


Abstract
The ease of generating high-throughput data has enabled investigations into organismal complexity at the systems level through the inference of networks of interactions among the various cellular components (genes, RNAs, proteins and metabolites). The wider scientific community, however, currently has limited access to tools for network inference, visualization and analysis because these tasks often require advanced computational knowledge and expensive computing resources. We have designed the network portal (http://networks.systemsbiology.net) to serve as a modular database for the integration of user uploaded and public data, with inference algorithms and tools for the storage, visualization and analysis of biological networks. The portal is fully integrated into the Gaggle framework to seamlessly exchange data with desktop and web applications and to allow the user to create, save and modify workspaces, and it includes social networking capabilities for collaborative projects. While the current release of the database contains networks for 13 prokaryotic organisms from diverse phylogenetic clades (4678 co-regulated gene modules, 3466 regulators and 9291 cis-regulatory motifs), it will be rapidly populated with prokaryotic and eukaryotic organisms as relevant data become available in public repositories and through user input. The modular architecture, simple data formats and open API support community development of the portal.
INTRODUCTION
Underlying the phenotype of any organism is the network of interactions among its constituent parts encoded in its genome. Systems biology is becoming a mature discipline for studying this biological complexity through the use of high-throughput instruments for data production and powerful computing technologies for their analysis. A central part of this effort is the reverse engineering of gene-regulatory networks through the integration of diverse genome-wide measurements such as gene-expression changes, transcription-factor occupancy and protein–protein interactions (1–3).
Biological insights revealed by gene-regulatory networks include identification of regulators and regulatory motifs driving particular responses, assignment of unannotated genes to biological processes, identification of coordinated regulation amongst cellular processes, description of overall network architecture for an organism, prediction of gene expression in new genetic or environmental conditions and development of hypotheses for how perturbation of regulators or motifs could manipulate metabolic flux or other phenotypes. Indeed, exploration of these networks has provided unprecedented insights into the biology of diverse organisms, including regulation of metabolism in bacteria (4), oxidative stress response in Archaea (5) and vertebrate immune cell specification (6). Further, comparative analysis of regulatory networks from multiple species allows insights into evolutionary changes in the roles of individual regulators, the regulation of homologous genes and pathways and overall network architecture features such as connectivity and density (7,8).
While there are many algorithms for gene-regulatory-network inference (1,2,9) as well as many tools for the exploration and analysis of networks (10), the complexity of these powerful tools generally makes them inaccessible to the wider scientific community. Specifically, network inference can be prohibitively difficult for many users because it is usually not automated or integrated with network-analysis tools, it requires extensive computational power and it demands that the user have access to a massive amount of high-quality data. Existing resources for storage of network information, such as RegulonDB (11), RegPrecise (12), IntegromeDB (13), DBTBS (14), CoryneRegNet (15), MTBregList (16), InnateDB (17) and BiologicalNetworks (18) are often limited to a few model organisms, only store existing network models, or are tailored for specific purposes. Similar trends during the genomic era led to the development of universal resources including NCBI Entrez (19), Gene Expression Omnibus (GEO) (20) and KEGG pathway (21).
We have developed the network portal to democratize access to the inference, storage, exploration and visualization of gene-regulatory networks. The network portal is connected to an automated network-inference pipeline, which can generate networks for any organism (prokaryotic or eukaryotic) whose genome is available using gene-expression data from public databases or custom user files. The network-inference pipeline is modular to allow use of different algorithms and currently runs the cMonkey (22) and Inferelator (23) algorithms. cMonkey integrates gene-expression data together with genomic, proteomic and functional associations in order to identify co-regulated group of genes under subsets of conditions. Inferelator then identifies the transcription factors and environmental conditions with the most probable regulatory influences on these groups of genes. Inferred networks are stored in a relational database. Network analysis is made possible by multiple novel tools for visualization, basic and advanced search interfaces and easy-to-use filters to explore and analyze regulation and gene function. The standardization within the network portal will facilitate community development of data, algorithms and software, allowing users to perform collaborative analysis of raw and processed data.
MATERIALS AND METHODS
Architecture
Architecture overview
The network portal is composed of four integrated layers. The data layer collects genomic information and gene-expression data for each organism. This layer provides the input for the algorithm layer. Algorithm output is stored in a PostgreSQL [http://www.postgresql.org] database and served by a Solr/Django-powered web interface [https://www.djangoproject.com]. The analysis and visualization layer allow users to query and explore networks at different levels, create and save workspaces for in-depth analysis of networks, and broadcast data via the Gaggle (24)/Firegoose (25) framework to third-party desktop and web applications (Figure 1).
Figure 1.
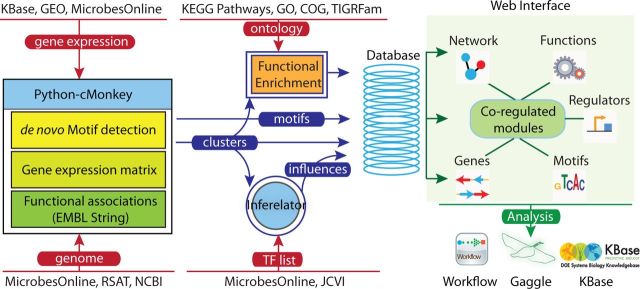
The network portal Framework. The network portal currently implements the Python–cMonkey algorithm for network inference. Publicly available gene-expression data and genomic information is collected from various databases along with functional associations from EMBL STRING. Conditionally co-regulated clusters of genes (modules) and motifs discovered by cMonkey are stored in the database. The most probable influences on these modules are identified by Inferelator, using a TF list collected from MicrobesOnline and JCVI-CMR. A Django-based Web interface dynamically creates module-centered views for Network, Functions, Genes, Regulators and Motifs. Further investigations of the networks can be performed by using interoperability and automation frameworks provided by Gaggle and Workflow, respectively.
Database schema
Network information is stored in a PostgreSQL relational database. Each species is association with genome information and one or more inferred networks. Modules, which are sets of co-regulated genes, are associated with regulatory sequence motifs, conditions where gene expression within the module is coherent, influential regulatory or environmental factors and functional enrichment (Supplementary Figure S1). Database releases are publicly available at the Network Portal Github repository [https://github.com/baliga-lab/network_portal].
Web interface
The network portal web interface is built with Python and the Django framework. Key word and faceted search is provided by Apache Solr [http://lucene.apache.org/solr]. Other software technologies used include JQuery [http://jquery.com], NetworkX [http://networkx.github.io], Cytoscape Web (26) and R [http://www.r-project.org]. Full source code for the network portal is available on Github at [https://github.com/baliga-lab/network_portal].
The network portal database and web interface will be updated and new downloads will be made available every 6 months as we build network models for new organisms.
Data sources
The network portal integrates genomic information and upstream promoter sequences from NCBI GenBank (19) and RSAT (27), operon predictions from MicrobesOnline (28), known and predicted protein–protein interactions from EMBL STRING (29) and functional associations from Prolinks (30) and Predictome (31). Functional enrichment analyses are based on gene annotations from the Gene Ontology (32), KEGG (21), TIGR (33) and Cluster of Orthologous Groups (COG) (34) databases. Lists of transcription factors for each organism for use with the Inferelator algorithm are collected from MicrobesOnline and JCVI CMR (33) based on GO (32) and COG (34) annotations.
Gene-expression data was collected from MicrobesOnline (28), GEO (20), DISTILLER (35) and Baliga lab datasets. All downloaded data are quality-checked computationally and manually for data integrity, normalization and redundancy. Gene-expression matrices for each organism were scanned for duplicate entries and converted into log2 ratios (Redundancy filter). Missing gene names or alternate gene names in these matrices were fixed by constructing a synonyms table (ProbeName filter). The data matrix was filtered to only include columns and rows that have enough measurements over all the conditions and genes (NoChange filter). Furthermore, values for each row in the data matrix were centered on their median and scaled by their standard deviation in preparation for the network inference (CenterScale filter). The transcription factor list that was used as the list of putative regulatory influences for the Inferelator algorithm was assembled using transcription factor annotations that are supported by GO and COG annotations. This list was further manually curated to remove TFs with poorly defined annotations.
Network-inference pipeline
Data processing
The automated network-inference pipeline features automatic data download from sources including MicrobesOnline, GEO and KBase. For network inference, gene-expression data is organized into matrices of log2 ratios. Such matrices can be used directly when available or computed by comparing each sample with a reference. Genes without significant expression change (<1.5-fold) in any of the experiments are removed. The expression level of each gene is normalized to mean = 0 and SD = 1 as described previously (22).
Regulatory network inference
The first step in the automated inference pipeline is clustering of conditionally co-regulated genes using cMonkey (22). We ported the cMonkey algorithm from R to Python with enhancements in modularity and performance for use with the network portal (W.J. Wu et al. in preparation). Python cMonkey integrates gene-expression data with de novo motif prediction and other functional associations such as operon predictions, protein–protein interactions and genomic neighborhood information to identify groups of genes that are co-regulated under a subset of the experimental conditions (co-regulated modules).
Second, the most probable regulatory influences from transcription factors or environmental factors on each co-regulated module are identified by Inferelator using linear regression and model shrinkage techniques (23). We have shown previously that Inferelator can predict gene expression responses of 80% of Halobacterium salinarum genes (36). Positive and negative influences on modules are deposited into the database.
In addition to cMonkey/Inferelator, many other powerful network-inference algorithms are available (1,2,9). To allow users access to these other algorithms, our architecture is designed to be modular. The central units of network models are co-regulated modules, their member genes and regulators with influences on these modules. Most regulatory network-inference algorithms provide output compatible with this framework (see Supplementary Table S1). Therefore, developers can easily integrate different algorithms using our API, and users will be able to select which inference tool to use.
Functional enrichment
We integrated KEGG pathway, Gene Ontology, TIGRFam and COG annotations to maximize data content. We use hypergeometric P-values to identify significant overlaps between co-regulated module members and genes assigned to a particular functional annotation category. P-values are corrected for multiple comparisons using Benjamini–Hochberg correction and filtered for P-values ≤ 0.05.
RESULTS
Available species
To demonstrate the flexibility of the network portal, we built regulatory networks for organisms from different phylogenies and with varying genome complexity and available amounts of gene expression data. The first release of the network portal includes two Archaea and 11 Bacteria, including three Firmicutes, six Proteobacteria, a Cyanobacterium and a Bacteroidetes species (Table 1). Even though the current version of the database includes only prokaryotic species, it is important to emphasize that the underlying database structure, network inference and web interface are also compatible with eukaryotic species and they will be included in the database as soon as they become available (37).
Table 1.
Organisms currently in the network portal
| Organism | Domain: phylum | Number of genes | Number of gene-expression comparisons | Gene-expression data source | Number of regulators |
|---|---|---|---|---|---|
| Halobacterium sp. NRC-1 | Archaea: Euryarchaeota | 2701 | 1661 | (23,36) | 125 |
| Methanococcus maripaludis S2 | Archaea: Euryarchaeota | 1863 | 58 | (38) | 57 |
| Bacteroides thetaiotaomicron VIP-5482 | Bacteria: Bacteroidetes | 4902 | 324 | MicrobesOnline | 263 |
| Clostridium acetobutylicum ATCC 824 | Bacteria: Firmicutes | 3995 | 111 | MicrobesOnline | 259 |
| Bacillus cereus ATCC 14579 | Bacteria: Firmicutes | 5501 | 151 | MicrobesOnline | 376 |
| Bacillus subtilis subsp. subtilis str. 168 | Bacteria: Firmicutes | 4313 | 138 | MicrobesOnline | 319 |
| Synechococcus elongatus PCC 7942 | Bacteria: Cyanobacteria | 2717 | 129 | GEO | 90 |
| Rhodobacter sphaeroides 2.4.1 | Bacteria: Proteobacteria | 4341 | 165 | MicrobesOnline | 231 |
| Pseudomonas aeruginosa PAO1 | Bacteria: Proteobacteria | 5646 | 635 | MicrobesOnline | 475 |
| Escherichia coli K-12 | Bacteria: Proteobacteria | 4497 | 868 | (35) | 310 |
| Campylobacter jejuni subsp. jejuni | Bacteria: Proteobacteria | 1711 | 114 | MicrobesOnline | 39 |
| Geobacter sulfurreducens PCA | Bacteria: Proteobacteria | 3519 | 87 | MicrobesOnline | 156 |
| Desulfovibrio vulgaris str. Hildenborough | Bacteria: Proteobacteria | 3661 | 383 | MicrobesOnline | 128 |
Advanced search features
The network portal is powered by an Apache Solr search engine. Solr provides fast, faceted full-text search capabilities for querying a large set of network features from an integrated database. Queries can be executed based on unique genomic, functional and network parameters as well as ranges of values. Multi-faceted advanced searching allows selection of specific organisms and the ability to perform queries either at the gene level (for ‘name’, ‘locus tag’ and ‘function’ fields) or module level (for ‘gene members’, ‘regulators’, ‘functions’ and ‘residual’ ranges).
Search results are organized based on species and presented with a quick feature overview including annotations, regulatory influences and module information. Further exploration of the search results is possible by following links. Moreover, users can visualize the relationships among selected search results in a network diagram with one click using the Cytoscape Web interface (21). See Supplementary Materials use Cases 1 and 2 for examples of the search features to explore network biology.
Visualizations
The module page shows co-expression profiles (Figure 2A), member genes (Supplementary Figure S2A), transcription factors and environmental factors as regulatory influences (Figure 2A and Supplementary Figure S2B), and de novo identified motifs (Figure 2B). A network view of the module created using Cytoscape Web (26) enables interactive exploration (Figure 2C). In this view, module member genes, motifs and regulatory influences are represented as peripheral nodes connected to core module nodes via edges. For each module, regulatory influences are listed in tables (Supplementary Figure S2B).
Figure 2.
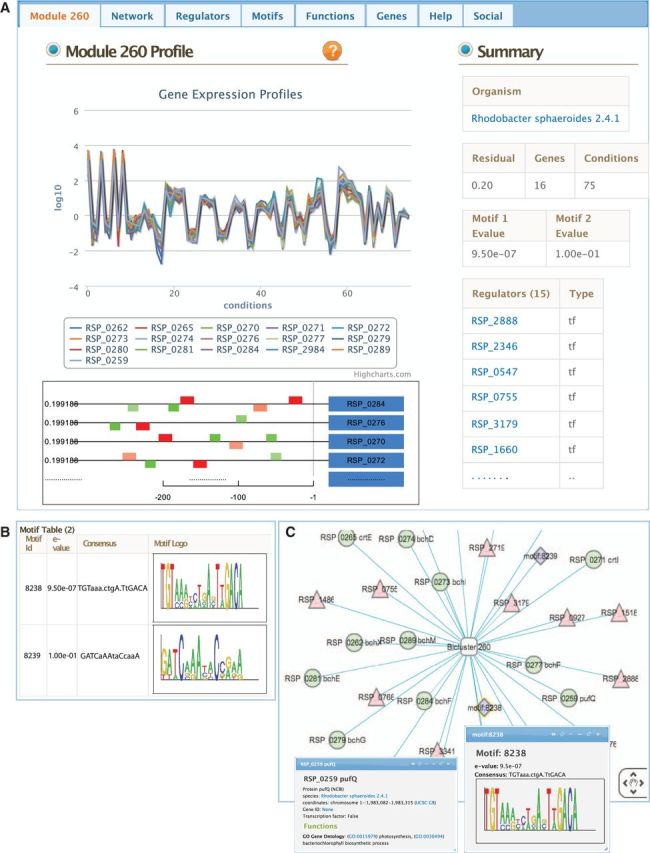
An example module page. (A) The landing page for each module presents a summary view of the module, including an interactive plot of gene-expression profiles across conditions, motif locations upstream of the member genes and summary statistics. Tabs located on top of the page provide access to other visualization tools. (B) The motif table shows motif logos for de novo identified upstream regulatory motifs and E-value statistics. For selected organisms, a link to analyze the motifs using RegPredict is provided. (C) Interactive network visualization is created by using Cytoscape Web. An edge connects the module and each of its gene members, motifs and regulators. Clicking on a node opens up overlay window with detailed information.
Transcription factor binding motifs help to elucidate regulatory mechanism. cMonkey integrates the MEME Suit (39) for de novo motif detection. Motifs for each module are listed as logo images along with prediction statistics (E-values) and the location of motifs within the upstream sequences of the module member genes (Figure 2A). Motifs can be broadcast to RegPredict (http://regpredict.lbl.gov/regpredict) in order to compare conservation in similar species. This integrated motif prediction and comparative analysis provides an additional checkpoint for regulatory motif prediction confidence.
Identification of functional enrichment for the module members is important in associating predicted motifs and regulatory influences with pathways. Over-represented functional ontology terms from KEGG, GO, TIGRFAM and COG are presented for each module along with hypergeometric P-values and the number of module genes assigned to each term. See Supplementary Materials Use Case 3 for an example of the use of functional enrichment information in exploration of gene function.
Gene-landing pages present genomic, functional and regulatory information for individual genes. A circular visualization displays connections between the selected gene and genes in the same modules, with edges drawn between the respective coordinates of the whole genome. The gene page also lists functional ontology assignments, module membership and motifs associated with these modules. Genes in the network inherit regulatory influences from the modules to which they belong, and the regulatory influences table lists influence name, type and target module. If the gene is a transcription factor, its target modules are displayed in a table with residual values and number of genes (See Supplementary Material Movie 1, for example, Use Case of gene-landing pages).
Interoperation with other desktop and web applications through Gaggle
We previously developed the Gaggle framework for exchanging data among independent desktop programs (e.g. Cytoscape, MeV, Firefox, R) and web resources (e.g. EMBL STRING, KEGG, STAMP, MicrobesOnline, RegTransBase, RegPrecise, KBase and DAVID) (24). We fully integrated the network portal with the Gaggle framework to extend analysis capabilities and interoperability with other resources that are not included in the portal itself. The Firefox extension Firegoose can capture multiple data types (i.e. NameList and matrix) and then broadcast this data to other resources, and data from outside web and desktop applications can be broadcasted into the network portal Workspace. See Supplementary Materials Use Case 4 for a case study in obtaining additional data about genes of interest using the Firegoose to connect to outside webpages.
Gaggle workspace
The network portal also acts as a gateway for the Gaggle Workspace (N. Jiang et al., submitted for publication). Gaggle Workspace provides a central space for managing data and an entry point for network analysis. Workspace can be used to integrate user-uploaded data and data available in the network portal. For each organism, it provides Cytoscape network and MeV gene-expression files in the form of java web starts, enabling cross-platform analysis. Gaggle/Firegoose can be used to capture information such as module member gene lists into the Workspace and to broadcast data to other applications or web resources. One of the unique features of the Workspace is the ability to save the state of the analysis, including analysis steps, associated data and the results for later access or sharing. Watch Movie 2 for a case study using Workspace in Supplementary Material.
Integration of outside resources
The network portal is designed to be extensible and modular to give users flexibility to choose different tools for inference and analysis and for developers to integrate their resources. As a proof of concept, we integrated RegPredict motif analysis into the network portal modules for Desulfovibrio vulgaris. RegPredict carries out module inference by searching a known position weight matrix (PWM) against genomes of closely related prokaryotes. The RegPredict link sends the user from a de novo identified motif within the network portal to the RegPredict website, allowing comparison of two independent motif detection methods. This seamless integration enables further exploration of predicted motifs to check their evolutionary conservation across multiple taxonomically related genomes.
DISCUSSION
The network portal improves the availability of regulatory information by implementing network-inference algorithms and novel visualization tools. The first release provides gene-regulatory network models for 13 microbial species of medical, biotechnological and environmental importance. The network portal will be rapidly expanded to include the >100 organisms for which there is already sufficient gene expression data available in public databases for robust regulatory network inference. As more networks become available, network comparisons become possible among species that vary by phylogenetic relationship, environmental niche or metabolic and phenotypic features.
Moreover, the network portal promotes cross-platform data analysis and collaboration among researchers with distinct areas of expertise. To this end, the network portal framework integrates the Gaggle framework and will allow developers to add other inference algorithms. Further, the new Workspace application enables users to upload data, capture information from the web and save analysis states, and future releases will add capabilities to create projects, workflows, favorites and bookmarks and share these features with collaborators.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Christopher Plaisier and Aaron Brooks for critical reading of the manuscript and helpful suggestions.
FUNDING
Funding for open access charge: Enabling a Systems Biology Knowledgebase with Gaggle and Firegoose [DE-FG02-04ER63807]; ENIGMA, Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory (Office of Science, Office of Biological and Environmental Research of the US Department of Energy under Contract No. DE-AC02-05CH11231).
Conflict of interest statement. None declared.
REFERENCES
- 1.Poultney CS, Greenfield A, Bonneau R. Integrated inference and analysis of regulatory networks from multi-level measurements. Methods Cell Biol. 2012;110:19–56. doi: 10.1016/B978-0-12-388403-9.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 2010;8:717–729. doi: 10.1038/nrmicro2419. [DOI] [PubMed] [Google Scholar]
- 3.Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Kellis M, Collins JJ, Stolovitzky G. Wisdom of crowds for robust gene network inference. Nat. Methods. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaur A, Van PT, Busch CR, Robinson CK, Pan M, Pang WL, Reiss DJ, DiRuggiero J, Baliga NS. Coordination of frontline defense mechanisms under severe oxidative stress. Mol. Syst. Biol. 2010;6:393. doi: 10.1038/msb.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazurie A, Bonchev D, Schwikowski B, Buck GA. Evolution of metabolic network organization. BMC Syst. Biol. 2010;4:59. doi: 10.1186/1752-0509-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, Seringhaus MR, Wang LY, Gerstein M, Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- 9.Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA, Dubchak I. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 2010;38:W299–W307. doi: 10.1093/nar/gkq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Z, Wang J, Zhang B. NetGestalt: integrating multidimensional omics data over biological networks. Nat. Methods. 2013;10:597–598. doi: 10.1038/nmeth.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salgado H, Peralta-Gil M, Gama-Castro S, Santos-Zavaleta A, Muniz-Rascado L, Garcia-Sotelo JS, Weiss V, Solano-Lira H, Martinez-Flores I, Medina-Rivera A, et al. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res. 2013;41:D203–D213. doi: 10.1093/nar/gks1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novichkov PS, Brettin TS, Novichkova ES, Dehal PS, Arkin AP, Dubchak I, Rodionov DA. RegPrecise web services interface: programmatic access to the transcriptional regulatory interactions in bacteria reconstructed by comparative genomics. Nucleic Acids Res. 2012;40:W604–W608. doi: 10.1093/nar/gks562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baitaluk M, Kozhenkov S, Dubinina Y, Ponomarenko J. IntegromeDB: an integrated system and biological search engine. BMC Genomics. 2012;13:35. doi: 10.1186/1471-2164-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sierro N, Makita Y, de Hoon M, Nakai K. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 2008;36:D93–D96. doi: 10.1093/nar/gkm910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pauling J, Rottger R, Tauch A, Azevedo V, Baumbach J. CoryneRegNet 6.0–Updated database content, new analysis methods and novel features focusing on community demands. Nucleic Acids Res. 2012;40:D610–D614. doi: 10.1093/nar/gkr883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacques PE, Gervais AL, Cantin M, Lucier JF, Dallaire G, Drouin G, Gaudreau L, Goulet J, Brzezinski R. MtbRegList, a database dedicated to the analysis of transcriptional regulation in Mycobacterium tuberculosis. Bioinformatics. 2005;21:2563–2565. doi: 10.1093/bioinformatics/bti321. [DOI] [PubMed] [Google Scholar]
- 17.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–D1233. doi: 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozhenkov S, Dubinina Y, Sedova M, Gupta A, Ponomarenko J, Baitaluk M. BiologicalNetworks 2.0–an integrative view of genome biology data. BMC Bioinform. 2010;11:610. doi: 10.1186/1471-2105-11-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinform. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, Thorsson V. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinform. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bare JC, Shannon PT, Schmid AK, Baliga NS. The Firegoose: two-way integration of diverse data from different bioinformatics web resources with desktop applications. BMC Bioinform. 2007;8:456. doi: 10.1186/1471-2105-8-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas-Chollier M, Defrance M, Medina-Rivera A, Sand O, Herrmann C, Thieffry D, van Helden J. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39:W86–W91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, et al. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010;38:D396–W400. doi: 10.1093/nar/gkp919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 2004;5:R35. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellor JC, Yanai I, Clodfelter KH, Mintseris J, DeLisi C. Predictome: a database of putative functional links between proteins. Nucleic Acids Res. 2002;30:306–309. doi: 10.1093/nar/30.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davidsen T, Beck E, Ganapathy A, Montgomery R, Zafar N, Yang Q, Madupu R, Goetz P, Galinsky K, White O, et al. The comprehensive microbial resource. Nucleic Acids Res. 2010;38:D340–D345. doi: 10.1093/nar/gkp912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemmens K, De Bie T, Dhollander T, De Keersmaecker SC, Thijs IM, Schoofs G, De Weerdt A, De Moor B, Vanderleyden J, Collado-Vides J, et al. DISTILLER: a data integration framework to reveal condition dependency of complex regulons in Escherichia coli. Genome Biol. 2009;10:R27. doi: 10.1186/gb-2009-10-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 37.Danziger SA, Ratushny AV, Smith JJ, Saleem RA, Wan Y, Arens CE, Armstrong AM, Sitko K, Chen WM, Chiang JH, et al. Molecular mechanisms of system responses to novel stimuli are predictable from public data. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt938. October 31 (doi:10.1093/nar/gkt938; epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon SH, Turkarslan S, Reiss DJ, Pan M, Burn JA, Costa KC, Lie TJ, Slagel J, Moritz RL, Hackett M, et al. A systems level predictive model for global gene regulation of methanogenesis in a hydrogenotrophic methanogen. Genome Res. 2013;23:1839–1851. doi: 10.1101/gr.153916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]