


Abstract
The National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) database is a collection of annotated genomic, transcript and protein sequence records derived from data in public sequence archives and from computation, curation and collaboration (http://www.ncbi.nlm.nih.gov/refseq/). We report here on growth of the mammalian and human subsets, changes to NCBI’s eukaryotic annotation pipeline and modifications affecting transcript and protein records. Recent changes to NCBI’s eukaryotic genome annotation pipeline provide higher throughput, and the addition of RNAseq data to the pipeline results in a significant expansion of the number of transcripts and novel exons annotated on mammalian RefSeq genomes. Recent annotation changes include reporting supporting evidence for transcript records, modification of exon feature annotation and the addition of a structured report of gene and sequence attributes of biological interest. We also describe a revised protein annotation policy for alternatively spliced transcripts with more divergent predicted proteins and we summarize the current status of the RefSeqGene project.
INTRODUCTION
The National Center for Biotechnology Information (NCBI) initiated the Reference Sequence (RefSeq) project in the spring of 1999 with the public release of 3446 human transcript and protein records (1). Since that small beginning, the RefSeq project has realized significant growth in the number and type of sequence records provided and in the number and taxonomic breadth of the organisms represented. NCBI’s RefSeq project now provides sequence records for genomes, transcripts and proteins for viruses, microbes, organelles and eukaryotic organisms. The genomic records provided for mammals include: (i) annotated nuclear and mitochondrial genomes; (ii) non-transcribed pseudogenes; (iii) haplotype-specific regions, such as the human leukocyte antigen-A (HLA-A) encoding regions and (iv) RefSeqGene records (described below), which are created for a subset of human genes to provide stable coordinate systems needed by clinical testing laboratories. Transcript records may be protein-coding, non-coding (ncRNA), or structural RNAs.
RefSeq records are generated for mammals and other higher eukaryotes in several ways. A gene may be represented by transcript, protein or genomic RefSeqs (with an NM, NR, NP or NG accession prefix, hereafter referred to as ‘known’ RefSeqs) generated by automatic and manual processing of public sequence data maintained by members of the International Nucleotide Sequence Database Consortium (INSDC) (2). An additional source is NCBI’s eukaryotic genome annotation pipeline, which provides predicted model RefSeq records (with an XM, XR or XP accession prefix, hereafter referred to as ‘model’ RefSeqs). Some RefSeq records may also be based on data imported from expert databases, such as the International Immunogenetics Information System (IMGT) (3), miRBase (4) and pseudogene.org (5). Curation by NCBI staff, focused particularly on the mammalian RefSeq branch, and collaboration with external groups helps in maintaining the quality of the collection and other resources it affects, including NCBI’s Gene resource. We collaborate with official nomenclature groups (6–8), the Consensus CDS (CCDS) project (9), the Genome Reference Consortium (10), the Locus Reference Genomic initiative (11), UniProt (12) and others. Curation and collaboration contribute new and updated sequence records, appropriate gene and protein names, feature annotation, relevant literature and help to ensure correct gene placement and to address sequence quality concerns. These activities in turn improve the outcome of the eukaryotic genome annotation pipeline.
In this article, we report on recent growth of the mammalian RefSeq dataset, changes to NCBI’s eukaryotic genome annotation pipeline and newly expanded annotation that provides supporting evidence and more biologically relevant information in a RefSeq record.
GROWTH AND ACCESS
A comprehensive RefSeq file transfer protocol (FTP) release is provided bi-monthly (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/) and can be downloaded as either the complete dataset or as subsets organized by major taxonomic group, such as mammals or molecule type. New and updated RefSeq records are provided daily and a full release of transcript and protein records for select mammals as well as the human RefSeqGene complement (described below), is available weekly. RefSeq FTP release 61, distributed in September 2013 included more than 41 million sequence records from over 29 000 organisms. The largest subset of the RefSeq release consists of microbial (primarily bacterial) genome and protein records, which are processed differently from eukaryotic RefSeq records and are not the focus of this report. Additional information on the growth of the complete dataset, or of specific subsets, is available on the RefSeq web site (www.ncbi.nlm.nih.gov/refseq/statistics/) and from the FTP site (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/release-statistics/).
Table 1 summarizes the yearly growth of the RefSeq transcript datasets for human and other mammals that have a nuclear genome assembly; the relative proportion of select categories including model transcripts, curated known transcripts and ncRNAs (both model and known RefSeqs) is also reported. Compared with previous releases, the increase in number of mammalian taxa and the number of mammalian transcript records from release 54 (July 2012) to release 60 (July 2013) is a consequence of improvements to the eukaryotic genome annotation pipeline (described below) that result in a large increase in model RefSeqs. Considering human data separately, a gradual growth trend is observed over time. Decreases in the total number of human RefSeq transcripts and genes between some years (such as following the July 2009 release 36) can be attributed to several factors including incremental changes to the annotation pipeline that resulted in fewer predicted models and/or improved gene-level tracking and curation efforts to remove low-quality records or re-classify a protein-coding gene to a pseudogene. The recent larger increase in total transcript records for mammalian species is not as pronounced for the human subset, in part because the data is derived from annotation that predates the use of RNA-Seq evidence in the pipeline. In addition, model RefSeq records comprise a comparatively small portion of human RefSeq transcripts as a result of our emphasis on curating human sequence data—in release 60, 79% of the RefSeq transcript records for human were curated by NCBI staff. RefSeq release 60 included transcripts for 26 266 genes, of which 43% had more than one alternatively spliced transcript record. Table 1 also indicates a continued expansion of ncRNA representation which is achieved through a combination of computed genome annotation, curation and collaboration with authoritative sources such as miRBase (4).
Table 1.
Annual growth of mammalian and human RefSeq transcript records
| Releasea | Taxab | Mammalian records |
Human records |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total transcripts | Percent modelsc | Percent curatedd | Percent ncRNAe | Total transcripts | Total genesf | Percent modelsc | Percent curatedd | Percent ncRNAe | ||
| 1 | 5 | 126 980 | 68 | 7 | <1 | 38 556 | na | 50 | 22 | <1 |
| 6 | 10 | 79 686 | 41 | 17 | <1 | 28 176 | na | 23 | 42 | <1 |
| 12 | 19 | 158 111 | 65 | 11 | <1 | 29 490 | na | 18 | 50 | 1 |
| 18 | 28 | 263 628 | 77 | 7 | 5 | 40 342 | 28 514 | 38 | 39 | 2 |
| 24 | 35 | 338 204 | 80 | 7 | 8 | 38 709 | 29 398 | 34 | 46 | 14 |
| 30 | 37 | 340 968 | 77 | 9 | 9 | 45 511 | 27 741 | 41 | 45 | 16 |
| 36 | 42 | 346 976 | 74 | 12 | 8 | 43 589 | 29 071 | 30 | 60 | 13 |
| 42 | 42 | 425 170 | 76 | 12 | 9 | 46 111 | 29 954 | 27 | 63 | 15 |
| 48 | 43 | 470 979 | 76 | 12 | 9 | 46 912 | 27 619 | 20 | 70 | 25 |
| 54 | 45 | 515 900 | 76 | 12 | 9 | 44 951 | 26 440 | 10 | 79 | 23 |
| 60 | 59 | 1 263 067 | 90 | 5 | 6 | 47 619 | 26 266 | 11 | 79 | 24 |
aRelease numbers listed correspond to ∼12 month intervals beginning from the first release in June 2003. The number of human transcripts in release 60 (July 2013) reflects the November 2012 genome annotation of three assemblies (GRCh37.p10, HuRef and CHM1_1.0) plus records added through ongoing curation activities.
bThe number of distinct NCBI Taxonomy IDs included in the RefSeq vertebrate_mammalian FTP directory that have a publicly available nuclear genome records. Twelve taxa are represented by un-annotated ENCODE genomic region records only. Mammals for which only a mitochondrial genome sequence is available are excluded. Data reported in Table 1 were extracted from archived reports available at ftp://ftp.ncbi.nlm.nih.gov/refseq/release/release-catalog/ using files named as ‘RefSeq-release##.catalog.gz’.
cThe percent of total transcripts that are model RefSeqs (with XM or XR accession prefix) generated by NCBI’s eukaryotic annotation pipeline. The percent known RefSeqs (with NM or NR prefix) can be inferred from this value (100% − percent model RefSeqs = percent known RefSeqs).
dThe percent of total transcripts that are known RefSeq records that have been curated by NCBI staff and are annotated with a ‘validated’ or ‘reviewed’ status in the COMMENT block of the RefSeq record. Validated records have undergone sequence review by NCBI staff, whereas a reviewed record includes curation of descriptive information, such as names, publications and a RefSeq summary in addition to sequence review. Known RefSeq records that have not been curated are not included; thus, the number of model records and curated records do not sum to 100%.
eThe percent of total transcripts that are not protein coding. This includes model or known long non-coding RNAs (lncRNA), small RNAs (e.g. microRNA, snoRNA, etc.), ribosomal RNAs and transcribed pseudogenes. Transfer RNAs, which are annotated on genomic records using tRNAscan but not tracked with RefSeq accessions, are not included.
fThe number of human genes per release was derived using FTP files named as ‘release##.accession2geneid.gz’. This file was not provided prior to release 14.
In addition to FTP access, RefSeq data are freely available from the NCBI web sites using queries, as BLAST databases, or through NCBI’s programing utilities (13). Subscribers to the refseq-announce mail list receive periodic updates about major changes and a summary of the content of each RefSeq FTP release (http://www.ncbi.nlm.nih.gov/mailman/listinfo/refseq-announce/). Some announcements are also provided in other NCBI forums, including the NCBI Newsletter, NCBI staff Twitter account and on the RefSeq web site (http://www.ncbi.nlm.nih.gov/refseq/), where other documentation about the project can be accessed.
UPDATE ON THE EUKARYOTIC GENOME ANNOTATION PIPELINE
The NCBI eukaryotic annotation pipeline adds value to the growing number of genome assemblies deposited into the INSDC databases by producing consistent annotation on RefSeq copies of the submitted assemblies. The evidence-based pipeline generates genome annotation based on alignments of known RefSeq transcripts and proteins, INSDC transcripts and UniProtKB/Swiss-Prot proteins with some supplemental HMM modeling based on the GENSCAN algorithm (14) used for genomes with less primary data. Extensive redevelopment of the pipeline over the last few years, including greater automation and parallel computing, resulted in a considerable increase in pipeline throughput. These improvements have allowed us to release a total of 42 genome annotations between January and August 2013, including 32 vertebrate genomes of which 22 are mammals. This number is nearly double the number of annotations produced in the entirety of 2012 (Figure 1). NCBI annotation priorities and a list of the genomes currently or recently annotated, with links to annotation release-specific resources, can be found on the eukaryotic annotation status page: http://www.ncbi.nlm.nih.gov/genome/annotation_euk/status/.
Figure 1.
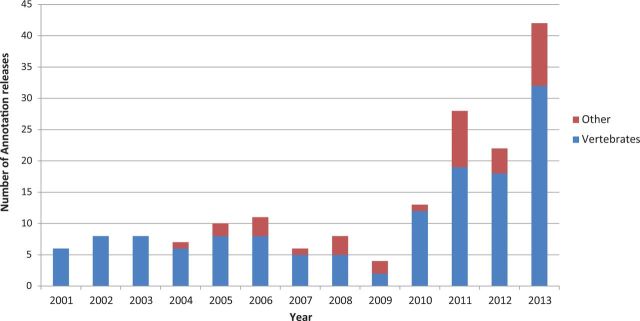
Number of vertebrate and other eukaryotic genome annotations released by NCBI per year since 2001. Additional information about recently annotated genomes is available at http://www.ncbi.nlm.nih.gov/genome/annotation_euk/status/#recent.
In 2013, the pipeline was further improved by the addition of a dataflow that uses RNAseq sequences deposited in NCBI’s Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) for gene prediction. The computational challenge posed by the vast amount and short length of reads generated by next-generation technologies was addressed in several ways: only single representatives of identical sequences retrieved from SRA are aligned, alignments with near-identical boundaries and same splice sites are collapsed and alignments representing very rare introns, likely to be background noise, are filtered out. At each step, information is recorded about the samples and number of reads represented by each alignment, so the level of support can be used to evaluate alignments and gene predictions.
The addition of short reads to the pool of evidence used for gene prediction permits the identification of more protein-coding and ncRNA alternative splice variants by the NCBI gene prediction software, Gnomon, and consequently, the supplementation of the RefSeq set. A recent policy change allows a gene to be represented by a mixture of known and predicted model RefSeqs providing a model RefSeq has an alternative splice pattern and all introns are supported by a single transcript or protein or by a single RNAseq sample (Figure 2). Consequently, model RefSeq annotation was modified to include computed transcript variant and, as relevant, protein isoform names that begin with ‘X’ to distinguish them from the analogous names provided for known RefSeqs, although for model RefSeqs a ‘Transcript Variant’ description comment is not included. These changes in the annotation pipeline account for the recent larger rate of increase in the number of mammalian records included in the RefSeq dataset. Other changes to model transcript and protein records include the addition of a ‘Genome Annotation Data’ structured comment that, among other things, identifies the model record as a product of a specific NCBI annotation release. In addition, the scope of supporting evidence reported as a note on the gene feature now includes RNAseq data (e.g. XM_005267559.1).
Figure 2.

Both known and model RefSeq records may be associated with the same locus. A portion of the ‘Genomic regions, transcripts, and products’ section of the Gene record for human GSTZ1 (NCBI GeneID 2954) is shown. Chromosome 14 coordinates corresponding to annotation of assembly GRCh13.p13 (NC_000014.8), NCBI annotation release 105 are shown at the top. The gene is associated with three known RefSeq transcripts (e.g. NM_145870.2, NM_145871.2 and NM_001513.3) and three model transcripts (e.g. XM_005267557.1, XM_005267558.1 and XM_005267559.1). The first exon of the overlapping POMT2 gene is also visible in this display. Supplementing curated RefSeqs (NM, NR, or NP prefixes) with model RefSeqs (XM, XR and XP accessions) enables better representation of alternative splice variants and exons.
UPDATE ON MAMMALIAN TRANSCRIPT AND PROTEIN RECORDS
To make evidence that supports the primary sequence and other curation decisions more discernible, changes were implemented to a subset of human and other vertebrate RefSeq records. RefSeq records that are in scope for curation (with the NM, NR and NP prefixes) may now include one or more structured comments labeled ‘Evidence Data’ or ‘RefSeq Attributes’ (Figure 3). Policy changes were also made that affect exon feature annotation as well as the decision to represent a transcript as non-coding versus protein-coding.
Figure 3.
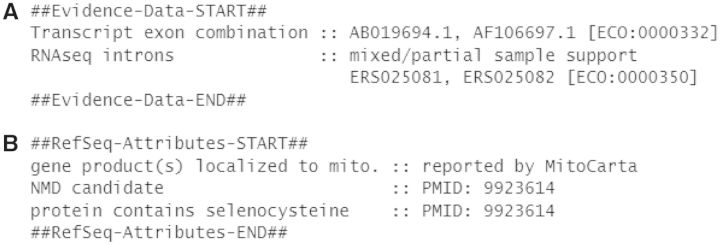
Structured comments provide information on supporting evidence and biological attributes. A portion of the COMMENT section of the NM_006440.4 record is displayed, illustrating the two structured comments. (A) The Evidence Data comment reports supporting evidence for the exon combination represented in the record. (B) The RefSeq Attributes comment reports biological attributes. Each comment type includes the attribute category on the left and supporting evidence on the right. Structured comments include special formatting and are bracketed by START and END to support parsing.
Reporting transcript supporting evidence
Evidence for the exon combination represented by a RefSeq transcript record is now reported in a structured comment with the header ‘Evidence Data’ (Figure 3A). Evidence is provided based on interpretation of Splign (15) alignments between the reference genome assembly and RefSeq transcripts, GenBank cDNA, expressed sequence tag (EST) and RNAseq reads available from the SRA; it is only provided when these primary data are available and high quality alignments to the reference genome exist. Any transcript reported as evidence must clearly place better at that genomic location than any other, match all splice sites found in the RefSeq alignment to the genome and not contain additional splice sites for the transcript or for the coding sequence (CDS), depending on evidence category. If RefSeq and transcript alignments to the reference genome are partial (e.g. due to a genome assembly issue), then supporting transcripts must have an alignment coverage >99% compared with the RefSeq transcript. Evidence categories are reported with up to two supporting identifiers (accession.version or RNAseq BioSample identifier) and the Evidence Code Ontology (ECO; http://www.evidenceontology.org/) identifier. The following categories are reported currently:
Transcript exon combination—the complete exon combination of the RefSeq transcript is supported by at least one cDNA or EST that spans all splice sites when aligned to the reference genome. This category is reported with the ECO ID ECO:0000332, e.g. NM_005589.3.
CDS exon combination—the exon combination of the RefSeq transcript is supported by at least one cDNA or EST that spans all splice sites within the annotated CDS feature. This category is reported with the ECO ID ECO:0000331, e.g. NM_002075.2.
RNAseq introns, single sample—all intron positions (as observed when the RefSeq transcript is aligned to the reference genome) are supported by a single RNAseq sample. This category is reported with the ECO ID ECO:0000348, e.g. NM_001278586.1.
RNAseq introns, mixed/partial sample support—some or all of the observed intron positions of the RefSeq transcript are supported by a combination of more than one RNAseq sample. This category is reported with the ECO ID ECO:0000350, e.g. NM_005589.3.
Transcript is intronless—intronless transcript alignments support an intronless RefSeq transcript. This category is reported with the ECO ID ECO:0000345, e.g. NM_205823.2.
Records annotated with the Evidence Data structured comment can be retrieved from the Nucleotide database using the comment label or category term (e.g. ‘evidence data [properties]’ or ‘transcript exon combination [properties]’). The Evidence Data structured comment is now reported on over 106 000 mammalian transcript records, including over 38 000 records for human (Table 2).
Table 2.
Number of mammalian and human transcript records annotated with evidence support, by evidence type categories
| Evidence type | Number of transcript recordsa |
|
|---|---|---|
| Mammals | Human | |
| transcript exon combination | 96 124 | 33 378 |
| CDS exon combination | 2985 | 1486 |
| Intronlessb | 1968 | 734 |
| RNAseq, single samplec | 33 446 | 23 251 |
| RNAseq, mixed/partialc | 12 918 | 11 056 |
| Total distinct transcript records | 106 895 | 38 911 |
aCounts as of 10 September 2013.
bThis evidence category is supported by a combination of curation and alignment evaluation and is under-reported.
cRNAseq data was used as an input reagent in calculating genome annotation for nine organisms at this time.
Reporting RefSeq attributes
The following biological attributes of the gene, transcript or protein are now reported, along with supporting information, on RefSeq transcript and protein records in a structured comment with the header ‘RefSeq Attributes’ (Figure 3B). This information is computationally identified, imported from external data sources (as indicated below) or added by NCBI staff after review of the evidence. Attributes are applied if there is evidence that the characteristic exists at least some of the time (e.g. in a particular tissue or experimental condition).
Bicistronic transcript—one transcript that may produce two distinct proteins from non-overlapping open reading frames (ORFs) based on review of published reports, e.g. NM_021267.3.
CDS uses downstream in-frame AUG—an upstream in-frame AUG codon exists but a curation decision was made to not use it for the annotated coding sequence based on curator review of publications, sequence conservation and considerations of a strong history of community use or signal peptide and protein domains. The upstream alternate AUG codon is annotated as a miscellaneous feature, e.g. NM_138448.3.
Imprinted gene—the transcript is expressed only from the paternal or maternal chromosome based on published reports, e.g. NM_016352.3.
Inferred exon combination—the exon combination of the annotated CDS feature is inferred by curators based on partial transcripts, protein homology, sequence analysis and/or publications. This attribute is currently stored by NCBI staff and is underreported. A single transcript spanning the exon combination was not available at the time of review, e.g. NM_133379.4.
Gene product(s) localized to mitochondrion—one or more product of this gene may be localized to the mitochondrion. Reported on all transcript variants for a gene based on a combination of nomenclature rules, homology data, publications and data imported from MitoCarta (16), e.g. NM_018394.3.
Non-AUG initiation codon—the annotated CDS uses a non-AUG translation initiation codon based on a published report or curator inference based on sequence conservation, e.g. NM_021182.1.
Nonsense-mediated messenger RNA (mRNA) decay—the transcript is a candidate for nonsense-mediated mRNA decay (NMD) (17) but is considered protein-coding based on published evidence for the protein or support from conservation, e.g. NM_018790.3. RefSeq defines a transcript as an NMD candidate when the last base of the stop codon is located >50 nt upstream of the terminal splice acceptor site.
PolyA required for stop codon—sequence conservation supports a protein-coding gene and for this species the stop codon is completed by polyadenylation. These loci may be unitary pseudogenes rather than protein-coding genes, e.g. NM_001145051.2.
Protein contains selenocysteine—the protein product contains a selenocysteine amino acid that is encoded by UGA, typically a translation termination codon, based on published reports or conservation, e.g. NM_000581.2.
Readthrough transcript—the transcript shares exons with two or more distinct genes, indicated by the NCBI GeneIDs displayed, e.g. NM_007203.4.
Ribosomal slippage—the transcript uses a programmed translational frameshift to encode the protein based on published reports for the gene or an ortholog, e.g. NM_204916.1.
Undergoes RNA editing—the transcript may undergo RNA editing based on a published report describing the gene or an ortholog. Reported on all transcript variants for the gene, e.g. NM_000826.3.
Unitary pseudogene—the locus is a pseudogene in this species but has a functional ortholog in at least one other species based on published reports or sequence review. The functional ortholog is indicated, e.g. NR_003227.1.
Records annotated with the RefSeq Attributes structured comment can be retrieved by querying the Nucleotide database with ‘refseq attributes[properties]’. Individual categories can be retrieved using the specific phrase (e.g., ‘undergoes RNA editing[properties]’). Currently, the RefSeq attribute structured comment is displayed on more than 7400 mammalian transcript records, including more than 3000 human records.
The majority of the above attributes are stored by RefSeq curation staff during the course of reviewing the sequence and publication data associated with a gene. Although most attributes are simply informative, curation of others including ‘protein contains selenocysteine’ and ‘ribosomal slippage’ plays a critical role in ensuring correct sequence representation and in turn, correct genome annotation. Transcripts that contain selenocysteine (Sec) use the UGA codon (typically a translation termination signal) to incorporate the Sec residue. Ribosomal slippage requires a ribosome to shift from the initiating reading frame to one of the two other frames. Neither occurrence follows standard rules of protein synthesis. Consequently, curation is required because standard computational tools cannot distinguish the dual functionality of the UGA codon or predict a programmed frameshift. Currently, the curated RefSeq dataset includes records for 267 selenoprotein-containing genes from 17 mammals (including the 25 known human genes) and 33 ribosomal slippage genes from 10 mammals.
Exon feature annotation
Exon features are annotated on human and mouse RefSeq transcripts based on Splign alignments to the reference assembly. Exon numbers are no longer provided. Our previous convention was to number the exons annotated on each transcript based on the set of exons represented in all RefSeq transcripts for the gene; exon numbers were not transcript-specific and were not stable because they were recalculated if a new RefSeq transcript record was added to the gene. Thus, adding a new record that included a more distal 5′ exon would result in renaming all of the exons on the other RefSeq records for the gene. The instability of exon numbers reported on RefSeq transcript records led to confusion and could have negative consequences if exon numbers were treated as a stable nomenclature in scientific communications. RefSeqGene genomic records continue to provide exon number information for the subset of transcripts selected as cDNA reference standards (described below).
Protein annotation criteria
A protein is annotated on a transcript based on an accumulated knowledge of translation, published data, conservation and consideration of the location and length of the ORF. Predicting a protein product encoded by an alternatively spliced transcript may be straightforward when the predicted protein is translated from the same start codon, is in the same reading frame and is of similar length to a ‘canonical’ form. Uncertainty in prediction arises when upstream ORFs are present that may have an inhibitory effect on downstream translation or when alternative splicing has a significant effect on the CDS compared with the canonical form. For example, alternative splicing may produce a variant that shares the translation initiation codon with the canonical form, but a downstream frameshift either introduces a premature stop codon that may render the transcript a candidate for NMD, or generates a C-terminus diverged from the canonical form.
The RefSeq Project is increasing the representation of proteins in certain situations given findings in ribosome profiling (18) that suggest translation initiation can occur at multiple sites on a given transcript. We are no longer treating upstream ORFs that have a strong Kozak signal and encode proteins of more than 35 amino acids as inhibitory based on newer studies that indicate upstream ORFs (uORFs) appear to negatively regulate but not fully inhibit translation of the downstream primary ORF (19,20). Non-coding RefSeq transcripts associated with protein-coding genes are under review to determine if translation of a downstream ORF is likely. Transcripts that may be subject to NMD will be represented as protein-coding if a downstream in-frame ORF that can encode a protein at least 50% the length of the canonical form exists (and is not predicted to be subject to NMD). Predicted proteins that are <50% the length of the canonical protein for the gene may still be represented in some cases when considering additional criteria such as shared translation initiation codon, epigenomic evidence for novel promoters, publications and sequence conservation. Although the arbitrary nature of a length threshold is recognized, our previous approach did not sufficiently consider ribosomal leaky scanning, re-initiation pathways or other translational regulatory factors such as mRNA structure and was overly conservative. Although this revised policy will result in annotation of a larger number of predicted alternate proteins, some alternatively spliced transcripts will continue to be represented as non-coding because no ORF meets our quality criteria.
REFSEQGENE STATUS
RefSeqGene (http://www.ncbi.nlm.nih.gov/refseq/rsg) is the subset of the RefSeq project that provides human gene-specific genomic sequence records to serve as stable reference standards for clinical reporting. The sequence and its feature annotations are established in collaboration with the Locus Reference Genome (LRG) project (http://lrg-sequence.org) (21), the CCDS project (9) and gene-specific experts. A genomic sequence is identified to represent a gene including sufficient sequence to include possible core regulatory regions (usually about 5 kb upstream and 2 kb downstream of the terminal exons); the RefSeqGene record always represents the gene in the sense strand. Transcript alignments are packaged with the RefSeqGene record to facilitate mapping coordinates from the RefSeqGene to reference standard transcripts. RefSeq transcripts that are selected as reference standards for feature annotation of RefSeqGene records (including exon features) are identified in the COMMENT section of the RefSeq record (see NM_000820.2 for an example). Exons of a RefSeqGene are named in two stages. Until an LRG reference standard sequence is established, the exons are annotated according to the RefSeq transcripts selected to determine exon placement. For example, RefSeqGene NG_029703.1 includes exon numbers for the GAS6 gene based on NM_000820.2, but not on other available transcripts for the gene (NM_001143945.1 and NM_001143946.1). As more stakeholders are included as the LRG reference standard is established, the set of reference transcripts may be updated and the exon numbers may be updated to their final state. Versions of the RefSeqGene record with an LRG identifier (e.g. LRG_1) match the LRG record exactly with respect to sequence, exons and annotated transcripts. The relationships between RefSeqGene and public LRG sequences are reported daily to RefSeqGene’s ftp site (ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/RefSeqGene/LRG_RefSeqGene); at the time of writing this manuscript, <10% of RefSeqGene sequences had been implemented as LRG.
RefSeqGene records are annotated with variations in NCBI’s dbSNP (22) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) databases, so the graphical display (e.g. http://www.ncbi.nlm.nih.gov/nuccore/NG_012772.3?report=graph) can be used to explore the location and effect of medically important variation. RefSeqGene records are retrieved from the Nucleotide database using the query ‘refseqgene[keyword]’ and are exported to the ftp site (ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/RefSeqGene/) weekly. The RefSeqGene web site provides a report of the ∼5100 genes currently represented, along with names of related disorders and links to access more information in Gene, LRG, the Genetic Test Registry (GTR) (23) and Online Mendelian Inheritance in Man (OMIM) (24). NCBI’s remapping service (http://www.ncbi.nlm.nih.gov/genome/tools/remap#tab=rsg) supports mapping coordinates among the genome assemblies, RefSeqGene, LRG and RefSeq transcript records.
FUTURE DIRECTIONS
The set of RefSeq attributes described herein will be expanded. For example, future goals include reporting attributes for regulatory uORFs, indicating when RefSeq transcripts for a gene originate from inferred (epigenomics support) or published alternate promoters, multifunctional ‘moonlighting’ proteins, polymorphic pseudogenes (where a locus is a pseudogene or encodes a protein product depending on population) as well as providing information about curator uncertainty with regard to the protein product (e.g. an alternative transcript with a short predicted protein or transcripts with long 5′-UTRs that contain uORFs). We are currently focusing on expanding attribute reporting based on observations or analysis of the transcript and protein sequence. Mitochondrial localization information is provided with the initial attribute set because: (i) this impacts a large number of proteins; (ii) the mitochondrial transit peptide may be mis-reported as a signal peptide by some prediction programs and (iii) the information is more readily available. We are not currently planning to report other subcellular localization data as it is frequently difficult to determine exactly which protein (when there are multiple alternative splice variants) is actually localized to the reported location. In addition, an additional category of Evidence Data is planned that will report evidence in support of the annotated start codon, based partially on the incorporation of ribosome profiling data into RefSeq curation workflow.
Significant growth in the RefSeq mammalian (and vertebrate) collection is anticipated. Improvements to the NCBI eukaryotic genome annotation pipeline will expand representation of the number of taxa, the number of alternatively spliced transcripts and the total number of exons. Criteria to expand the use of RNAseq data are being developed for several purposes, including its use as a source of primary evidence for extending UTRs and for creating one- to two-exon ncRNA records lacking polyadenylation support, where the transcript is supported by more than one RNA-Seq sample.
Other development plans include providing detailed genome annotation reports on the evidence data used by the annotation pipeline, the quality of the resulting annotation and information regarding what annotation changed following annotation of a new assembly version or re-annotation of an existing assembly. Development to restructure and improve the organism-oriented data downloads from the Genomes FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/) is in progress. In addition, we are currently working to refine and expand on NCBI’s calculation of RefSeq orthologs and paralogs, as reported in the HomoloGene and Gene resources, and on further curation of this content.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank our collaborators, especially the HUGO Gene Nomenclature Committee, the Mouse Genome Database, Rat Genome Database, UniProt and CCDS curators at the University of California Santa Cruz and the Wellcome Trust Sanger Institute for many fruitful discussions regarding correct genome annotation, gene location and nomenclature. We also thank the numerous individual scientists who have contacted us over the years to suggest an improvement. We sincerely value your input to help improve the RefSeq database content.
REFERENCES
- 1.Pruitt KD, Katz KS, Sicotte H, Maglott DR. Introducing RefSeq and LocusLink: curated human genome resources at the NCBI. Trends Genet. 2000;16:44–47. doi: 10.1016/s0168-9525(99)01882-x. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura Y, Cochrane G, Karsch-Mizrachi I. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson J, Halliwell JA, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res. 2013;41:D1222–D1227. doi: 10.1093/nar/gks949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karro JE, Yan Y, Zheng D, Zhang Z, Carriero N, Cayting P, Harrrison P, Gerstein M. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007;35:D55–D60. doi: 10.1093/nar/gkl851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray KA, Daugherty LC, Gordon SM, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2013. Nucleic Acids Res. 2013;41:D545–D552. doi: 10.1093/nar/gks1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bult CJ, Eppig JT, Blake JA, Kadin JA, Richardson JE. The mouse genome database: genotypes, phenotypes, and models of human disease. Nucleic Acids Res. 2013;41:D885–D891. doi: 10.1093/nar/gks1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dwinell MR, Worthey EA, Shimoyama M, Bakir-Gungor B, DePons J, Laulederkind S, Lowry T, Nigram R, Petri V, Smith J, et al. The Rat Genome Database 2009: variation, ontologies and pathways. Nucleic Acids Res. 2009;37:D744–D749. doi: 10.1093/nar/gkn842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, Chen HC, Agarwala R, McLaren WM, Ritchie GR, et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091. doi: 10.1371/journal.pbio.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalgleish R, Flicek P, Cunningham F, Astashyn A, Tully RE, Proctor G, Chen Y, McLaren WM, Larsson P, Vaughan BW, et al. Locus Reference Genomic sequences: an improved basis for describing human DNA variants. Genome Med. 2010;2:24. doi: 10.1186/gm145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 15.Kapustin Y, Souvorov A, Tatusova T, Lipman D. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol. Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. Genome-wide annotation and quantitation of translation by ribosome profiling. In: Frederick MA, editor. Current Protocols in Molecular Biology. John Wiley & Sons, Inc.; 2013. Chapter 4, Unit 4, p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somers J, Poyry T, Willis AE. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013;45:1690–1700. doi: 10.1016/j.biocel.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natil Acad. Sci. USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacArthur JAL, Morales J, Tully RE, Astashyn A, Gil L, Bruford EA, Dalgleish R, Larsson P, Flicek P, Maglott DR, et al. Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence variants. Nucleic Acids Res. in press doi: 10.1093/nar/gkt1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubinstein WS, Maglott DR, Lee JM, Kattman BL, Malheiro AJ, Ovetsky M, Hem V, Gorelenkov V, Song G, Wallin C, et al. The NIH genetic testing registry: a new, centralized database of genetic tests to enable access to comprehensive information and improve transparency. Nucleic Acids Res. 2013;41:D925–D935. doi: 10.1093/nar/gks1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009;37:D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]