


Abstract
Although microRNAs (miRNAs), other non-coding RNAs (ncRNAs) (e.g. lncRNAs, pseudogenes and circRNAs) and competing endogenous RNAs (ceRNAs) have been implicated in cell-fate determination and in various human diseases, surprisingly little is known about the regulatory interaction networks among the multiple classes of RNAs. In this study, we developed starBase v2.0 (http://starbase.sysu.edu.cn/) to systematically identify the RNA–RNA and protein–RNA interaction networks from 108 CLIP-Seq (PAR-CLIP, HITS-CLIP, iCLIP, CLASH) data sets generated by 37 independent studies. By analyzing millions of RNA-binding protein binding sites, we identified ∼9000 miRNA-circRNA, 16 000 miRNA-pseudogene and 285 000 protein–RNA regulatory relationships. Moreover, starBase v2.0 has been updated to provide the most comprehensive CLIP-Seq experimentally supported miRNA-mRNA and miRNA-lncRNA interaction networks to date. We identified ∼10 000 ceRNA pairs from CLIP-supported miRNA target sites. By combining 13 functional genomic annotations, we developed miRFunction and ceRNAFunction web servers to predict the function of miRNAs and other ncRNAs from the miRNA-mediated regulatory networks. Finally, we developed interactive web implementations to provide visualization, analysis and downloading of the aforementioned large-scale data sets. This study will greatly expand our understanding of ncRNA functions and their coordinated regulatory networks.
INTRODUCTION
Eukaryotic genomes encode thousands of short and long non-coding RNAs (ncRNAs), such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), pseudogenes and circular RNAs (circRNAs). These RNA molecules are emerging as key regulators of diverse cellular processes, including proliferation, apoptosis, differentiation and the cell cycle (1–7).
Although many studies that address these ncRNAs have focused on defining their protein-coding gene regulatory functions, increasing numbers of researchers are assessing the regulatory interactions between ncRNAs classes, as well as the relationships between RNA-binding proteins (RBP) and ncRNAs. Several well-characterized lncRNAs (e.g. HOTAIR) exert their functions cooperatively with RBPs (e.g. EZH2) in cancers (8,9). Multiple classes of ncRNAs (lncRNAs, circRNAs, pseudogenes) and protein-coding mRNAs function as key competing endogenous RNAs (ceRNAs) and ‘super-sponges’ to regulate the expression of mRNAs in plants and mammalian cells (4,6,7,10–14). However, the understanding of ceRNA mechanisms and its consequences are in their infancy, and further experimental evidences and large-scale bioinformatic efforts for ceRNAs are needed. Despite these intriguing studies of individual miRNA-ncRNA and protein–RNA interactions, generalizing these findings to thousands of RNAs remains a daunting challenge.
Recent advances in high-throughput sequencing of immunoprecipitated RNAs after cross-linking (CLIP-Seq, HITS-CLIP, PAR-CLIP, CLASH, iCLIP) provide powerful ways to identify biologically relevant miRNA-target and RBP–RNA interactions (5,15,16). The application of CLIP-Seq methods has reliably identified Argonaute (Ago) and other RBP binding sites (5,15,16). We and others have used CLIP-seq data generated from HEK293 cells to characterize miRNA-mRNA and miRNA-lncRNA interactions (17–21). With the increasing amount of CLIP-Seq data available, there is a great need to integrate these large-scale data sets to explore the miRNA-pseudogene, miRNA-circRNA and protein–RNA interactions and to further construct ceRNA regulatory networks involving mRNAs and ncRNAs.
To facilitate the annotation, visualization, analysis and discovery of these interaction networks from large-scale CLIP-Seq data, we have updated starBase (17) to version 2.0 (starBase v2.0) (Figure 1). In starBase v2.0, we performed a large-scale integration of public RBP binding sites generated by high-throughput CLIP-Seq technology and provided the most comprehensive RBP data set for various cell types that are presently available. By analyzing millions of Ago and other RBP binding sites, we constructed the most comprehensive miRNA-lncRNA, miRNA-pseudogene, miRNA-circRNA, miRNA-mRNA and protein–RNA interaction networks.
Figure 1.

A system-level overview of the starBase v2.0 core framework. A total of 108 data sets of CLIP-seq experiments were compiled to achieve various RBP target sites. Interactions between miRNAs and target genes were predicted and used to construct miRNA-mediated ceRNA networks. Functional predictions of miRNAs and associated genes were achieved by enrichment analysis of 13 functional genomic annotations. All results generated by starBase were deposited in MySQL relational databases and displayed in the visual browser and web pages.
MATERIALS AND METHODS
Integration of Ago and other RBP binding sites from published CLIP data
HITS-CLIP, PAR-CLIP, iCLIP and CLASH data were retrieved from the Gene Expression Omnibus (22), the supplementary data of original references or directly from authors on request (Supplementary Table S1). Although Ago PAR-CLIP raw data were preprocessed with the FASTX-Toolkit v0.0.13 and reanalyzed using PARalyzer v1.1 (23), other CLIP-identified binding sites clusters/peaks were used directly. All binding sites coordinates were converted to hg19, mm9/mm10 and ce6/ce10 assemblies, respectively, by using the UCSC LiftOver Tool (24).
Ago CLIP-supported miRNA target prediction from public database
Conserved miRNA families were defined as those labeled with ‘highly conserved’ or ‘conserved’ in TargetScan Release 6.2 (25). miRNA IDs from miRBase Release 20 were used (26). Genomic coordinates of these conserved miRNAs target sites predicted by TargetScan (25), miRanda/mirSVR (27), PITA (28), Pictar 2.0 (19) and RNA22 (29) were collected and converted to hg19, mm9/mm10 and ce6/ce10 assemblies using LiftOver, respectively. The resulting coordinates were intersected with the previously described Ago CLIP clusters using BEDTools v2.16.2 (30). The target sites that overlap with any entry of the Ago CLIP clusters were considered as CLIP-supported sites.
MicroRNA target scanning in annotated transcripts
Human gene annotations were acquired from GENCODE v17 (31). Protein-coding transcripts were defined as those with ‘protein_coding’ gene biotype and ‘protein_coding’ transcript biotype. The lncRNAs transcripts were defined as those with ‘processed_transcript’, ‘lincRNA’, ‘3prime_overlapping_ncrna’, ‘antisense’, ‘non_coding’, ‘sense_intronic’ or ‘sense_overlapping’ gene biotype. Small non-coding RNA (sncRNA) transcripts were defined as those with ‘snRNA’, ‘snoRNA’, ‘rRNA’, ‘Mt_tRNA’, ‘Mt_rRNA’, ‘misc_RNA’ or ‘miRNA’ gene biotype. Pseudogene transcripts were defined as those with ‘polymorphic_pseudogene’, ‘pseudogene’, ‘IG_C_pseudogene’, ‘IG_J_pseudogene’, ‘IG_V_pseudogene’, ‘TR_V_pseudogene’ or ‘TR_J_pseudogene’ gene biotype.
Mouse and Caenorhabditis elegans gene annotations were extracted from Ensembl Gene Release 72 and LiftOver to mm9/mm10 and ce6/ce10, respectively. Protein-coding, lncRNAs, sncRNAs and pseudogenes were classified using a similar method. Human, mouse and C. elegans circRNA annotations were downloaded from circBase v0.1 (6).
These transcripts were scanned to find conserved miRNAs target sites using miRanda v3.3a with the ‘-strict’ parameter. The target sites that overlap with any entry of the aforementioned AGO CLIP clusters were considered as the CLIP-supported target sites.
Identification of ceRNA pairs with hypergeometric test
A hypergeometric test (14) is executed for each ceRNA pair separately, which is defined by four parameters: (i) N is the total number of miRNAs used to predict targets; (ii) K is the number of miRNAs that interact with the chosen gene of interest; (iii) n is the number of miRNAs that interact with the candidate ceRNA of the chosen gene; and (iv) c is the common miRNA number between these two genes. The test calculates the P-value by using the following formula:
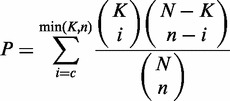 |
Multiple miRNAs belonging to the same family were combined into one, and the hypergeometric test counted every miRNA family only once, even if it had multiple binding sites at the same 3′-UTR of protein-coding genes or transcript of non-coding genes. All P-values were subject to false discovery rate (FDR) correction (32).
Enrichment analysis for functional terms
GO ontology data (33) for the NCBI RefSeq genes were downloaded from the NCBI ftp site. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (34) were downloaded from the KEGG database. The protein analysis through evolutionary relationships (PANTHER) pathways was downloaded from the PANTHER database (35). The Reactome (36) and other pathways were downloaded from the molecular signatures database (MSigDB) (37). Enrichment analysis for these pathways in the data set was determined using a hypergeometric test with Bonferroni and FDR correction (32).
Other RBP binding sites in annotated transcripts
The aforementioned RBP CLIP clusters were used to intersect with the coordinates of all annotated transcripts to find their RBP binding sites.
Other annotation data sets
All refSeq genes were downloaded from the UCSC bioinformatics Web sites (38). Other known ncRNAs were downloaded from the Ensembl database (39) or the UCSC Web sites (38) The human (UCSC hg19), mouse (UCSC mm9/mm10) and C. elegans (UCSC ce6/ce10) genome sequences were downloaded from the UCSC bioinformatics Web sites (38).
DATABASE CONTENT
The genome-wide binding map of Ago and other RBPs
To depict a comprehensive binding map of Ago and other RBP, we integrated 108 published CLIP-seq data generated from various tissues or cell lines under different treatments in 37 independent studies (detailed in ‘Materials and Methods’, Supplementary Table S1). For the Ago protein, a total of 1 007 618, 26 833 and 4842 unique binding site clusters were compiled in human, mouse and C. elegans, respectively (Table 1). These clusters were used in the following analysis to obtain CLIP-supported miRNA target sites of high confidence. Millions of binding site clusters of 42 other RBPs were also achieved (Supplementary Table S1 and Table 1).
Table 1.
The data sets that are incorporated into starBase v2.0
| Species | Experiments | RBPs | Cell lines/ tissues | ABSs | RBSs | miRNA-mRNA | miRNA-ncRNA | ceRNA | protein–RNA |
|---|---|---|---|---|---|---|---|---|---|
| Human | 85 | 36 | 18 | 1 007 618 | 8 206 884 | 423 975 | 35 459 | 11 439 | 242 017 |
| Mouse | 21 | 11 | 16 | 26 833 | 1 857 199 | 64 749 | 234 | 829 | 51 542 |
| C. elegans | 2 | 2 | 2 | 4842 | 1360 | 12 883 | 140 | 2 | 411 |
These statistics show the numbers of sequencing experiments (CLIP-Seq), RNA-binding proteins (RBPs) covered in these experiments, cell lines or tissues used in these experiments, Ago binding sites (ABSs), other RNA-binding protein binding sites (RBSs), miRNA-mRNA interactions, miRNA-ncRNA interactions, ceRNA pairs and protein–RNA interactions that are incorporated into starBase. These data are from three organisms: human (hg19), mouse (mm9) and C. elegans (ce6).
The annotation and identification of miRNA-mRNA and miRNA-ncRNA interactions
To inspect genome-wide interactions between miRNAs and their target genes, we retrieved the conserved miRNA target sites predicted by five algorithms (TargetScan, miRanda, Pictar2, PITA and RNA22) from public databases, which were intersected with the aforementioned Ago CLIP clusters to gain CLIP-supported sites. Using this approach, we characterized ∼500 000 interactions between 818 conserved miRNAs and 20 480 protein-coding genes.
We also investigated the potential regulatory relationships between miRNAs and non-coding RNAs. We performed conserved miRNA target site scanning on the transcripts of lncRNAs, sncRNAs, pseudogenes and circRNAs using miRanda, and filtered the resulting candidates with the previously described Ago CLIP clusters. Although less than CLIP-supported miRNA-mRNA interactions, the thousands of CLIP-supported miRNA-ncRNA interactions suggested that miRNAs might regulate other ncRNAs as well.
The annotation and identification of miRNA-mediated ceRNA regulatory networks
To construct and characterize the miRNA-mediated ceRNA network, a workflow was developed to identify the ceRNA pairs (Figure 1). First, CLIP-supported miRNA-mRNA, miRNA-lncRNA, miRNA-circRNA and miRNA-pseudogene interactions were combined. Next, hypergeometric test was used to predict ceRNA pairs among mRNAs, lncRNAs, circRNAs and pseudogenes. Finally, all ceRNA pairs with FDR<0.05 were imported into mySQL database and displayed in a web page. In this study, we identified approximately 10 000 ceRNA pairs from CLIP-supported miRNA target sites. Surprisingly, many nodes of ceRNA networks are lncRNAs, circRNAs and pseudogenes. Several experimentally validated ceRNAs were recaptured in our starBase v2.0, e.g. PTEN ceRNA: DCBLD2 (P < 0.001), JARID2 (P < 0.005), LRCH1 (P < 0.00005), TNRC6A (P < 0.00005) (12–14).
WEB INTERFACE
The web-based exploration of miRNA-mRNA, miRNA-ncRNA and protein–RNA regulatory relationships
Multiple web interfaces are applied to display three types of regulatory relationships. As an example, we explore miRNA-mRNA interactions to introduce the platform application. In the query page of miRNA-mRNA interactions, users can enter a gene symbol and select one miRNA to browse their relationships. The number of supporting experiments can be adjusted to control the stringency of the predictions. All relationships will be displayed in the results page if users do not submit a constraint. Once users click on a non-zero number in the table, more details are shown. For instance, users can click the target location to link to the deepView genome browser (17) and view data across the entire genome (Supplementary Figure S1). More information about the platform application is described in the relevant web interfaces.
The web-based exploration of ceRNA regulatory networks
In the query page, users enter a gene symbol that is used for ceRNAs prediction. The option of ‘minimum common miRNA number’ denotes the minimum number of miRNAs shared by the input gene and its ceRNA candidates. In the results page, users can click on one of the tabular FDR values for details.
The web-based functional annotation of genes from miRNA-mediated regulatory networks
For miRNA function predictions, there are five options on the query page, and the option ‘Select one or multiple microRNAs’ is required. In contrast from the options earlier in text, it allows users to select one or more miRNAs in the drop-down list. The results page shows the enrichment analysis for 13 functional prediction categories. The running parameters, selected miRNA target genes and every outcome in the 13 categories of function prediction are available for users to download. For ceRNA functions prediction, the query page also presents users five options and gene symbol is required to enter. The results page is similar to the miRNA function predictions page.
EXAMPLE APPLICATIONS
In the following section, example applications of starBase v2.0 are illustrated.
The targetome of hsa-miR-21-5p
Assume that we are interested in the targetome of hsa-miR-21-5p. Given the constraints requiring target sites to have a number of supporting experiments no less than one and to be predicted by at least three of the five programs (Supplementary Figure S2A), our platform returns 173 CLIP-supported hsa-miR-21-5p target sites in 155 protein-coding genes among which ZNF367, RHOB and PELI1 rank top three by read numbers (Supplementary Figure S2B). These results coincide with data in an experimentally validated database named miRTarBase (40), suggesting that these genes are likely targeted by hsa-miR-21-5p.
The identification of ‘super-sponges’ of miRNAs
Inspired by the observation that CDR1as circRNA (6,7) acts as a miR-7 super-sponge that contains multiple target sites from the same miRNA at the same transcript or 3′-UTR, we tested whether the other class of ncRNAs and protein-coding genes hosted in our database also can act as miRNA super-sponges. We can recapitulate the known CDR1as circRNA as a miR-7 super-sponge using the miRNA-circRNA interactions web page (Supplementary Figure S3A and Supplementary Data). The results page sorted by the number of miRNA target sites showed that CDR1as circRNA contains 52 miR-7 target sites overlapped with CLIP-Seq data (Supplementary Figure S3B). The same strategy was applied to search potential super-sponges among mRNAs, lncRNAs and pseudogenes, resulting in tens of candidates, such as XIST and HOXD-AS1 lncRNA genes and ONECUT2 and CDK6 mRNAs (Supplementary Figure S3C).
ceRNAs of the oncogenes
Recently, the ceRNA hypothesis has been proposed (3) and efforts have been made to decipher the roles of ceRNA cross talk in regulating cancer-associated genes such as tumor suppressor PTEN (12–14). Requiring a minimum common miRNA number of ten and a FDR threshold of 0.05 on the ‘ceRNA Network’ web page (Supplementary Figure S4A), our platform produced a ceRNA network involving PTEN and 123 genes, among which the published PTEN ceRNAs LRCH1, TNRC6A and SMAD5 (12–14) were recaptured (Supplementary Figure S4B).
We were also able to predict a batch of other cancer-associated genes that were entangled within the highly sophisticated networks of ceRNAs. For example, NFIB, an oncogene upregulated in small cell lung cancer (41) and estrogen receptor-negative breast cancer (42), was found in multiple ceRNA pairs with other well-described cancer-related genes, such as MLL and KDSR (Supplementary Figure S4C).
CONCLUSIONS
By analyzing a large set of Ago and RBP binding sites derived from all available CLIP-Seq experimental techniques (PAR-CLIP, HITS-CLIP, iCLIP, CLASH), we have shown extensive and complex RNA–RNA and protein–RNA interaction networks.
Compared with the previous version of starBase (v1.0) and other databases, the distinctive features of starBase v2.0 include the following: (i) starBase v2.0 is the first database that provides the miRNA-pseudogene interaction networks; (ii) starBase v2.0 drafts the first interaction maps between miRNAs and circRNAs; (iii) unlike other databases or tools (12,14,43) that predict ceRNA regulatory networks using computationally predicted miRNA targets, starBase v2.0 provides an enhanced resolution to determine ceRNA functional networks based on miRNA-target interactions overlapping with high-throughput CLIP-Seq data; (iv) starBase v2.0 provides the most comprehensive miRNA-lncRNA interactions to date; and (v) starBase v2.0 provides a variety of interfaces and graphic visualizations to facilitate analysis of the massive and heterogeneous CLIP-Seq, RBP binding sites, miRNA targets and ceRNA regulatory networks in normal tissues and cancer cells.
FUTURE DIRECTIONS
As CLIP-Seq technology is applied to a broader set of species, cell lines, tissues, conditions and RBPs, we will continuously maintain and update the database. starBase will continue to expand the storage space and improve the computer server performance for storing and analyzing these new data, and improve the database to accept new user data uploads. In addition, we intend to integrate the cancer genomics data from the Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) into starBase to improve our understanding of miRNA-mediated regulatory networks in developmental, physiological and pathological processes.
AVAILABILITY
starBase v2.0 is freely available at http://starbase.sysu.edu.cn/. The starBase data files can be downloaded and used in accordance with the GNU Public License and the license of primary data sources.
FUNDING
Funding for open access charge: Ministry of Science and Technology of China, National Basic Research Program [No. 2011CB811300].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Ministry of Science and Technology of China, National Basic Research Program [No 2011CB811300]; the National Natural Science Foundation of China [31230042, 30900820, 81070589, 31370791]; the funds from Guangdong Province [S2012010010510, S2013010012457]; Project of Science and Technology New Star in ZhuJiang Guangzhou city [2012J2200025]; Fundamental Research Funds for the Central Universities [2011330003161070]; China Postdoctoral Science Foundation [200902348]. This research is supported in part by the Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team.
REFERENCES
- 1.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yates LA, Norbury CJ, Gilbert RJ. The long and short of microRNA. Cell. 2013;153:516–519. doi: 10.1016/j.cell.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konig J, Zarnack K, Luscombe NM, Ule J. Protein-RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genet. 2011;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 6.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 7.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 8.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl Acad. Sci. USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007;39:1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 12.Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, Rajbhandari P, Bansal M, Guarnieri P, Silva J, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147:370–381. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darnell RB. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ascano M, Hafner M, Cekan P, Gerstberger S, Tuschl T. Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA. 2012;3:159–177. doi: 10.1002/wrna.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khorshid M, Rodak C, Zavolan M. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011;39:D245–D252. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012;40:D180–D186. doi: 10.1093/nar/gkr1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jalali S, Bhartiya D, Lalwani MK, Sivasubbu S, Scaria V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One. 2013;8:e53823. doi: 10.1371/journal.pone.0053823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paraskevopoulou MD, Georgakilas G, Kostoulas N, Reczko M, Maragkakis M, Dalamagas TM, Hatzigeorgiou AG. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–D245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 29.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 30.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 33.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–DD114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37:D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, Rosenbloom KR, Rhead B, Raney BJ, Pohl A, Pheasant M, et al. The UCSC Genome Browser Database: update 2009. Nucleic Acids Res. 2009;37:D755–D761. doi: 10.1093/nar/gkn875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–DD169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, et al. Nuclear factor I/B is an oncogene in small cell lung cancer. Gene Dev. 2011;25:1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon HG, Hwang KT, Kim JA, Kim HS, Lee MJ, Jung EM, Ko E, Han W, Noh DY. NFIB is a potential target for estrogen receptor-negative breast cancers. Mol. Oncol. 2011;5:538–544. doi: 10.1016/j.molonc.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu K, Yan Z, Li Y, Sun Z. Linc2GO: a human LincRNA function annotation resource based on ceRNA hypothesis. Bioinformatics. 2013;29:2221–2222. doi: 10.1093/bioinformatics/btt361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.