


Abstract
Microbial genome sequencing is one of the longest-standing areas of biological database development, but high-throughput, low-cost technologies have increased its throughput to an unprecedented number of new genomes per year. Several thousand microbial genomes are now available, necessitating new approaches to organizing information on gene function, phylogeny and microbial taxonomy to facilitate downstream biological interpretation. MetaRef, available at http://metaref.org, is a novel online resource systematically cataloguing a comprehensive pan-genome of all microbial clades with sequenced isolates. It organizes currently available draft and finished bacterial and archaeal genomes into quality-controlled clades, reports all core and pan gene families at multiple levels in the resulting taxonomy, and it annotates families’ conservation, phylogeny and consensus functional information. MetaRef also provides a comprehensive non-redundant reference gene catalogue for metagenomic studies, including the abundance and prevalence of all gene families in the >700 shotgun metagenomic samples of the Human Microbiome Project. This constitutes a systematic mapping of clade-specific microbial functions within the healthy human microbiome across multiple body sites and can be used as reference for identifying potential functional biomarkers in disease-associate microbiomes. MetaRef provides all information both as an online browsable resource and as downloadable sequences and tabular data files that can be used for subsequent offline studies.
INTRODUCTION
High-throughput sequencing has become an invaluable tool for scientific investigation, and computational resources are still adapting to the ubiquity and scale of modern genomic resources. This has been particularly true for microbial sequencing, where new methods must be developed to organize tens of thousands of genomes, hundreds of isolates per species and a pan-genome that comprises millions of gene families. Many databases catalogue microbial sequences, but few yet place these sequences within their phylogenetic and functional context.
For example, many microbial clades have now been shown to have surprisingly large pan-genomes in contrast to the size of their core genomes (1). Any one Escherichia coli isolate typically contains ∼4700 genes, but only some 2000 of these are found in all E. coli, and the pan-genome selected from all strains in the species now greatly exceeds 8000 genes (2). Yet current microbial genome resources rarely indicate the core or pan-genomes of a clade of interest, nor do they conversely assess the phylogenetic distribution of individual gene families.
No comprehensive tool is thus available for interrogation of microbial clade-specific gene sequences, families or functional annotations. Here, we describe the algorithms, database and online interface for such a framework, initially detailing over 10 million genes from ∼2800 microbial genomes. All information is both browsable interactively through an online web interface and downloadable for offline analysis, and the underlying data are automatically updated every 6 months.
OVERVIEW OF THE METAREF DATABASE
MetaRef is a novel online resource reporting a systematic sequence-based catalogue of the diversity and characteristics of the gene repertoires of all microbial clades with available whole-genome sequence reference information. It identifies and reports each gene family present in at least one genome, organizing them into pan, core or marker families, reconciles their functional annotations when possible, and systematically surveys their presence in multiple body sites of the human microbiome.
MetaRef serves as a comprehensive and convenient resource for tasks in microbial genome investigations, comparative genomics, and quantitative microbial ecology and offers several features not provided by existing systems. The MetaRef microbial gene family system (defined at 80% full-length nucleotide identity) includes every annotated reading frame in currently sequenced genomes, including singleton and uncharacterized genes. The underlying gene family clustering algorithm is defined hierarchically so as to scale efficiently to the increasing number of sequenced genomes (currently >2800); many existing catalogues such as COG (3) and KEGG KOs (4) neither match this scale nor typically annotate more than a fraction of genes in any one genome. Unsupervised approaches such as KEGG OC (5), PHOG (6) and OMA (7) face scalability issues and are often only appropriate for finished genomes. The largest current databases, such as MBGD (8), eggNOG (9) and UniRef (10), extend these systems to all proteins in a larger number of input genomes, but uniformly at the cost of a loss of transferable information—all provide comprehensive definitions of microbial gene families, but none link these families to phylogenetic and functional information for downstream interpretation.
The novel resource provides information useful for at least two different classes of investigator. For microbiologists focusing on individual microbial clades (e.g. a genus or a species), MetaRef provides a pre-computed pan-genome comprising core families consistently present in the clade, marker families present uniquely in the clade, and pan families present only in some clade members. This constitutes a tree-of-life-wide resource for phylogenetic analysis (e.g. by aligning and concatenating MetaRef core families) with several additional features unique to the MetaRef system including consensus-based functional characterization and pre-computed conservation/diversity scores. Second, for investigators focusing on large-scale comparative genomics or ecology, MetaRef also provides a catalogue of pre-identified marker genes for microbiome taxonomic profiling (11), and the means to assess the relevance of any microbial gene families with respect to their symbiotic healthy relationship with the human body, as the system reports the abundance and prevalence of each family in each clade in six body sites of the healthy human microbiome integrating shotgun metagenomic data produced by the Human Microbiome Project (HMP) (12). No existing resources for microbial genomics provide both this level of clade-specific detail and whole-microbiome analysis.
MetaRef thus provides a new resource based on automatic and unsupervised complete gene clustering, processing a highly scalable number of sequenced microbial genomes. Both final and draft genomes are included in this process; open reading frame (ORF) calls are the only necessary annotations. The MetaRef web interface has been developed to provide a variety of clade-centric and gene family-centric interactive views to access and explore the sequenced microbial diversity. All MetaRef information, including sequence data, annotations and metagenomic mapping, is also available for download in plain text format for offline processing. While MetaRef provides novel aspects such as clade-specificity for the gene family system andmetagenomic surveys of each gene family considered, it also combines four features currently not present simultaneously in other resources: (i) it comprehensively processes all available microbial genomic data; (ii) it provides both online and downloadable sequence information; (iii) it efficiently scales to the increasing throughput of reference genomes; and (iv) it leverages functional and phylogenetic information to gain insights into each gene family.
DATABASE GENERATION, CONTENT AND DEFINITIONS
The MetaRef database and online resource are built upon publicly available microbial genomic and metagenomic information automatically acquired and processed on a regular basis (Figure 1). The computational pipelines we implemented process the ∼3000 microbial genomes available (from IMG (13)) with the associated taxonomic and gene information and the ∼3.5TB of shotgun metagenomic data of 757 human microbiome samples (from the HMP (14)). These pipelines include PhyloPhlAn (15) for taxonomic curation and a novel clustering pipeline for pan gene classification (11,16) (see Supplementary Methods). The second phase of computational processing comprises metagenome mapping using a Bowtie-based strategy (17), a novel taxonomic and phylogenetic tree visualization (GraPhlAn, publicly available at http://huttenhower.sph.harvard.edu/graphlan), and the derivation of consensus functional annotations. The MetaRef online interface is a dynamic website with a back-end MySQL relational database, developed using Django (version 1.4.3 https://www.djangoproject.com); (see Supplementary Methods for implementation details).
Figure 1.

MetaRef comprises automated processing of available microbial genomic information, downloadable phylogenetic and functional annotations, and the MetaRef web portal. The implemented computational pipeline first processes the available microbial reference genomes with the associated taxonomic metadata (currently from IMG) to produce the comprehensive MetaRef sequence database of all gene families. The database is then integrated with the available functional annotations and shotgun metagenomic data (currently from the HMP project) to provide clade-centric and family-centric views of the current microbial diversity.
The MetaRef system: definition of marker, core, and pan genes and families
The phylogenetic diversity within each microbial taxonomic clade is captured by cataloguing the overall repertoire of distinct ORFs contained in the pan-genome (18,19). MetaRef naming conventions refer to individual microbial ORF sequences as genes, and groups of genes related by some homology criterion as families. Pan genes are defined as ORFs present at least once in at least one genome of a clade of interest. Pan genes are usually grouped into homologous groups, called pan families, which capture a combination of paralogy, orthology and horizontal gene transfer relationships through sequence clustering requiring a full gene length similarity threshold (20). Among the pan genes forming a pan family, the ORF minimizing the dissimilarity with respect to all the other pan genes is called the pan centroid.
Some pan families are conserved within all organisms of a clade and are thus defined as core families (Figure 2). When a core family for a clade (say the Staphylococcus aureus species) is not conserved in its direct ancestor clade (in this case the genus Staphylococcus), it is of biological interest as a potentially conserved function distinguishing the clade from sibling clades (e.g. Staphylococcus epidermidis and other Staphylococcus spp.) and is called a crown family (Figure 2). A crown family can still be simultaneously present in unrelated branches of the taxonomy, possibly as a consequence of evolutionarily successful horizontal gene transfer events or complex gene loss patterns. However, when a crown family appears to be uniquely present in a given clade, we define it as a marker family (11), containing genes that represent specific sequences and, possibly, specific functions universally distinguishing the clade within the full microbial phylogeny (Figure 2).
Figure 2.
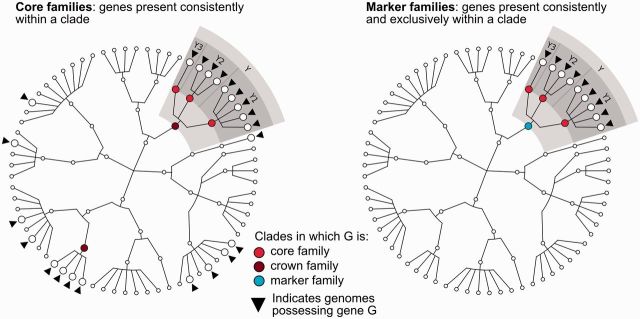
Core and marker family definitions. MetaRef defines three types of families made up of individual genes, represented here on a cartoon taxonomic tree in which leaf nodes represent strains/genomes. Core families contain genes consistently present within a clade, crown families are cores for which the clade is the lowest common ancestor, marker families are the subset of core families uniquely present only in a specific clade, and pan families comprise all genes found at least once in a sequenced microbial genome. The two trees exemplify these definitions for a gene G present in the genomes marked with a small black triangle. Specifically, on the left panel, G is a crown gene for clade Y as it is present in all its leaves and it is not a core gene in any ancestor of Y. The presence of G outside clade Y does not affect the definition of Y as a crown gene, and for clade Y1, Y2 and Y3, gene G is a core gene. On the right panel, gene G is instead a marker gene for clade Y as G is never present in genomes outside clade Y.
MetaRef families consist of the union of all pan, core, crown and marker families that determine the full microbial gene repertoire. MetaRef families can in the largest cases contain hundreds or thousands of genes, in which case it is convenient to use a unique representative sequence for the whole family, referred to as the family centroid (as for the pan families introduced above). Singleton families consisting of only one gene cannot fall into any of these categories and are excluded from core, crown and marker family definitions. Overall, MetaRef comprises ∼5 M families, 3.6 M of which are core families and ∼1 M are markers as reported in Table 1 together with the statistics of some representative clades.
Table 1.
MetaRef family statistics for representative phyla, genera and species
| Species | Genomes | Genes | MetaRef families | Tot. Pan families | Tot. core families | Tot. marker families | |
|---|---|---|---|---|---|---|---|
| All microbes | 1184 | 2818 | 10 880 874 | 5 006 295 | 376 947 | 3 600 814 | 1 028 534 |
| Proteobacteria | 463 | 1162 | 4 560 201 | 1 802 529 | 408 957 | 1 078 082 | 315 490 |
| Bacteroidetes | 88 | 124 | 474 420 | 294 243 | 236 924 | 48 040 | 9279 |
| Actinobacteria | 148 | 272 | 999 822 | 582 201 | 38 028 | 435 935 | 108 238 |
| Firmicutes | 302 | 838 | 2 600 128 | 890 666 | 211 005 | 452 615 | 227 046 |
| Staphylococcus | 11 | 92 | 248 936 | 26 206 | 16 204 | 9125 | 877 |
| Streptococcus | 32 | 132 | 277 611 | 52 673 | 27 072 | 23 171 | 2430 |
| Bacteroides | 22 | 45 | 206 420 | 70 775 | 45 843 | 22 774 | 2158 |
| Strep. pneumoniae | 42 | 93 431 | 4341 | 2733 | 1579 | 29 | |
| Staph. aureus | 72 | 198 655 | 5301 | 3137 | 1973 | 191 |
Singleton families are not reported.
Organization of the MetaRef interactive web resource
The MetaRef website provides two basic routes for data browsing: clade-centric and gene family-centric. The clade-centric view presents data based on the hierarchically structured taxonomy clades. The gene family-centric view provides biological information of individual MetaRef gene families in great detail. We recognize the intrinsic limitation of online low-throughput data navigation; therefore, throughout the site, the underlying data reported on screen can always be downloaded in plain text and FASTA format for further offline analysis.
The clade-centric view is organized according to the hierarchical (parent–child) relationships between clades and is rooted at two domain levels (Bacteria and Archaea) for which MetaRef currently catalogues 2706 and 112 genomes, respectively. The clade-centric webpage for a given query clade (example in Supplementary Figure S1) indicates the full taxonomic lineage of that clade and links to pages describing all related clades. When the query clade is at the family, genus or species level, a summary table of the core/pan genes of all sequenced genomes in the clade is reported. Links provided from this table connect to the gene family-centric view for further gene-specific inquiry.
To indicate how much sequencing data are currently available in the query clade, the total number of genomes in the clade and its direct child clades are reported and hyperlinked to genome pages. A circular genome tree of the query clade is provided to help visualize the number and distribution of genomes (the leaf nodes) sequenced in the clade and its direct child clades. When the query clade is itself a genome, more information is reported, including a table that summarizes the number of genes in that genome that are core at which ancestor clade and how many genes are unique markers of the genome. Additionally, a download link gives access to a detailed tab-delimited table describing all genes in the genome. The original external source of the genome is also provided for more inquiry.
The family-centric view (Figure 3) focuses on biological information for each individual MetaRef gene family. Information reported on the gene family page includes functional annotations aggregated from individual gene members and external functional databases; summary plots of the prevalence and abundance of the gene as found at the major HMP body sites; and reports on homologs of the query gene family found outside of its clade. Access to taxonomic clades and MetaRef families is also possible through keyword search (see Supplementary Methods).
Figure 3.

Summary of the main MetaRef panels reported in the family-centric view. For each gene family, MetaRef reports taxonomic, phylogenetic and conservation information as well as available functional information, additional consensus annotations, and the family’s prevalence and abundance in human-associated microbiomes. The information can further be analysed interactively (by exploring sub-branches of the microbial phylogeny) and offline (by downloading sequence FASTA files and gene annotation tables for the family).
Consensus functional annotation of MetaRef families
MetaRef gene families were defined solely using sequence homology with stringent criteria (80% full-length identity, see Supplementary Methods). The genes clustered into a family were therefore likely to be genes that would carry out the same biological functions. However, we observed that functional annotation of individual members within a family were, at times, inconsistent, often stemming from the fact that individual genomes were annotated by different methods at different times, with varying criteria and terminology. Consolidating annotation information from individual gene members within a family helps improve consistency of annotation interpretation across genes and provides putative annotation corrections in many cases. Additionally, MetaRef directly links a gene family with corresponding taxonomic clades, which is crucial when focusing on the property of a function encoded by a gene restricted to a specific group of microbes sharing a common ancestor (e.g. studying a given gene family in E. coli rather than on all microbes possessing the gene).
The current MetaRef implementation provides a simple and conservative effort towards consensus annotation by tallying identical annotations from all available annotation schemes, including free-text deflines, KO, COG and EC assignments. Each family's resulting table is dynamically and automatically generated from the consensus of its gene members. By reviewing the tally table, functions of some unknown/hypothetical protein genes have become clearer and recognizable. For example, a 71 gene core family of S. aureus (reported in Supplementary Figure S2) is consistently annotated as a putative siderophore biosynthesis protein subunit, although eight individual gene members were previously only annotated as hypothetical proteins. In another case (Supplementary Figure S2), two DNA mismatch repair proteins in a core family are labelled as HexA, whereas all the other members of the family (70) are labelled as MutS, suggesting a misannotation. This potential inconsistency has been confirmed by ad hoc sequence-based analysis, which showed that both of the former two proteins possess the MutS_1, MutS_2 and MutS_3 superfamilies and the ABC MutS1 domains and that they differ from several consistently annotated MutS proteins by only one peptide along the full-length of 872 peptides. These examples confirm the usefulness of MetaRef efforts towards large-scale sequence-based consensus annotation.
MetaRef characterization of metagenomic samples
Comprehensive studies of human-associated microbial communities are still in their first generation and have yet to be well-integrated with microbial isolate sequence resources such as the Human Microbiome Project’s (HMP) Data Analysis and Coordination Center (21) (http://hmpdacc.org). MetaRef includes what we believe to be the first comprehensive gene family-centric reference generated from a large cohort of healthy human subjects: using our pre-computed database of family centroids along with the metagenomes sequenced by the HMP, we have compiled baseline data characterizing the relative abundance and prevalence of all gene families across all major body sites in healthy individuals. Investigators studying site-specific pathologies, for example, can use this data to efficiently characterize differences in functional potential (at the individual gene level) between healthy- and disease-associated microbial communities (22) by looking for significant differences in observed prevalence/abundance of detected genes (23). It has been shown (11) that marker centroids can be reliably used to estimate the presence and relative abundance of their associated clades. As a result, a single alignment pass of a collection of pathology-associated metagenomic sequence reads against the full collection of MetaRef centroids—which is then compared to analogous data compiled from a cohort of healthy subjects at the same body site—can be interrogated to detect both taxonomic and functional differences between health- and disease-associated microbiota.
To construct our baseline reference data, we first used Bowtie (17) to align all sequence reads from 757 shotgun metagenomic HMP samples—covering all five studied body areas (airways, GI tract, mouth, skin and urogenital tract, including their various subsites)—against 5 006 294 family centroid sequences (Figure 4). For each sample, we counted all matches to each centroid, then converted these raw counts to RPKMs (24) to normalize with respect both to differences in inter-sample sequencing depth and differences in individual centroid lengths. Using these normalized counts, we calculated, for each sample, the relative abundance of each centroid as a percentage of all normalized counts. For all body sites, we computed the mean relative abundance (over all non-zero samples) of each centroid detected at that site, to generate a single statistic estimating the typical relative abundance of each centroid at that site. The presence or absence of each centroid at each site was also used to compute prevalence statistics: for each centroid and each body site in which it was found, we report the percentage of samples in which that centroid was detected. Body-site-based relative abundance and prevalence statistics are summarized (as box plots and bar plots, respectively) on individual centroid pages on the MetaRef website (cf. Figure 3). Although for completeness of the downloadable resource we aligned all 757 HMP samples against the centroid collection, the prevalence and abundance data provided by MetaRef reflects the subset of 630 samples that passed HMP’s quality-control procedures for the six body sites considered. Please see the Supplementary Methods for a discussion of our alignment strategy, precise details of the alignment operations, and a summary of the results of validation tests we conducted to ensure that our results were sufficiently representative of the sampled microbial communities.
Figure 4.
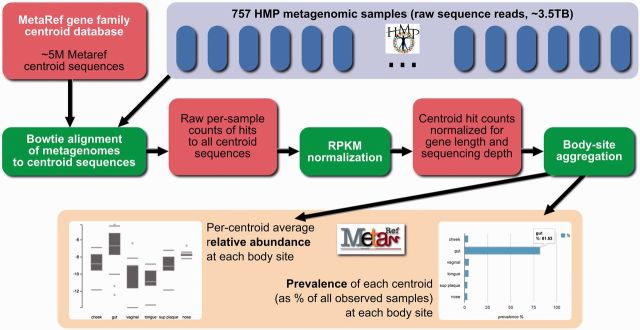
Overview of MetaRef's statistical processing of microbial gene families in the healthy human body-site-specific microbiome. Raw sequence samples from HMP metagenomes are aligned against all MetaRef family centroids; the number of reads aligning to each centroid in each sample is tallied; read counts are transformed via RPKM to normalize with respect to centroid length and sampling depth; centroid RPKMs for all samples associated with each body site are aggregated into measures of prevalence and relative abundance for all centroids detected at that site.
The number of centroids detected in each sample that passed QC ranged from 2563 (from a sample taken from the posterior vaginal fornix: when healthy, such samples exhibit the lowest-diversity microbe communities of all studied body sites (25)) to 4 128 821 (from a distal gut sample, the most complex of all human-associated microbial communities (25)), with a median of 94 135 (from an oral sample). As examples, one core gene (encoding a sigma factor subunit of RNA polymerase) for Bacteroides ovatus—a bacterium commonly found in the human gut (26,27)—was observed in 81.53% of all GI-tract samples, and in <5% of samples sequenced from all other body sites. Its average relative abundance within gut samples was more than two orders of magnitude higher than its relative abundance in all other body sites. Similarly, a core gene (encoding a cell division protein) for S. epidermidis—a common commensal colonist of the human nasal passage (28,29)—was seen in ∼25% of all nasal samples, but <3% of samples from other body sites; its average relative abundance in nasal samples was again at least two orders of magnitude greater than in samples from any other body site. Comprehensive prevalence/abundance statistics for all family centroids (including markers) have been made available for direct download from the MetaRef website, and graphical reports for individual centroids can be browsed directly (Supplementary Figure S3). These can be conveniently used as a robust collection of healthy baseline data for any research involving the human microbiome, whether the focus be on taxonomic composition, functional potential or both.
DISCUSSION
With MetaRef.org, we provide the most comprehensive and up-to-date database of a non-redundant reference gene catalogue that not only delivers information on gene family conservation, but also captures phylogeny and consensus functional annotation. It is easily accessible as an online browsable resource allowing quick functional and phylogenetic explorations of gene families, and its contents are also available by direct download for large-scale bioinformatics applications. MetaRef is committed to providing a comprehensive microbial gene family resource, and thus includes all systematically available finished and draft genomes from the time of creation, automatic detection of taxonomic inconsistencies and suggestions of plausible corrections, and is designed with regular automated updates at increasing scale in mind. We anticipate future versions including additional sources of microbial genomes and expanding rapidly as isolate sequencing becomes increasingly ubiquitous.
With microbial sequence data in all its diversity being strongly on the rise (30–32), a new approach such as MetaRef is needed to simultaneously organize information on gene function, phylogeny and microbial taxonomy throughout the tree of life to facilitate downstream biological interpretation. Going forward, we also plan to increase the use of and connectivity to other genome databases, particularly those providing extended annotation schemes in order to continue the harmonization process for annotations. We will also automatically incorporate additional phylogenetic lineages currently being sequenced (32,33) as well as additional meta'omic datasets (34–37), and based on current performance we expect the system to scale smoothly in anticipation of the rapid increase of available genomes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [38–40].
FUNDING
National Institutes of Health [R01HG005969 to C.H.]; National Science Foundation [DBI-1053486 to C.H.]; Danone Research [PLF-5972-GD to Wendy Garrett]; This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract [No.: HHSN272200900018C], and with funds from the National Human Genome Research Institute, National Institutes of Health, Department of Health and Human Services [U54HG004969 to the Broad Institute]. The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007-2013) under REA grant agreement [PCIG13-GA-2013-618833 to N.S.]. Funding for open access charge: Startup fund at the Centre for Integrative Biology (University of Trento) (to N.S.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We would like to thank Roman Stolyarov for his contributions to software components underlying MetaRef.
REFERENCES
- 1.Medini D, Serruto D, Parkhill J, Relman DA, Donati C, Moxon R, Falkow S, Rappuoli R. Microbiology in the post-genomic era. Nat. Rev. Microbiol. 2008;6:419–430. doi: 10.1038/nrmicro1901. [DOI] [PubMed] [Google Scholar]
- 2.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 3.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 4.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakaya A, Katayama T, Itoh M, Hiranuka K, Kawashima S, Moriya Y, Okuda S, Tanaka M, Tokimatsu T, Yamanishi Y, et al. KEGG OC: a large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res. 2013;41:D353–D357. doi: 10.1093/nar/gks1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta RS, Meacham C, Samad B, Neyer C, Sjolander K. Berkeley PHOG: PhyloFacts orthology group prediction web server. Nucleic Acids Res. 2009;37:W84–W89. doi: 10.1093/nar/gkp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–D294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchiyama I, Mihara M, Nishide H, Chiba H. MBGD update 2013: the microbial genome database for exploring the diversity of microbial world. Nucleic Acids Res. 2013;41:D631–D635. doi: 10.1093/nar/gks1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 11.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012;9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segata N, Bornigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 2013;4:2304. doi: 10.1038/ncomms3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segata N, Huttenhower C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS ONE. 2011;6:e24704. doi: 10.1371/journal.pone.0024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl Acad. Sci. USA. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 21.Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, et al. A catalog of reference genomes from the human microbiome. Science. 2010;328:994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 25.Relman DA. Microbiology: learning about who we are. Nature. 2012;486:194–195. doi: 10.1038/486194a. [DOI] [PubMed] [Google Scholar]
- 26.Hamady ZZ, Scott N, Farrar MD, Lodge JP, Holland KT, Whitehead T, Carding SR. Xylan-regulated delivery of human keratinocyte growth factor-2 to the inflamed colon by the human anaerobic commensal bacterium Bacteroides ovatus. Gut. 2010;59:461–469. doi: 10.1136/gut.2008.176131. [DOI] [PubMed] [Google Scholar]
- 27.Karlowsky JA, Walkty AJ, Adam HJ, Baxter MR, Hoban DJ, Zhanel GG. Prevalence of antimicrobial resistance among clinical isolates of Bacteroides fragilis group in Canada in 2010-2011: CANWARD surveillance study. Antimicrob. Agents Ch. 2012;56:1247–1252. doi: 10.1128/AAC.05823-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.den Heijer CD, van Bijnen EM, Paget WJ, Pringle M, Goossens H, Bruggeman CA, Schellevis FG, Stobberingh EE. Prevalence and resistance of commensal Staphylococcus aureus, including meticillin-resistant S aureus, in nine European countries: a cross-sectional study. Lancet Infec. Dis. 2013;13:409–415. doi: 10.1016/S1473-3099(13)70036-7. [DOI] [PubMed] [Google Scholar]
- 29.Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. mBio. 2010;1:e00129-10. doi: 10.1128/mBio.00129-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8:e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013;31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 33.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 34.Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta'omics for microbial community studies. Mol. Syst.Biol. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carvalhais LC, Dennis PG, Tyson GW, Schenk PM. Application of metatranscriptomics to soil environments. J. Microbiol. Meth. 2012;91:246–251. doi: 10.1016/j.mimet.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Trans. Med. 2011;3:106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28:304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]