synechocystis PCC6803 were also found to interact with each other.
Although PNPase was found to associate with RNase E in an increasing number of bacteria, its recognition site in RNase E so far has been identified only in two species, E. coli and C. crescentus (Vanzo et al. 1998; Lee and Cohen 2003; Yang et al. 2008; Aït-Bara and Carpousis 2010; Erce et al. 2010; Hardwick et al. 2011). In E. coli, RNase E interacts with PNPase via a fragment of 23 residues (GAAGGHTATHHASAAPARPQPVE, residues 1039–1061) (Callaghan et al. 2004; Nurmohamed et al. 2009), while in C. crescentus, the site is demonstrated to be the GWW motif (EKPRRGWWRR, residues 889–898) (Hardwick et al. 2011, 2012). We showed that a highly conserved motif of cyanobacterial RNase E within the noncatalytic domain of AnaRne could bind to AnaPnp. This motif consists of nine residues (RRRRRRSSA), being the shortest PNPase recognition site identified to date. In terms of residue composition, these PNPase recognition sites display great variation. Accordingly, the regions on E. coli and C. crescentus PNPases that determine RNase E recognition are also distinct (Nurmohamed et al. 2009; Hardwick et al. 2012). These differences probably reflect the long coevolution history of PNPase–RNase E interactions in different species. One remarkable feature for the PNPase binding sites identified in E. coli, C. crescentus, and Anabaena is that they all reside at the very C-terminal end of RNase E. Such an arrangement may save more space in RNase E for other components to be assembled into the complex using the noncatalytic domain as a scaffold, because PNPase as a homotrimer is relatively large in size (Shi et al. 2008; Hardwick et al. 2012).
As shown in Figure 1B, the PNPase binding site in the RNase E of Anabaena PCC7120 and Synechocystis PCC6803 is strongly conserved in all known cyanobacterial RNase E homologs. We also noticed that the PNPases from all known cyanobacteria share a very high sequence similarity (>60% identities in overall amino acids). Combining the high conservation for both PNPase and PNPase binding sites in RNase Es even among evolutionarily distant cyanobacterial species, we postulate that PNPase and RNase E had already functioned as a ribonuclease complex in the common ancestor of modern cyanobacteria and that the molecular mechanism by which they interact with each other has been well maintained during evolution. Chloroplasts in higher plants, originated from ancient cyanobacteria through endosymbiosis, have RNase E and PNPase as well (Baginsky et al. 2001; Schein et al. 2008; Erce et al. 2009). A recent study shows that chloroplasts occurred much later than the common ancestor of modern cyanobacteria in evolution (Falcón et al. 2010), thus chloroplast RNase E and PNPase might also form a complex similar to that in cyanobacteria. However, after examining the chloroplast RNase E sequences from Arabidopsis thaliana or Oryza sativa, we did not detect a motif similar to the PNPase binding site in cyanobacterial RNase E. Moreover, it was reported that PNPase in Arabidopsis functions as a homo-oligomer, not in complex with RNase E (Baginsky et al. 2001). Therefore, it seems that the interdependence between RNase E and PNPase in chloroplasts is not essential and may have been lost during evolution.
Polyadenylation at the 3′-end is the prerequisite for normal metabolism of many RNA species in prokaryotes (Mohanty and Kushner 2011). In E. coli, RNA polyadenylation, which is able to stimulate substrate decay by the degradosome, is carried out primarily by the nondegradosomal enzyme poly(A) polymerase (Blum et al. 1999; Mohanty and Kushner 1999; Khemici and Carpousis 2004). However, in the cyanobacterium Synechocystis PCC6803, no poly(A) polymerase exists, and RNA polyadenylation is carried out by PNPase—the same enzyme for RNA phosphorolysis (Rott et al. 2003). Our finding of cyanobacterial RNase E and PNPase associating with each other suggests that the activities of RNA cleavage, polyadenylation, and phosphorolysis are tightly integrated. Thus, the cyanobacterial PNPase–RNase E complex may function as a very efficient RNA decay machine in vivo. The question of how different activities of this complex are coordinated for cyanobacterial RNA metabolism could be an interesting subject for future studies.
In E. coli and some other organisms, in addition to PNPase, RNase E-based RNA degradation complexes usually contain several other major components, mostly RNA helicase and glycolytic enzymes (Aït-Bara and Carpousis 2010; Erce et al. 2010; Hardwick et al. 2011). The role of glycolytic enzymes in the complex is still unclear, while RNA helicase, which unwinds double-strand RNA, is important for degradation of structured RNAs by RNase E and PNPase (Coburn et al. 1999; Khemici et al. 2005). The PNPase–RNase E complex in cyanobacteria probably also needs the help of RNA helicases in order to efficiently digest substrates with stable structures. However, perhaps due to technical limitations, only PNPase was coisolated with RNase E from Anabaena cells in this study. Note that among the four conserved and three variable subregions in AnaRne, only C4 has been identified as the PNPase binding site. The roles of the other subregions remain unknown. These conserved subregions may serve as recognition sites for other components in the RNA degradation complex. C2, mostly consisting of positively charged arginine residues, could be involved in binding to the negatively charged RNA substrates. V1, V2, and V3 may either be the sites for certain species-specific interactions or just serve as the linkers joining those conserved subregions. The study reported here should pave the way for further elucidation of the RNA metabolism complex in cyanobacteria.
MATERIALS AND METHODS
Bioinformatic analysis
By the time we did the analysis, the data of a total of 60 cyanobacterial genomes were available in the GenBank database. The RNase E homologs encoded by the genomes were identified by BLAST analysis using the query of Synechocystis PCC6803 RNase E. Sequence alignment for cyanobacterial RNase E homologs was performed in the program MEGA5 (Tamura et al. 2011) using the program's default parameters. The alignment for the noncatalytic domains of cyanobacterial RNase Es produced by MEGA was refined manually.
Plasmid construction
The oligonucleotides used in this study are listed in Supplemental Table 1. All the constructed plasmids were verified by sequencing.
TABLE 1.
Recombinant proteins expressed in E. coli in this study
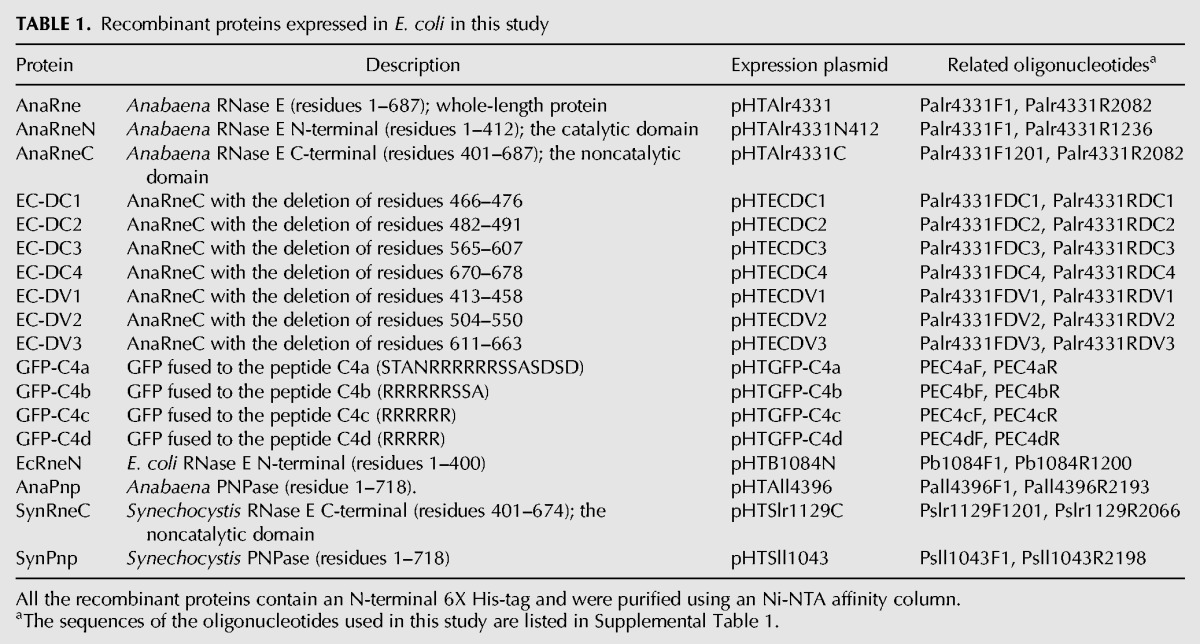
To construct the plasmids for expression of the recombinant proteins (i.e., EcRneN, AnaRne, AnaRneN, AnaRneC, EC-DC4, AnaPnp, SynRneC, and SynPnp), the genomic regions encoding the target proteins were amplified and cloned into the expression vector pET28a (Invitrogen) with NdeI and XhoI (Table 1). The AnaRneC expression plasmid, pHTEC, was used as the template to generate the plasmids for the expression of AnaRneC variants using the QuickChange Site-Directed Mutagenesis Kit (Stratagene).
The plasmids for the expression of the C4 motif and its variants fused to GFP were made as follows. First, the ORF of gfp was amplified using the oligonucleotides of Plgfp_F3 and Plgfp_R717 from the plasmid p8760 (Santini et al. 2001) and cloned into pET28a with NheI and BamHI, resulting in the plasmid pGFP2H. Four pairs of complementary oligonucleotides were annealed to generate the fragments encoding C4a (STANRRRRRRSSASDSD), C4b (RRRRRRSSA), C4c (RRRRRR), and C4d (RRRRR). These fragments, bearing BamHI- and XhoI-compatible ends, were subsequently inserted into pGFP2H with BamHI and XhoI, resulting in the plasmids pGFP-C4a, pGFP-C4b, pGFP-C4c, and pGFP-C4d, respectively (Table 1).
In Synechocystis PCC6803, the promoter of nsrB is specifically responsive to Ni2+ through the regulation of the two-component system NsrSR (López-Maury et al. 2002). We found this system also works in Anabaena PCC7120 (data not shown). In this study, we constructed nickel-inducible plasmids (i.e., pRLNiHG, pRLNiHG-AnaRneC, pRLNiAnaRneC-GH) to express the His-tagged GFP protein and its AnaRneC fusions in Anabaena PCC7120. The plasmid construction procedures are as follows. First, the nsrSR cluster together with the promoter of nsrB from Synechocystis PCC6803 were amplified using the oligonucleotides Psll0798R1541 and Psll0797F124m. Then, the PCR fragment was cloned into the E. coli-Anabaena shuttle vector pRL25N (Zhang et al. 2013) with BamHI and XhoI, generating the plasmid pRLNi. Subsequently, the gfp-encoding region from p8760 was amplified using the oligonucleotides of PgfpF14 and pgfpR754a, digested with SalI and NotI, and ligated into pRLNi linearized with XhoI and NotI, resulting in the plasmid pRLNiHG. The upstream oligonucleotide PgfpF14 bears an 8X His-tag encoding region, thus making pRLNiHG suitable for expressing GFP with an N-terminal 8X His-tag. Similarly, the gfp gene from p8760, amplified using the oligonucleotides of PgfpF1m and PgfpR754bd, was cloned into pRLNi with XhoI and NotI, resulting in the plasmid pRLNiGH. The downstream oligonucleotide PgfpR754bd bears an 8X His-tag encoding region, enabling pRLNiGH to express GFP with a C-terminal 8X His-tag. To generate the plasmid pRLNiHG-AnaRneC, the coding region of AnaRneC was amplified using the oligonucleotides of Palr4331F1201a and Palr4331R2064 and inserted into pRLNiHG with XhoI and NotI. Similarly, to generate the plasmid pRLNiAnaRneC-GH, the coding region of AnaRneC amplified using the oligonucleotides of Palr4331F1201b and Palr4331R2061 was digested by SalI and inserted into pRLNiGH through XhoI.
Copurification of protein(s) associated with AnaRneC from Anabaena cells
The plasmids pHGFP, pNiHGEC, and pNiECGH were introduced into the wild-type Anabaena PCC7120 by conjugation (Wolk et al. 1988). The resulting strains were grown in liquid BG11 medium to OD700 = 0.5. Then, NiSO4 was added into the medium to 3 µM to induce the expression of hGFP, hGFP-AnaRneC, and AnaRne-GFPh, respectively. The expression level of the target proteins, which can be reflected by the intensity of intracellular GFP fluorescence, was checked by fluorescent microscopy every 4 h. After 12 h, when the target proteins were moderately induced, 100 mL of each culture were collected and homogenized in the lysis buffer (1X PBS, 1 mM PMSF, 1 mM DTT, pH7.4). The cell lysate was centrifuged at 13,000g for 15 min at 4°C, and the supernatant (∼5 mL) was transferred into a tube containing 200 µL Ni-NTA beads previously equilibrated with the lysis buffer. After incubation on the horizontal shaker at 4°C for 1 h, the beads were separated by centrifugation at 500g for 5 min followed by three washes in the washing buffer (1× PBS, 20 mM imidazole, pH 7.4). Finally, the proteins bound to the beads were eluted into 0.5 mL of the elution buffer (1× PBS, 0.5 M imidazole, pH 7.4) and analyzed by SDS-PAGE or Western blot.
Expression and purification of recombinant protein
The E. coli BL21 (DE3) cells transformed with the protein expression plasmid were grown in LB medium at 37°C. When cells reached the density of OD600 = 0.5, 1 mM IPTG was added into the culture. After 4 h of induction, cells were harvested by centrifugation and resuspended in the lysis buffer (500 mM NaCl, 20 mM Tris-HCl, 10 mM imidazole, pH 7.9). Cells were broken with a high-pressure homogenizer, and the target protein in the lysate was purified using the Ni-NTA affinity column (Qiagen) according to the product manual. The purified protein eluted from the Ni-NTA column was dialyzed into the protein storage buffer (20 mM Tris-HCl, 20 mM KCl, 100 mM NaCl, 25% glycerol, pH 7.5) and preserved at −80°C. Protein purity was estimated by SDS-PAGE, and protein concentration was quantified by Bradford assay using BSA as the standard.
Antibody production and immunoblotting analyses
The recombinant antigen protein was expressed in E. coli and isolated using the Ni-NTA column. The antigen purity was further improved by SDS-PAGE gel purification, in which the antigen protein was separated in SDS-PAGE gel and recovered by electroelution. The purified antigen was injected into rabbit for antibody production. The specificity of the obtained polyclonal antibodies was assessed by Western blot assays with the total cellular protein of Anabaena PCC7120.
The assays of regular Far-Western blot and Far-Western dot-blot were performed as described with minor modifications (Wu et al. 2007). In the regular Far-Western blot, AnaRneC and related proteins were migrated on a native-PAGE and electrotransferred onto a nitrocellulose membrane. Then, the membrane was blocked with 5% skimmed milk in TBS (50 mM Tris, 150 mM NaCl, pH 7.4) for 1 h, followed by 2 h of incubation with 2 µM AnaPnp in TBS containing 1% skimmed milk. After the incubation, the membrane was washed three times with TBS-T (0.1% Tween-20 in TBS), 5 min each. Subsequently, the membrane was sequentially incubated with the anti-AnaPnp polyclonal antibodies (1:1000) for 1 h and with HRP-conjugated goat-anti-rabbit secondary antibodies (1:5000, Jackson ImmunoResearch), as in standard Western blot protocols. The immunoblotting signals were developed using the BioReady ECL kit (MicLab Biotechnology). In the Far-Western dot-blot assays, the AnaRneC and related proteins were spotted in parallel onto two nitrocellulose membranes directly at 1 µL (20 pmol) per dot. Subsequently, one membrane was incubated with the solution of AnaPnp and the other was not. Then, both membranes were immunodetected as described in the regular Far-Western blot assay. The membrane without AnaPnp incubation was a control to test whether impurities in the dotted proteins also react with the antibodies against AnaPnp. Micrococcal nuclease (100 units/mL, with 5 mM CaCl2) or the synthetic C4 peptide (4, 10, or 20 µM) was added in the step of AnaPnp incubation when indicated. The nonapeptide of C4 was synthesized by Shanghai Science Peptide Biological Technology Co., Ltd.
Bacterial two-hybrid assay
The BacterioMatch II Two-hybrid System (Agilent) was used to detect protein–protein interactions in E. coli. The coding sequence of AnaRneC amplified using the oligonucleotides Palr4331F1201bt and Palr4331R2064a was inserted into the bait vector pBT with NotI and XhoI, resulting in the plasmid pBT-AnaRneC. The two-hybrid plasmids were constructed as follows. The coding sequence of AnaPnp amplified using the oligonucleotides Pall4396F1a and Pall4396R2193a was inserted into the target vector pTRG with NotI and XhoI, resulting in the plasmid pTRG-AnaPnp. The plasmids pBT-AnaPnp and pTRG-AnaRneC were constructed by swapping the EcoRI-XhoI fragments in pBT-AnaRneC and pTRG-AnaPnp. For the two-hybrid assay, the bait and target plasmid pairs were cotransformed into the host strain XL1-Blue MRF′ Kan, and the transformants were grown on the selective and the nonselective solid media, respectively. Compared to the nonselective medium, the selective medium additionally contains 0.05 mM IPTG, 5 mM 3-AT, and 12.5 µg/mL streptomycin. The growth of the transformants on both the selective and nonselective media is the indication of interaction between the bait and the target proteins encoded by the plasmids. The transformant containing pBT-LGF2 and pTRG-Gal11P (supplied in the kit) was used as the positive control and that containing the empty vector pBT and pTRG was used as the negative control.
Protein complex analysis by gel electrophoresis
To test the interaction between AnaRneC and AnaPnp by native-PAGE gel, AnaRneC (0.5 μg) and AnaPnp (1.9 μg) were incubated at 25°C in a 20-μL mixture containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM CaCl2, and 100 units/mL micrococcal nuclease. The molar ratio between AnaRneC and AnaPnp in the mixture was approximately 1:3. After 30 min of incubation, the sample was analyzed by native-PAGE in running buffer of 25 mM Tris, 192 mM glycine, pH 8.3. The band of protein complex in the native gel was subsequently analyzed by a standard SDS-PAGE.
RNase E activity assay
The DNA template used for E. coli 9S RNA preparation was amplified using the oligonucleotides EcT79SF and EcT79SR. The in vitro transcription reaction was performed using the TranscriptAid T7 High Yield Transcription Kit (Fermentas). The RNA product was purified using the PCR Purification Kit (Qiagen). The enzymatic assay for RNase E proteins was performed at 30°C or 37°C in a 50-µL mixture containing 20 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 100 mM NaCl, 5% glycerol, 0.1% Triton X-100, 0.1 mM DTT, 0.4 µM of 9S RNA, and 0.4 µM of enzyme. Ten-microliter aliquots of the reaction mixture were taken out at time points 0, 30 min, and 60 min and immediately mixed with 5 µL of 3× loading buffer (87.5% formamide, 30 mM EDTA). After being heated at 85°C for 3 min, the samples were separated on a 6% PAGE gel containing 7 M urea. The resulting gel was visualized by ethidium bromide staining.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Ben F. Luisi at the University of Cambridge for helpful guidance in preparing E. coli 9S RNA. We thank Xiaozhen Huang for help in the enzymatic assay, Songbo Li for construction of some protein expression plasmids, and Shuang Wan for help in the bacterial two-hybrid assay and native-PAGE assay. This work was supported by the National Natural Science Foundation of China (31100038, 30970088) and the Fundamental Research Funds for the Central Universities of China (2011PY090).
REFERENCES
- Aït-Bara S, Carpousis AJ 2010. Characterization of the RNA degradosome of Pseudoalteromonas haloplanktis: Conservation of the RNase E-RhlB interaction in the gammaproteobacteria. J Bacteriol 192: 5413–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, Moreira RN, Pobre V, Reis FP, Saramago M, et al. 2010. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 34: 883–923 [DOI] [PubMed] [Google Scholar]
- Baginsky S, Shteiman-Kotler A, Liveanu V, Yehudai-Resheff S, Bellaoui M, Settlage RE, Shabanowitz J, Hunt DF, Schuster G, Gruissem W 2001. Chloroplast PNPase exists as a homo-multimer enzyme complex that is distinct from the Escherichia coli degradosome. RNA 7: 1464–1475 [PMC free article] [PubMed] [Google Scholar]
- Blum E, Carpousis AJ, Higgins CF 1999. Polyadenylation promotes degradation of 3′-structured RNA by the Escherichia coli mRNA degradosome in vitro. J Biol Chem 274: 4009–4016 [DOI] [PubMed] [Google Scholar]
- Callaghan AJ, Aurikko JP, Ilag LL, Günter Grossmann J, Chandran V, Kühnel K, Poljak L, Carpousis AJ, Robinson CV, Symmons MF, et al. 2004. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol 340: 965–979 [DOI] [PubMed] [Google Scholar]
- Carpousis AJ 2007. The RNA degradosome of Escherichia coli: An mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol 61: 71–87 [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM 1994. Copurification of E. coli RNAase E and PNPase: Evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 76: 889–900 [DOI] [PubMed] [Google Scholar]
- Coburn GA, Miao X, Briant DJ, Mackie GA 1999. Reconstitution of a minimal RNA degradosome demonstrates functional coordination between a 3′ exonuclease and a DEAD-box RNA helicase. Genes Dev 13: 2594–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack RS, Genereaux JL, Mackie GA 1993. RNase E activity is conferred by a single polypeptide: Overexpression, purification, and properties of the ams/rne/hmp1 gene product. Proc Natl Acad Sci 90: 9006–9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erce MA, Low JK, March PE, Wilkins MR, Takayama KM 2009. Identification and functional analysis of RNase E of Vibrio angustum S14 and two-hybrid analysis of its interaction partners. Biochim Biophys Acta 1794: 1107–1114 [DOI] [PubMed] [Google Scholar]
- Erce MA, Low JK, Wilkins MR 2010. Analysis of the RNA degradosome complex in Vibrio angustum S14. FEBS J 277: 5161–5173 [DOI] [PubMed] [Google Scholar]
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Bréchemmier-Baey D, Putzer H 2005. Ribonucleases J1 and J2: Two novel endoribonucleases in B.subtilis with functional homology to E.coli RNase E. Nucleic Acids Res 33: 2141–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón LI, Magallón S, Castillo A 2010. Dating the cyanobacterial ancestor of the chloroplast. ISME J 4: 777–783 [DOI] [PubMed] [Google Scholar]
- Hambraeus G, von Wachenfeldt C, Hederstedt L 2003. Genome-wide survey of mRNA half-lives in Bacillus subtilis identifies extremely stable mRNAs. Mol Genet Genomics 269: 706–714 [DOI] [PubMed] [Google Scholar]
- Hardwick SW, Chan VS, Broadhurst RW, Luisi BF 2011. An RNA degradosome assembly in Caulobacter crescentus. Nucleic Acids Res 39: 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick SW, Gubbey T, Hug I, Jenal U, Luisi BF 2012. Crystal structure of Caulobacter crescentus polynucleotide phosphorylase reveals a mechanism of RNA substrate channelling and RNA degradosome assembly. Open Biol 2: 120028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie Y, Ito Y, Ono M, Moriwaki N, Kato H, Hamakubo Y, Amano T, Wachi M, Shirai M, Asayama M 2007. Dark-induced mRNA instability involves RNase E/G-type endoribonuclease cleavage at the AU-box and SD sequences in cyanobacteria. Mol Genet Genomics 278: 331–346 [DOI] [PubMed] [Google Scholar]
- Kaberdin VR, Bizebard T 2005. Characterization of Aquifex aeolicus RNase E/G. Biochem Biophys Res Commun 327: 382–392 [DOI] [PubMed] [Google Scholar]
- Kaberdin VR, Miczak A, Jakobsen JS, Lin-Chao S, McDowall KJ, von Gabain A 1998. The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc Natl Acad Sci 95: 11637–11642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaberdin VR, Singh D, Lin-Chao S 2011. Composition and conservation of the mRNA-degrading machinery in bacteria. J Biomed Sci 18: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khemici V, Carpousis AJ 2004. The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol Microbiol 51: 777–790 [DOI] [PubMed] [Google Scholar]
- Khemici V, Poljak L, Toesca I, Carpousis AJ 2005. Evidence in vivo that the DEAD-box RNA helicase RhlB facilitates the degradation of ribosome-free mRNA by RNase E. Proc Natl Acad Sci 102: 6913–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khemici V, Poljak L, Luisi BF, Carpousis AJ 2008. The RNase E of Escherichia coli is a membrane-binding protein. Mol Microbiol 70: 799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Cohen SN 2003. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol Microbiol 48: 349–360 [DOI] [PubMed] [Google Scholar]
- Lee M, Yeom JH, Jeon CO, Lee K 2011. Studies on a Vibrio vulnificus functional ortholog of Escherichia coli RNase E imply a conserved function of RNase E-like enzymes in bacteria. Curr Microbiol 62: 861–865 [DOI] [PubMed] [Google Scholar]
- Lehnik-Habrink M, Lewis RJ, Mäder U, Stülke J 2012. RNA degradation in Bacillus subtilis: An interplay of essential endo- and exoribonucleases. Mol Microbiol 84: 1005–1017 [DOI] [PubMed] [Google Scholar]
- Liou GG, Chang HY, Lin CS, Lin-Chao S 2002. DEAD box RhlB RNA helicase physically associates with exoribonuclease PNPase to degrade double-stranded RNA independent of the degradosome-assembling region of RNase E. J Biol Chem 277: 41157–41162 [DOI] [PubMed] [Google Scholar]
- López-Maury L, García-Domínguez M, Florencio FJ, Reyes JC 2002. A two-component signal transduction system involved in nickel sensing in the cyanobacterium Synechocystis sp. PCC 6803. Mol Microbiol 43: 247–256 [DOI] [PubMed] [Google Scholar]
- Miczak A, Kaberdin VR, Wei CL, Lin-Chao S 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci 93: 3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR 1999. Analysis of the function of Escherichia coli poly(A) polymerase I in RNA metabolism. Mol Microbiol 34: 1094–1108 [DOI] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR 2011. Bacterial/archaeal/organellar polyadenylation. Wiley Interdiscip Rev RNA 2: 256–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, Kawamoto H, Mizota T, Inada T, Aiba H 2004. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol Microbiol 54: 1063–1075 [DOI] [PubMed] [Google Scholar]
- Murashko ON, Kaberdin VR, Lin-Chao S 2012. Membrane binding of Escherichia coli RNase E catalytic domain stabilizes protein structure and increases RNA substrate affinity. Proc Natl Acad Sci 109: 7019–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsai R, Howell KA, Millar AH, O'Toole N, Small I, Whelan J 2007. Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana. Plant Cell 19: 3418–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmohamed S, Vaidialingam B, Callaghan AJ, Luisi BF 2009. Crystal structure of Escherichia coli polynucleotide phosphorylase core bound to RNase E, RNA and manganese: Implications for catalytic mechanism and RNA degradosome assembly. J Mol Biol 389: 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purusharth RI, Klein F, Sulthana S, Jäger S, Jagannadham MV, Evguenieva-Hackenberg E, Ray MK, Klug G 2005. Exoribonuclease R interacts with endoribonuclease E and an RNA helicase in the psychrotrophic bacterium Pseudomonas syringae Lz4W. J Biol Chem 280: 14572–14578 [DOI] [PubMed] [Google Scholar]
- Redon E, Loubière P, Cocaign-Bousquet M 2005. Role of mRNA stability during genome-wide adaptation of Lactococcus lactis to carbon starvation. J Biol Chem 280: 36380–36385 [DOI] [PubMed] [Google Scholar]
- Ross J 1995. mRNA stability in mammalian cells. Microbiol Rev 59: 423–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott R, Zipor G, Portnoy V, Liveanu V, Schuster G 2003. RNA polyadenylation and degradation in cyanobacteria are similar to the chloroplast but different from Escherichia coli. J Biol Chem 278: 15771–15777 [DOI] [PubMed] [Google Scholar]
- Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF 2001. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem 276: 8159–8164 [DOI] [PubMed] [Google Scholar]
- Schein A, Sheffy-Levin S, Glaser F, Schuster G 2008. The RNase E/G-type endoribonuclease of higher plants is located in the chloroplast and cleaves RNA similarly to the E. coli enzyme. RNA 14: 1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinger DW, Saxena RM, Cheung KJ, Church GM, Rosenow C 2003. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res 13: 216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbabian K, Jamalli A, Zig L, Putzer H 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28: 3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Yang WZ, Lin-Chao S, Chak KF, Yuan HS 2008. Crystal structure of Escherichia coli PNPase: Central channel residues are involved in processive RNA degradation. RNA 14: 2361–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva IJ, Saramago M, Dressaire C, Domingues S, Viegas SC, Arraiano CM 2011. Importance and key events of prokaryotic RNA decay: The ultimate fate of an RNA molecule. Wiley Interdiscip Rev RNA 2: 818–836 [DOI] [PubMed] [Google Scholar]
- Steglich C, Lindell D, Futschik M, Rector T, Steen R, Chisholm SW 2010. Short RNA half-lives in the slow-growing marine cyanobacterium Prochlorococcus. Genome Biol 11: pR54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ 1998. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 12: 2770–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk CP, Cai Y, Cardemil L, Flores E, Hohn B, Murry M, Schmetterer G, Schrautemeier B, Wilson R 1988. Isolation and complementation of mutants of Anabaena sp. strain PCC 7120 unable to grow aerobically on dinitrogen. J Bacteriol 170: 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li Q, Chen XZ 2007. Detecting protein–protein interactions by far western blotting. Nat Protoc 2: 3278–3284 [DOI] [PubMed] [Google Scholar]
- Yang J, Jain C, Schesser K 2008. RNase E regulates the Yersinia type 3 secretion system. J Bacteriol 190: 3774–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller ME, Csanadi A, Miczak A, Rose T, Bizebard T, Kaberdin VR 2007. Quaternary structure and biochemical properties of mycobacterial RNase E/G. Biochem J 403: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LC, Risoul V, Latifi A, Christie JM, Zhang CC 2013. Exploring the size limit of protein diffusion through the periplasm in cyanobacterium Anabaena sp. PCC 7120 using the 13 kDa iLOV fluorescent protein. Res Microbiol 164: 710–717 [DOI] [PubMed] [Google Scholar]