


Abstract
FINDbase (http://www.findbase.org) aims to document frequencies of clinically relevant genomic variations, namely causative mutations and pharmacogenomic markers, worldwide. Each database record includes the population, ethnic group or geographical region, the disorder name and the related gene, accompanied by links to any related databases and the genetic variation together with its frequency in that population. Here, we report, in addition to the regular data content updates, significant developments in FINDbase, related to data visualization and querying, data submission, interrelation with other resources and a new module for genetic disease summaries. In particular, (i) we have developed new data visualization tools that facilitate data querying and comparison among different populations, (ii) we have generated a new FINDbase module, built around Microsoft’s PivotViewer (http://www.getpivot.com) software, based on Microsoft Silverlight technology (http://www.silverlight.net), that includes 259 genetic disease summaries from five populations, systematically collected from the literature representing the documented genetic makeup of these populations and (iii) the implementation of a generic data submission tool for every module currently available in FINDbase.
INTRODUCTION
National and Ethnic Mutation Databases (NEMDBs) are structured and continuously updated data repositories recording the various spectra of causative genomic variations for any gene or disease in different populations and ethnic groups worldwide (1). The data content of these resources can be exploited mainly to study gene/mutation flow and admixture patterns, human demographic history and possibly to stratify national molecular diagnostic services (1) and can be nicely complement the data content of either central (or core) and/or locus-specific databases (LSDBs).
The first NEMBDs appeared online a little over a decade ago, and in 2006, our group established FINDbase (Frequency of INherited Disorders database; http://www.findbase.org), a worldwide database pertaining to frequencies of causative mutations, leading to inherited disorders in various populations and ethnic groups worldwide (2). This resource contains data only in an aggregated manner, i.e. allele frequencies without any sensitive personal data of their carriers, in order to maintain anonymity. Content-wise, FINDbase is the richest among the NEMDBs currently available and, it is considered as one of the key resources to retrieve population-specific information for clinically relevant genomic variations, as indicated by the number and origin of visitors.
Here, we present significant data content updates in an effort to make FINDbase even more useful and appealing to a broader user group. We also present some technological advances which expand the current battery of data visualization tools. Finally, we report the addition of a new module for genetic disease summaries and the implementation of a generic data submission tool to allow direct data submission to all FINDbase modules.
DATA CONTENT UPDATES
Existing FINDbase data content, resulting from the initial database launch (2006) and its 2010 data update (3) including population-specific data from the European Commission FP5 Cystic Fibrosis Thematic Network Consortium [http://www.cfnetwork.be; (4)], and the PAH [http://www.pahdb.mcgill.ca; (5)], SERPINA1 (6) and the globin genes LSDBs [http://globin.bx.psu.edu/hbvar; (7)] were curated and corrected where necessary.
Apart from data curation, we also continued to enrich FINDbase data collection with new population-specific data records from individual NEMDBs and the published literature. All entries were manually curated and incorporated into the main FINDbase data collection, which resulted into an increase of >30% in data content, representing 1480 new records from 46 populations and 15 ethnic groups and/or geographical regions. As with every FINDbase entry, the selection criteria for each FINDbase dataset, includes the inclusion of the most representative study for each population that involves sufficient number of patients and controls to avoid alteration of the calculated allele frequencies, in case multiple reports are included in the analysis. All published data entries were recorded against their corresponding unique PubMed ID, whereas in case of unpublished information (e.g. personal communication or aggregated LSDB datasets), we have exploited the microattribution approach (8,9), using the contributor’s unique ResearcherID (http://www.researcherid.com), an approach that not only provides incentives to potential data contributors to share their data with the broader scientific community but also allows unambiguous identification of curated data when data update or correction is needed.
FINDbase data compilation and representation are subject to copyright and usage principles to ensure that FINDbase and its contents remains freely available to all interested parties.
NEW FEATURES
Apart from the comprehensive data content curation, correction and update described above, several new features and modules have been incorporated into FINDbase. These include the development of additional data querying and visualization tools, a new module to document genetic disease summaries in various populations and the implementation of data submission tools for every FINDbase module. These new features are described below.
Development and implementation of new data querying and visualization tools
During the previous FINDbase update (3), we have developed and implemented interactive web-based data visualization and querying tools, to enable FINDbase users to combine large groups of similar data elements and identify hidden relationships between them. This tool met one of the significant challenges of handling and querying large and complex datasets, that is the effective presentation of and interaction with the data. In particular, we have built a web-based multimedia web front-end, based on a software tool launched by Microsoft, namely the PivotViewer (http://www.microsoft.com/silverlight/pivotviewer), in order to support a high-level visualization of FINDbase data collection and the data mining process.
We have recently implemented an additional visualization querying interface based on the Flare visualization toolkit (http://flare.prefuse.org), which provides two extra types of data visualization and query output, namely the Gene and Mutation Map and the Mutation Dependency Graph. These tools allow FINDbase users to query mutation distributions and correlations among populations (10).
The Gene and Mutation Map is based on a tree-map, which constitutes of an easy way of analyzing large amounts of data (http://www.cs.umd.edu/hcil/treemap). Tree-maps are based on a space-filling approach of showing hierarchies in which the rectangular screen space is divided into regions, and then each region is divided again for each level in the hierarchy. In the case of FINDbase data collection, the tree-map corresponds to mutation frequencies estimated for each population. Each rectangle represents a population’s mutation and a specific color corresponds to each population. The area covering each node encodes the frequency of rare alleles. Each time the user clicks on a node, the occurrence of the selected mutation is shown over all populations. Moreover, when a user hovers the mouse over a rectangle, Gene and Mutation Map displays additional information pertaining to the selected mutation such as the population, the gene name, the mutation, and the mutation’s frequency (Figure 1A).
Figure 1.

Summary of the recently implemented FINDbase data querying and visualization tools. Sample query to indicate the occurrence of the HBB:c.92 + 6T > C causative mutation in various populations. (A) The Gene and Mutation Map shows, in boxes of different size (depending on the frequency of the variant) and color, the various populations in which this causative mutation leading to β-thalassemia is identified. The user can directly see the details of each record by hovering the mouse over each box. (B) The Mutation Dependency Graph shows all populations that are associated with the selected genomic variant. In both cases, data output indicates that this causative mutation is more prevalent in South European and Middle Eastern populations.
Similarly, the Mutation Dependency Graph visualizes the dependencies that occur among different populations on the basis of a selected genomic variant. In computer science, as well as in mathematics, a dependency graph is a directed graph (or set of nodes connected by edges) representing dependencies of several objects towards each other. From this graph, it is possible to derive an evaluation order or the absence of an evaluation order based on the given dependencies from the dependency graph. In the case of the Mutation Dependency graph, the different edges are populations that are clustered based on the presence and/or the frequency of a certain genomic variant. Population names are placed along a circle. A link between populations indicates that these populations have the same genomic variant in common. When a user hovers the mouse over a specific population, the corresponding incident links are highlighted. Red links show all the populations that are associated with the selected genomic variant. By clicking on a specific population, the user can see all the relevant dependencies of that population concerning the selected mutation (Figure 1B).
Although seemingly complex, the Gene and Mutation Map together with the Mutation dependency graphs provide the means of establishing relationships among different populations on the basis of certain genome variants and together with the basic data querying option provided by the PivotViewer significantly enhance the battery of the data visualization tools available to explore FINDbase data content.
Introduction of a new module to document genetic disease summaries
Apart from the documentation of detailed clinically relevant genomic variant allele frequency spectra in various NEMDBs, it is equally important to comprehensively summarize all genetic disorders found in a specific population (1). The Israeli NEMDB was among the first NEMDBs that were developed with this principle (11). However, the initial database platform, namely the ETHNOS NEMDB software (12) based on which several NEMDBs were established, was very primitive in nature and provided only limited data querying and visualization capacity. We have therefore decided to rebuild the underlying database structure and architecture such that it would not only significantly enhance the data querying capacity, but would also make the resulting datasets compatible with the entire FINDbase data collection.
Consistent with the previous update, we have chosen to build and introduce in the existing FINDbase data collection, a new module for genetic disease summaries. As with the existing FINDbase modules for causative mutations and pharmacogenomic biomarkers, the component services of this module also follow the Service Oriented Architecture [SOA; (13,14)] and the querying interface is built around PivotViewer, based on Microsoft Silverlight technology (Figure 2A). In this module, database records include the population, the genetic disease, the gene name, online Mendelian Inheritance in Man (OMIM) ID and chromosome on which it resides that are all included in the query interface. When the user clicks on the record, that resembles a box, the details for the selected record are shown in the menu on the right (Supplementary Figure S1A). The name of the genetic disease is a hyperlink, and when selected, the entire genetic disease summary description appears in a new window, where additional information on the genetic basis of the disease and corresponding reference(s) for this population are provided (Supplementary Figure S1B).
Figure 2.
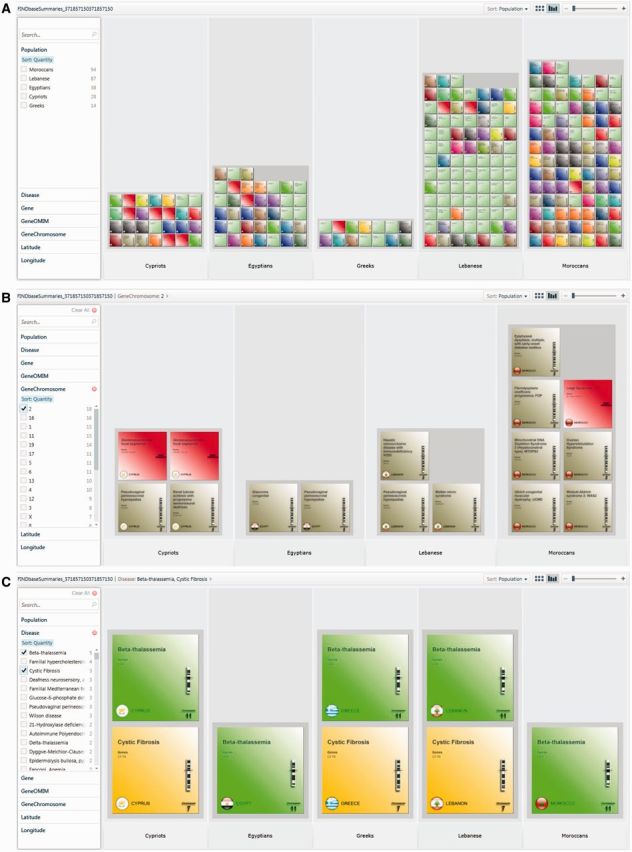
Overview of the new FINDbase genetic disease summaries module. (A) Overview of the entire FINDbase genetic disease summary module data collection, based on Microsoft’s PivotViewer. The querying interface is shown on the left (see detail in Supplementary Figure S4) and the output option can be selected at the top-right corner of the screen. The different entries, presented as display items, are shown as colored boxes, depending on the chromosomal location of the gene that is related to a genetic disease. (B) Sample query: ‘Display those genetic diseases that result from mutations in genes residing on chromosome 2’. Query output includes 18 records in four populations as different display items. (C) Sample query: ‘Display all populations in which β-thalassemia and cystic fibrosis are prevalent’. Query output includes eight records in total, from which three records correspond to cystic fibrosis in the Cypriot, Greek and Lebanese populations and the remaining five records for β-thalassemia in all five populations (see text for details). Note that all display items are shown in the same color per genetic disease, resulting from mutations in the HB and CFTR genes that reside on chromosomes 11 and 7, respectively.
We have subsequently used this new FINDbase module to migrate the entire genetic disease summary data collection from five previously developed NEMDBs, namely the Hellenic (15), Lebanese, Cypriot, Egyptian and Moroccan. As with the existing modules, the user can smoothly and quickly arrange FINDbase data according to common characteristics that can be selected from the data query menu. Data are in the form of a card, with a chromosomal figure and chromosome number on the right, depending on the gene that when mutated leads to the specific genetic disorder (in case of genetic diseases that results from mutations in more than one genes, then a different color code is used), accompanied by a sidebar textbox with in-depth data concerning the particular genetic disease summary and population. Additional filtering allows the user to see specific genetic diseases that are present in different populations (Figure 2B) or those genetic diseases that result from mutated genes residing on specific chromosomes (Figure 2C).
The new FINDbase genetic disease summary module includes summaries from 259 genetic diseases for five populations and not only provides the means to expand the existing data collection by adding additional populations, but also to harmonize NEMDB development and minimize data content heterogeneity.
Implementation of a generic data submission environment
Data entry and modification in FINDbase is only possible for registered users. The initial FINDbase version (2) included a data submission tool that allowed direct data entry from registered users that had access rights as data curators and national coordinators. Since 2010, we have engaged into a major refit and significantly extended the existing FINDbase data submission environment. As before, FINDbase is developed such to support three different user profiles, namely administrators, national coordinators and data curators, providing scaled access rights for data submission according to their access rights. Administrators (i) have full access rights to all database functionalities and data, (ii) are responsible for the activation of user accounts and (iii) are assigned data entry and modification rights. One level below in the hierarchy are the national coordinators, who (i) have data entry and modification rights for a particular population and (ii) can perform data reallocation among data curators, if needed, registered to enter data in this particular population. As such, national coordinators are responsible for managing the overall development and maintenance of a NEMDB, contributing to FINDbase. Finally, data curators have data entry and modification rights only for those data entered by themselves and they cannot alter data entered by another curator. If a curator wishes to end his involvement in FINDbase, their data will be allocated to another data curator, by the national coordinator, who will then be responsible for their curation. All FINDbase registered users are logged in using their ResearcherIDs to enable the implementation of microattribution for data sharing (8,9).
The last few months, the data entry page has been redesigned in an effort to make it more user-friendly (Supplementary Figure S2 and Supplementary Table S1). Clinically relevant genomic variation allele frequency data entry is conducted via uploading an excel spreadsheet and a web-based editor subsequently extracts the data from the spreadsheet into the system. When a data curator uploads an excel spreadsheet, the submitted data are temporarily stored into the system (they are not displayed by the visualization tools until the administrator’s or the national coordinator’s approval). During the uploading stage, an automatic process that runs in the background performs validation rules on the submitted data to verify that they conform to the appropriate data types and if errors are detected, the system notifies and prompts the user to correct them in order to complete the uploading process. Upon successful submission, the administrator and the national coordinator corresponding to the country of the data curator’s origin are automatically notified to review the data submitted against certain content criteria (minimum number of chromosomes studied, justification of data submission, statistical analysis of the findings and means of genotyping). These users have full access to all the uploaded data and can modify them, if necessary, to maintain data accuracy and uniformity. Furthermore, a control for the detection of similar or duplicate entries is available. Each time this happens, the appropriate user is notified and can select the correct entry to be published in the whole visualization environment. Once the data are approved, they become part of the main FINDbase data collection.
Similarly, a dedicated data entry form is developed for uploading genetic disease summary data. At present, this data entry tool only support data entry from the administrator but we plan to extend this data submission tool to also support data submission from three different user profiles, namely administrators, national coordinators and data curators.
Interrelation with Café Variome
As of early 2007, FINDbase is affiliated with the Human Gene Mutation Database [HGMD; http://www.hgmd.org; (16)]. In particular, FINDbase data content has been bidirectionally linked with that of HGMD, in an effort to liaise clinically relevant genomic variation with their allele frequency data, where available. For the same reason, we have decided to share FINDbase data content with Café Variome (http://www.cafevariome.org). In particular, the entire FINDbase data collection has been shared using this platform, derived from the GEN2PHEN project. For example, the user can select FINDbase as the source database and the gene in the query menu (e.g. CYP2C19). The query output includes 119 entries as linked, to FINDbase, access in a tabular format (Supplementary Figure S3). The user can then select the record of choice that is hyperlinked to the respective record in FINDbase.
CONCLUSIONS AND FUTURE PERSPECTIVES
Since its establishment, FINDbase is a comprehensive resource of clinically relevant genomic variation allele frequency data, with the ultimate goal to constitute a reference repository of such information worldwide. At present, FINDbase consists of three distinct modules: (i) the ‘causative mutation’ module, which documents the mutation frequency spectra for each population, and hence the rare allele frequency is calculated on the basis of the frequency of this variant with respect to the total number of mutant alleles, including the uncharacterized mutants, (ii) the ‘pharmacogenomic biomarkers’ module, in which the rare allele frequency is documented as the frequency of the under-represented allele and (iii) the ‘genetic disease summaries’ module, which documents the genetic heterogeneity in each population.
Database access is free of charge and there are no registration requirements for data querying. Not only has FINDbase data collection been significantly enriched since its launch in 2006, but its battery of data querying and visualization tools has been significantly expanded using state-of-the-art web-based approaches. Since 2006 when FINDbase was officially announced (2) and its previous update (3), we have recorded over 125 000 accesses to the query page from unique IP addresses spanning over 120 countries worldwide (including .com, .org, .net and .gov URLs), while missing information and erroneous entries for existing variants are occasionally reported by database users, which helps to improve data quality and accuracy.
To meet with the future user requirements and the anticipated increased data influx from high throughput genotyping methods, FINDbase architecture will continue to be modernized. As a matter of fact, our efforts have been facilitated so far by our participation to the GEN2PHEN project (http://www.gen2phen.org; funded by the European Commission that has recently come to an end), and will continue unobstructed with partnership with another major European Commission-funded project, RD-Connect (http://www.rd-connect.eu), ensuring not only interaction with the field’s leading experts, but also funding that is secured for a further 5-year period. Also, FINDbase is a key component of the Genomic Medicine Alliance (http://www.genomicmedicinealliance.org), a newly established international initiative aiming to catalyze integration of genomic medicine in various countries, particularly from the developing world.
To enrich the existing data collection, we plan, as of early 2014, to include pharmacogenomic marker data from 18 European populations (namely 1936 pharmacogenomic markers in 235 pharmacogenes, screened for each population in an aggregated level), that will constitute a massive data influx in the pharmacogenomic biomarker FINDbase module. Also, we have already started to connect pharmacogenomic biomarker allele frequency data with related drugs, in an effort to correlate a drug’s efficacy or toxicity with a particular pharmacogenomic biomarker, in a certain population. This effort can subsequently enable regulatory agencies to develop guidelines to rationalize drug use in those populations, where certain pharmacogenomic biomarkers reach high frequencies (e.g. HLA-B*5701 has variable allele frequency worldwide, from <1% in sub-Saharan Africa to up to 20% in India, while in European populations HLA-B*5701 frequency varies between 1% and 7%; 17, Motslinger and coworkers, in preparation). Also, we envisage generating a tool to accommodate data submission of whole-exome and whole-genome data, from which causative mutation and pharmacogenomic biomarker allele frequencies will be automatically generated for a particular population, in an aggregated level to ensure data anonymity.
We also plan to expand the novel data querying and visualization functionalities, namely the Gene and Mutation Map and the Mutation Dependency Graph that are currently available only for the causative mutation FINDbase module, as described above, for FINDbase pharmacogenomic markers and genetic disease summaries modules.
Lastly, we plan to exploit the entire FINDbase data content, subdivided into three modules, to generate an ‘off-the-shelf’ web application that would allow not only the development of new NEMDBs but also data migration of existing NEMDBs, that would be further enriched with existing FINDbase data. This application is based on the established recommendations and guidelines to develop nation-wide projects to document the genetic heterogeneity in various countries (18) and, when operable and with the extended genetic data submission tool to support three different users, particularly national coordinators and data curators, will constitute the next generation of the ETHNOS software.
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online.
ACKNOWLEDGEMENTS
The authors are grateful to Dr Marianthi Georgitsi for fruitful discussions, and Theodoros Karlis and Ioanna Kohliadi for competent data mining and curation. Also, the authors are indebted to all FINDbase users worldwide for their valuable comments and suggestions, which helped in keeping the information as updated and complete as possible and also contributed to the continuous improvement of the database profile and contents.
FUNDING
The European Commission grants [MEDGENET (FP6-31968); GEN2PHEN (FP7-200754), RD-CONNECT (FP7-305444), SEE_DRUG (FP7-CT-2011-285950) to G.P.P.]; Golden Helix Foundation (UK). Funding for open access charge: RD-CONNECT [FP7-304555].
Conflict of interest statement. None declared.
REFERENCES
- 1.Patrinos GP. National and ethnic mutation databases: documenting populations’ genography. Hum. Mutat. 2006;27:879–887. doi: 10.1002/humu.20376. [DOI] [PubMed] [Google Scholar]
- 2.van Baal S, Kaimakis P, Phommarinh M, Koumbi D, Cuppens H, Riccardino F, Macek M, Jr, Scriver CR, Patrinos GP. FINDbase: a relational database recording frequencies of genetic defects leading to inherited disorders worldwide. Nucleic Acids Res. 2007;35:D690–D695. doi: 10.1093/nar/gkl934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgitsi M, Viennas E, Gkantouna V, van Baal S, Petricoin EF, Poulas K, Tzimas G, Patrinos GP. FINDbase: a worldwide database for genetic variation allele frequencies updated. Nucleic Acids Res. 2011;39:D926–D932. doi: 10.1093/nar/gkq1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum. Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 5.Scriver CR, Waters PJ, Sarkissian C, Ryan S, Prevost L, Cote D, Novak J, Teebi S, Nowacki PM. PAHdb: a locus-specific knowledgebase. Hum. Mutat. 2000;15:99–104. doi: 10.1002/(SICI)1098-1004(200001)15:1<99::AID-HUMU18>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 6.Zaimidou S, van Baal S, Smith TD, Mitropoulos K, Ljujic M, Radojkovic D, Cotton RG, Patrinos GP. A1ATVar: a relational database of human SERPINA1 gene variants leading to alpha1-antitrypsin deficiency. Hum. Mutat. 2009;30:308–313. doi: 10.1002/humu.20857. [DOI] [PubMed] [Google Scholar]
- 7.Patrinos GP, Giardine B, Riemer C, Miller W, Chui DH, Anagnou NP, Wajcman H, Hardison RC. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32:D537–D541. doi: 10.1093/nar/gkh006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giardine B, Borg J, Higgs DR, Peterson KR, Philipsen S, Maglott D, Singleton BK, Anstee DJ, Basak AN, Clark B, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat. Genet. 2011;43:295–301. doi: 10.1038/ng.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrinos GP, Cooper DN, van Mulligen E, Gkantouna V, Tzimas G, Tatum Z, Schultes E, Roos M, Mons B. Microattribution and nanopublication as means to incentivize the placement of human genome variation data into the public domain. Hum. Mutat. 2012;33:1503–1512. doi: 10.1002/humu.22144. [DOI] [PubMed] [Google Scholar]
- 10.Viennas E, Gkantouna V, Ioannou M, Georgitsi M, Rigou M, Poulas K, Patrinos GP, Tzimas G. Population-ethnic group specific genome variation allele frequency data: a querying and visualization journey. Genomics. 2012;100:93–101. doi: 10.1016/j.ygeno.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Zlotogora J, van Baal S, Patrinos GP. Documentation of inherited disorders and mutation frequencies in the different religious communities in Israel in the Israeli National Genetic Database. Hum. Mutat. 2007;28:944–949. doi: 10.1002/humu.20551. [DOI] [PubMed] [Google Scholar]
- 12.van Baal S, Zlotogora J, Lagoumintzis G, Gkantouna V, Tzimas I, Poulas K, Tsakalidis A, Romeo G, Patrinos GP. ETHNOS: a versatile electronic tool for the development and curation of National Genetic databases. Hum. Genomics. 2010;4:361–368. doi: 10.1186/1479-7364-4-5-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell M. SOA Modeling Patterns for Service-Oriented Discovery and Analysis. Chichester, United Kingdom: Wiley & Sons; 2010. [Google Scholar]
- 14.Valipour MH, AmirZafari B, Maleki KN, Daneshpour N. A Brief Survey of Software Architecture Concepts and Service Oriented Architecture. 2009 Proc 2nd IEEE International Conference on Computer Science and Information Technology, ICCSIT'09, Beijing, China, 34–38. [Google Scholar]
- 15.Patrinos GP, van Baal S, Petersen MB, Papadakis MN. The Hellenic national mutation database: a prototype database for mutations leading to inherited disorders in the Hellenic population. Hum. Mutat. 2005;25:327–333. doi: 10.1002/humu.20157. [DOI] [PubMed] [Google Scholar]
- 16.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolan D, Gaudieri S, Mallal S. Pharmacogenetics: a practical role in predicting antiretroviral drug toxicity? J. HIV. Ther. 2003;8:36–41. [PubMed] [Google Scholar]
- 18.Patrinos GP, Al Aama J, Al Aqeel A, Al-Mulla F, Borg J, Devereux A, Felice AE, Macrae F, Marafie MJ, Petersen MB, et al. Recommendations for genetic variation data capture in developing countries to ensure a comprehensive worldwide data collection. Hum. Mutat. 2011;32:2–9. doi: 10.1002/humu.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]