


Abstract
In recent years, human regulatory SNPs (rSNPs) have been widely studied. Here, we present database rSNPBase, freely available at http://rsnp.psych.ac.cn/, to provide curated rSNPs that analyses the regulatory features of all SNPs in the human genome with reference to experimentally supported regulatory elements. In contrast with previous SNP functional annotation databases, rSNPBase is characterized by several unique features. (i) To improve reliability, all SNPs in rSNPBase are annotated with reference to experimentally supported regulatory elements. (ii) rSNPBase focuses on rSNPs involved in a wide range of regulation types, including proximal and distal transcriptional regulation and post-transcriptional regulation, and identifies their potentially regulated genes. (iii) Linkage disequilibrium (LD) correlations between SNPs were analysed so that the regulatory feature is annotated to SNP-set rather than a single SNP. (iv) rSNPBase provides the spatio-temporal labels and experimental eQTL labels for SNPs. In summary, rSNPBase provides more reliable, comprehensive and user-friendly regulatory annotations on rSNPs and will assist researchers in selecting candidate SNPs for further genetic studies and in exploring causal SNPs for in-depth molecular mechanisms of complex phenotypes.
INTRODUCTION
Similar to the effect of SNPs on protein structure and function, the impact of SNPs on gene regulation has been considered for decades (1,2) and widely studied in recent years. Some recent findings imply an important role for regulatory SNPs (rSNPs) in the molecular mechanisms of complex diseases and other complex biological processes. For example, recent studies have shown that the majority of published GWAS-significant SNPs are intergenic or intronic (3), indicating that many risk SNPs may affect phenotypes in a non-coding manner, such as impacting gene regulation. Furthermore, experimental data generated by the Encyclopedia of DNA Elements (ENCODE) project have revealed the overlap between GWAS SNPs or SNPs in strong linkage disequilibrium (LD) with GWAS SNPs and regulatory regions (4,5).
In the past decade, efforts have been made to annotate the regulatory feature of SNPs in a genome scope to facilitate relevant studies. SNPs within transcription factor binding sites (TFBSs) or that affect TF-DNA binding affinity were predominantly considered rSNPs (6–10). Most previous rSNP databases have identified rSNPs with reference to computationally predicted regulatory elements, such as predicted TFBSs (rSNP_Guide) (6), predicted promoters (SNP@Promoter) (11), regions affecting RNA splicing (ssSNPTarget) (12), miRNA target regions (PolymiRTS (13,14), Patrocles (15) and miRNASNP (16)), or multiple types of regulatory elements (FESD (17), F-SNP (18), FASTSNP (19) and SNP Function Portal (20)). These databases have supported functional SNP studies but did not introduce high-throughput experimentally identified regulatory elements into the functional analysis of SNPs.
The ENCODE project studies regulatory elements from data of systematic high-throughput experiments (21) and has generated a significant amount of data for identifying various types of functional elements in the human genome sequence (22). Different types of regulatory elements may correspond with different regulation processes; for example, regulatory elements that characterize open chromatin (such as DNase I hypersensitive sites, DHSs) are associated with transcriptional regulation (23), and some other regulatory elements (such as TFBS (24), histone modification-marked sequences (25) and DNA methylation sequences (26)) are also involved in this process. Specifically, experimentally identified chromosome interacting regions provide information on distal transcriptional regulation (27). This type of regulation is difficult, if not impossible, to be predicted in silico. Furthermore, RNA-binding protein (RBP) associated regions identified by RNA immunoprecipitation (RIP) are related to the process of post-transcriptional regulation (28). The database RegulomeDB (29) utilizes ENCODE-generated experimental data that characterize chromatin accessibility. These experimental data and two other types of data (predicted regulatory elements and experimental eQTL evidence) were integrated into a cataloging and heuristic scoring system to represent the functional confidence of a variant. However, similar to the majority of previous databases, RegulomeDB predominantly focuses on SNPs involved in a single type of regulation. Indeed, there is currently no database that focuses on regulatory elements involved in distal transcriptional regulation. Additionally, in previous databases, annotations have been performed on single SNPs, and correlations between SNPs have not been well considered.
rSNPBase is an rSNP database that annotates the regulatory features of SNPs in the human genome with reference to experimentally supported regulatory elements. Regulatory elements that reflect proximal transcriptional regulation, distal transcriptional regulation and RBP-mediated post-transcriptional regulation were acquired from ENCODE data and then utilized to identify rSNPs. The corresponding genes potentially regulated by these regulatory elements were also analysed. Considering the importance of miRNA-mediated post-transcriptional regulation, rSNPs in mature miRNAs are also included in rSNPBase, and their relevant regulated genes were analysed with reference to experimentally supported miRNA-targeted gene databases. rSNPBase also includes non-rSNPs in strong LD (r2 > 0.8) with rSNPs. Furthermore, rSNPBase provides spatio-temporal labels and experimental eQTL labels of SNPs to further facilitate researchers in acquiring the exact data in which they are interested. rSNPBase is targeted to provide a more reliable, comprehensive and user-friendly regulatory annotation on rSNPs to facilitate researchers in selecting candidate SNPs for further genetic studies (especially QTL studies), identifying causal variants of certain phenotypes, and exploring in-depth molecular mechanisms.
DATA CONTENT AND DATA PROCESSING
Data content
rSNPBase includes rSNPs, LD proxies of rSNPs and genes that are potentially regulated by rSNPs. Experimentally supported regulatory elements were collected and utilized to annotate the regulatory feature of rSNPs. Regulation-related spatio-temporal information and experimental eQTL evidences are employed as data labels for the included SNPs. The data for rSNPBase (as of 1 August 2013) are shown in Table 1.
Table 1.
Data content of rSNPBase as of 1 August 2013
| Data type | Data description | Data statistics |
|---|---|---|
| rSNPs | Totala | 22 846 898 |
| Involved in proximal transcriptional regulation | 7 081 726 | |
| Involved in distal transcriptional regulation | 9 720 393 | |
| Involved in RBP-mediated post-transcriptional regulation | 15 782 798 | |
| Involved in miRNA-mediated post-transcriptional regulation | 928 | |
| LD proxies (non-rSNPs)b | 2 281 874 | |
| rSNP-related genes | 56 869 | |
| Spatio-temporal labels | Cell lines | 363 |
| Tissues | 74 | |
| Developmental stages | 5 | |
| eQTL labels | SNP-gene pairs | 2 428 727 |
aAn rSNP may be involved in multiple types of regulation.
bIn rSNPBase, SNPs (both rSNP and non-rSNP) in strong LD (r2 > 0.8) with an rSNP are defined as LD proxies. Here we only count the number of non-rSNPs of the LD proxies.
Data processing
Genome-wide human SNPs and genes were filtered and mapped by experimentally validated regulatory elements, which are involved in four types of regulation (proximal and distal transcriptional regulation and RBP-mediated and miRNA-mediated post-transcriptional regulation). As shown in Figure 1, rSNPBase hosts rSNPs that are within regulatory elements. Element-regulated genes were also analysed and hosted as genes potentially regulated by rSNPs. For each rSNP, SNPs (both rSNP and non-rSNP) in strong LD (r2 > 0.8) were analysed. Finally, spatio-temporal labels and eQTL labels were generated and labeled on all included SNPs.
Figure 1.

Data processing and data content of rSNPBase.
Generating regulatory elements involved in different types of regulation
Processed ENCODE production data that are associated with chromatin accessibility (including open chromatin, histone-marked regions, CpG islands and TFBSs), chromatin interactions and RBPs were downloaded from the UCSC Genome Browser (hg 19) (30) (http://genome.ucsc.edu/ENCODE/downloads.html) to generate experimentally validated regulatory elements involved in proximal and distal transcriptional regulation and RBP-mediated post-transcriptional regulation (the ENCODE data that are utilized in rSNPBase are shown in Supplementary Table S1). The same type of data was integrated, and redundant data were pruned. Specific for histone modification data, only regions marked by active-associated histones (H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27ac, H3K36me3, H3K79me2, H4K20me1 and H3K9me1) (31–33) were included in rSNPBase. Mature miRNAs were collected from miRBase (release 20) (34) as regulatory elements involved in miRNA-mediated post-transcriptional regulation.
Analysing rSNPs and corresponding genes involved in different types of regulation
Human SNPs from dbSNP (build 137) (35) were filtered using experimentally validated regulatory elements based on the genomic location to identify rSNPs. According to the involved regulation types, the regulatory element-filtered SNPs are defined as proximal transcriptional rSNPs (proximal-rSNPs), distal transcriptional rSNPs (distal-rSNPs), RBP-mediated post-transcriptional rSNPs (RBP-rSNPs) and miRNA-mediated post-transcriptional rSNPs (miRNA-rSNPs), all of which are termed rSNPs in rSNPBase. Human genes from Ensembl (GRCh37. P11) (36) were mapped by regulatory elements or analysed with reference to experimentally supported databases to identify genes corresponding with rSNPs.
Proximal transcriptional regulation is related to regulatory elements associated with DNA accessibility, and this type of regulation is largely dependent on the genomic proximity of the regulatory elements and transcript start site (TSS). Therefore, SNPs filtered by relevant regulatory elements were re-filtered by upstream and 5′ UTR regions of genes. The final double-filtered SNPs are defined as proximal-rSNPs, and their corresponding genes were identified with reference to their consequence types, which were cataloged by Ensembl (36). Distal transcriptional regulation-related regulatory elements were analysed from the ENCODE data of chromatin interactions. This type of data provides interacted TSS-fragment pairs that are distant in sequence but relatively close in space. For each TSS-fragment pair, the distal-rSNPs were identified from the distal fragment, and their corresponding genes were identified from the TSSs located in the paired region. Sometimes both interacting regions contain TSSs, rSNPs were then generated from both regions correspondingly. DNA regulatory elements related to RBP-mediated post-transcriptional regulation were mapped from RBP-associated RNA sequences generated by ENCODE. SNPs falling within these regulatory elements are defined as RBP-rSNPs. Genes that were mapped by RBP-associated RNA sequences correspond with this type of rSNP. SNPs within mature miRNAs recorded by miRBase are defined as miRNA-rSNPs and correspond with miRNA-targeted genes, which were obtained from the experimentally supported miRNA-targeted gene database miR2Disease (37) and miRTarBase (38).
Analysing LD proxies
Because of the genetic correlation between nearby SNPs, besides the analysis of a single SNP, rSNPBase also analysed LD correlations between SNPs. In the genome scale, the set of SNPs (both rSNPs and non-rSNPs) that are in strong LD (r2 > 0.8) with the rSNPs are defined as LD proxies of rSNPs. The LD data were compiled from both merged HapMap phases I + II + III genotype data for markers that are up to 200 kb apart (39) and integrated the 1000 Genomes project phase I release data (40,41), which were downloaded from the International HapMap Consortium and MaCH (42). Data from all populations that the two projects are involved in were all utilized to perform LD analyses.
Adding data labels
Due to the importance of eQTL evidence for deciphering gene regulation and the spatio-temporal specificity of gene regulation, rSNPBase provides eQTL labels and spatio-temporal labels for the included SNPs. eQTL attributes were collected from experimentally supported eQTL databases (43–45) and the eQTL browser (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/) (46–52) to provide association labels for SNPs. Tissue and developmental stage information were labeled according to cell type, from which regulatory elements were identified.
APPLICATIONS AND EXAMPLES
Data retrieving in rSNPBase could be SNP-centric or gene-centric. SNP-centric data retrieving is appropriate to analyse results from genetic studies, especially the results of high-throughput studies, and then provide evidence for further functional studies to identify causal SNPs and shed light on underlying molecular mechanisms. Gene-centric searches are useful in candidate SNP selections that are based on genes of interest. Specifically, rSNPBase provides various search options (such as regulation type, tissue and developmental stage, and eQTL evidence) for gene-centric searches in ‘Advanced search’ modules to facilitate data filtering.
Here, we present a process for data retrieval as an example. Jostins et al. (53) identified 110 SNPs that are significantly associated with inflammatory bowel disease. We acquired detailed descriptions of these SNPs from the GWAS catalog (3) (see http://www.genome.gov/GWASStudySNPS.cfm?id=6987 and Supplementary Table S1). The majority of these SNPs could not be mapped to a specific gene, which brings challenge for further functional studies (e.g., studies to identify casual variants and explore disease mechanisms). We retrieved the 110 SNPs via the ‘List search’ module in rSNPBase. The search results (http://rsnp.psych.ac.cn/result) showed that 87 of these SNPs were defined as ‘rSNP’, and 15 of the 23 non-rSNPs were defined as LD proxies of rSNPs. Additionally, 86 of the 110 SNPs had been identified as eQTLs in previous studies and nearly all of the eQTLs (83 of 86) were shown to be rSNPBase-defined rSNPs or LD proxies of rSNPs (Figure 2A). The concordance between our functional annotation and previous association analyses indicates the reliability of the annotation procedures. It also supports the hypothesis that risk SNPs may affect inflammatory bowel disease by altering gene expression. To facilitate further in-depth mechanism studies, detailed annotations, which are useful to propose hypothesis and drive new findings, are shown on the ‘rSNP report’ page (Figure 2B). Users can obtain a systematic view of each rSNP, its LD proxies, potential target genes and a detailed presentation of rSNP-related regulatory elements. For each regulatory element, the spatio-temporal labels on tissue and developmental stage are provided for convenient study design.
Figure 2.
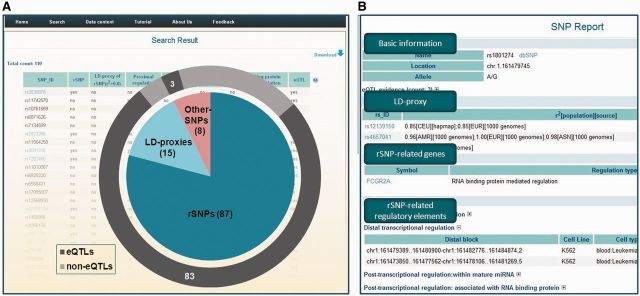
An example of data retrieving process.
We also searched the rSNPBase by significant SNPs identified by all published GWASs (collected via the GWAS (NHGRI) catalog as of 27 July 2013) (3). The results showed that among the 10 992 GWAS-identified significant SNPs, 6058 were rSNPs and 2361 were LD proxies of rSNPs. These rSNPs and LD proxies are likely to reveal regulatory mechanisms underlying diseases or other phenotypes. The systematic and detailed functional annotations in rSNPBase are expected to provide appropriate and powerful data references for the follow-up study of the significant SNPs reported by the published GWASs.
CONCLUSIONS
rSNPBase is a database that functionally annotates the regulatory features of SNPs in the human genome with reference to experimentally supported regulatory elements. It identifies rSNPs and their corresponding regulated genes from four regulation types: proximal transcriptional regulation, distal transcriptional regulation, RBP-mediated post-transcriptional regulation and miRNA-mediated post-transcriptional regulation. It also analyses LD correlations between SNPs to annotate the regulatory feature to SNP-set rather than a single SNP. The spatio-temporal labels and experimental eQTL evidence provided for each SNP in rSNPBase. Predictably, the number of both human SNPs and experimentally supported regulatory elements will continue to increase. Therefore, we will update the rSNPBase periodically to include new data and data types. For data on distal transcriptional regulation and RBP-mediated post-transcriptional regulation, the relevant experiments in the ENCODE project are only in a pilot phase and involve minor cell lines or RBPs. We will follow their progress and update rSNPBase promptly in compliance with the ENCODE data release policy. In summary, rSNPBase provides functional annotations of rSNPs in a wide range of regulation types with up-to-date experimental evidences. rSNPBase is targeted to assist researchers to have a deep understanding of the regulatory features of SNPs, and to support further genetic and molecular mechanism studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Chinese Academy of Sciences/State Administration of Foreign Experts Affairs (CAS/SAFEA) International Partnership Program for Creative Research Teams [Y2CX131003]; the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-J-8); the Strategic Priority Research Program (B) of the Chinese Academy of Sciences [XDB02030002]; Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences; National Natural Science Foundation of China [81201046 and 81101545]. Funding for open access charge: The Chinese Academy of Sciences/State Administration of Foreign Experts Affairs (CAS/SAFEA) International Partnership Program for Creative Research Teams [Y2CX131003].
Conflict of interest statement. None declared.
REFERENCES
- 1.Vasiliev GV, Merkulov VM, Kobzev VF, Merkulova TI, Ponomarenko MP, Kolchanov NA. Point mutations within 663-666 bp of intron 6 of the human TDO2 gene, associated with a number of psychiatric disorders, damage the YY-1 transcription factor binding site. FEBS Lett. 1999;462:85–88. doi: 10.1016/s0014-5793(99)01513-6. [DOI] [PubMed] [Google Scholar]
- 2.Bienvenu T, Lacronique V, Raymondjean M, Cazeneuve C, Hubert D, Kaplan JC, Beldjord C. Three novel sequence variations in the 5′ upstream region of the cystic fibrosis transmembrane conductance regulator (CFTR) gene: two polymorphisms and one putative molecular defect. Hum. Genet. 1995;95:698–702. doi: 10.1007/BF00209490. [DOI] [PubMed] [Google Scholar]
- 3.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponomarenko JV, Merkulova TI, Orlova GV, Fokin ON, Gorshkova EV, Frolov AS, Valuev VP, Ponomarenko MP. rSNP_Guide, a database system for analysis of transcription factor binding to DNA with variations: application to genome annotation. Nucleic Acids Res. 2003;31:118–121. doi: 10.1093/nar/gkg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molineris I, Schiavone D, Rosa F, Matullo G, Poli V, Provero P. Identification of functional cis-regulatory polymorphisms in the human genome. Hum. Mutat. 2013;34:735–742. doi: 10.1002/humu.22299. [DOI] [PubMed] [Google Scholar]
- 8.Andersen MC, Engstrom PG, Lithwick S, Arenillas D, Eriksson P, Lenhard B, Wasserman WW, Odeberg J. In silico detection of sequence variations modifying transcriptional regulation. PLoS Comput. Biol. 2008;4:e5. doi: 10.1371/journal.pcbi.0040005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macintyre G, Bailey J, Haviv I, Kowalczyk A. is-rSNP: a novel technique for in silico regulatory SNP detection. Bioinformatics. 2010;26:i524–i530. doi: 10.1093/bioinformatics/btq378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riva A. Large-scale computational identification of regulatory SNPs with rSNP-MAPPER. BMC Genomics. 2012;13(Suppl. 4):S7. doi: 10.1186/1471-2164-13-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim BC, Kim WY, Park D, Chung WH, Shin KS, Bhak J. SNP@Promoter: a database of human SNPs (single nucleotide polymorphisms) within the putative promoter regions. BMC Bioinform. 2008;9(Suppl. 1):S2. doi: 10.1186/1471-2105-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang JO, Kim WY, Bhak J. ssSNPTarget: genome-wide splice-site Single Nucleotide Polymorphism database. Hum. Mutat. 2009;30:E1010–E1020. doi: 10.1002/humu.21128. [DOI] [PubMed] [Google Scholar]
- 13.Bao L, Zhou M, Wu L, Lu L, Goldowitz D, Williams RW, Cui Y. PolymiRTS Database: linking polymorphisms in microRNA target sites with complex traits. Nucleic Acids Res. 2007;35:D51–D54. doi: 10.1093/nar/gkl797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebarth JD, Bhattacharya A, Chen A, Cui Y. PolymiRTS Database 2.0: linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic Acids Res. 2012;40:D216–D221. doi: 10.1093/nar/gkr1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiard S, Charlier C, Coppieters W, Georges M, Baurain D. Patrocles: a database of polymorphic miRNA-mediated gene regulation in vertebrates. Nucleic Acids Res. 2010;38:D640–D651. doi: 10.1093/nar/gkp926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong J, Tong Y, Zhang HM, Wang K, Hu T, Shan G, Sun J, Guo AY. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2012;33:254–263. doi: 10.1002/humu.21641. [DOI] [PubMed] [Google Scholar]
- 17.Kang HJ, Choi KO, Kim BD, Kim S, Kim YJ. FESD: a Functional Element SNPs Database in human. Nucleic Acids Res. 2005;33:D518–D522. doi: 10.1093/nar/gki082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee PH, Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–D824. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, Wang HH, Yao A, Chen YT, Hsu CN. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–W641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang P, Dai M, Xuan W, McEachin RC, Jackson AU, Scott LJ, Athey B, Watson SJ, Meng F. SNP Function Portal: a web database for exploring the function implication of SNP alleles. Bioinformatics. 2006;22:e523–e529. doi: 10.1093/bioinformatics/btl241. [DOI] [PubMed] [Google Scholar]
- 21.Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 22.Consortium EP, Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dekker J. Gene regulation in the third dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl Acad. Sci. USA. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson K. 1000 genomes: a world of variation. Circ. Res. 2011;108:534–536. doi: 10.1161/RES.0b013e31821470fe. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia K, Shabalin AA, Huang S, Madar V, Zhou YH, Wang W, Zou F, Sun W, Sullivan PF, Wright FA. seeQTL: a searchable database for human eQTLs. Bioinformatics. 2012;28:451–452. doi: 10.1093/bioinformatics/btr678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–262. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.John L, Jeffrey T, Mike S, Rebecca P, Edmund L, Saboor S, Richard H, Gary W, Fernando G, Nancy Y, et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, et al. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 48.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, et al. Genetics and beyond–the transcriptome of human monocytes and disease susceptibility. PLoS ONE. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]