


Abstract
Proper selection of the translation initiation site (TIS) on mRNAs is crucial for the production of desired protein products. Recent studies using ribosome profiling technology uncovered a surprising variety of potential TIS sites in addition to the annotated start codon. The prevailing alternative translation reshapes the landscape of the proteome in terms of diversity and complexity. To identify the hidden coding potential of the transcriptome in mammalian cells, we developed global translation initiation sequencing (GTI-Seq) that maps genome-wide TIS positions at nearly a single nucleotide resolution. To facilitate studies of alternative translation, we created a database of alternative TIS sites identified from human and mouse cell lines based on multiple GTI-Seq replicates. The TISdb, available at http://tisdb.human.cornell.edu, includes 6991 TIS sites from 4961 human genes and 9973 TIS sites from 5668 mouse genes. The TISdb website provides a simple browser interface for query of high-confidence TIS sites and their associated open reading frames. The output of search results provides a user-friendly visualization of TIS information in the context of transcript isoforms. Together, the information in the database provides an easy reference for alternative translation in mammalian cells and will support future investigation of novel translational products.
INTRODUCTION
In all kingdoms of life, mRNA translation represents the last step of the flow of genetic information and primarily defines the proteome. Translation is a complex process, consisting of initiation, elongation, termination and ribosome recycling (1). Initiation is considered to be the rate-limiting step and determines the overall rate of translation (2). In eukaryotes, the cap-dependent initiation mechanism accounts for the vast majority of cellular mRNA translation. During initiation, the 43 S pre-initiation complex (PIC) is recruited to the 5′ end m7G cap structure of mRNA with the aid of many translation initiation factors. It is generally accepted that PIC migrates along the 5′ untranslated region (5′ UTR) in an ATP-dependent process known as scanning, until it encounters a start codon, normally the first AUG. Following the start codon recognition, the 60S ribosomal subunit joins to form the 80S ribosome complex and elongation now begins. The scanning model implicates that the features of the 5′UTR have major influences on the start codon selection. Interestingly, non-AUG start codons, such as CUG, could also serve as initiators (3,4). In contrast, failed recognition of an initiation codon results in continuous scanning of the PIC and initiating at a downstream site, in a process known as leaky scanning (5). In addition to the cap-dependent mechanism, translation could also be initiated in a cap-independent manner. For instance, internal initiation can be mediated by a secondary structure within the 5′UTR known as an internal ribosome entry site (IRES) (6,7). This alternative translation initiation is believed to be regulated under different growth conditions. However, fundamental principles governing the selection of translation initiation sites (TIS) remain unclear.
The functional significance of alternative translation is multifaceted. First, selection of upstream TIS codons leads to generation of upstream open reading frames (uORFs), which directly regulate downstream protein synthesis from the main open reading frame (ORF) (8,9). Second, translation via alternative TIS sites produces protein isoforms differing in NH2-terminal sequences when these alternative initiators are in the same reading frame (10). Depending on the position of TIS sites relative to the annotated start codon, either the NH2-terminal extended or truncated isoforms will be produced. Third, totally different proteins will be generated if the alternative TIS sites are in different reading frames. Therefore, alternative translation reshapes the landscape of the proteome by increasing both diversity and complexity of translational products.
The existence of alternative TIS codons clearly indicates that the coding potential of a given genome is much richer than we previously thought. Given the physiological importance of alternative translation, there is an urgent need for techniques suitable for mapping global TIS positions. Early attempts applied machine-learning techniques to identify novel TIS sites on cDNA or genomic sequences based on sequence features summarized from the known start codons and their flanking sequences (11,12). However, in silico sequence analysis cannot precisely predict alternative TIS sites, in particular non-AUG codons. Recent development of ribosome profiling techniques allows monitoring ribosome dynamics with unprecedented resolution at the genome-wide scale (13). To capture translation initiation events, some variants of ribosome profiling have been developed by applying distinct translation inhibitors to freeze initiating ribosomes. Ingolia et al.used harringtonine to stall the first 80S ribosome complex during initiation (14). Because harringtonine cannot completely block the initiating ribosomes, TIS identification relies on a support vector machine (SVM) learning technique trained with prior TIS information (14). In this instance, additional filtering steps were required to increase the accuracy of identified TIS positions. The extensive computational processing of the sequencing data likely introduces bias in identifying non-canonical TIS sites. Fritsch et al.used puromycin to enrich ribosomes near the start codon because initiating ribosomes are less sensitive to this translation inhibitor (15). The identification of TIS sites once again relies on a machine learning technique based on neural networks (15). In addition, it only focuses on a region covering the 5′UTR and the first 30 nt of the coding region, which could miss TIS sites downstream of the annotated start codon.
To circumvent the drawbacks mentioned above and improve the accuracy of TIS identification, we developed global translation initiation sequencing (GTI-Seq) that permits precise TIS identification at the nucleotide resolution (16). The rationale of GTI-seq is to use a translation inhibitor lactimidomycin (LTM) that is capable of completely freezing the initiating ribosomes. Unlike the commonly used translation inhibitor cycloheximide, LTM preferentially acts on the initiating ribosomes because it only binds to the empty E-site of the ribosome that is normally occupied by deacylated tRNA during elongation. Indeed, ribosome footprints associated with LTM were highly enriched at the annotated start codon. The single nucleotide resolution permits frame shifting analysis of ORFs. Importantly, GTI-Seq uses a straightforward computational approach in mapping global TIS sites, minimizing possible bias introduced by data processing. From GTI-seq data sets, a few novel alternative TIS codons have been experimentally validated. The accuracy of TIS identification was further supported by evolutionary conservation between human and mouse genomes.
A high precision map of global TIS positions will ignite numerous interests in deciphering physiological functions of alternative translation. To facilitate mechanistic investigation of alternative translation for individual genes, we designed a comprehensive TIS database (TISdb) built on multiple high-resolution GTI-seq data sets. A web search interface is provided with certain filtering options to narrow down the search results. The information of TIS-associated ORFs as well as reading frames is presented as an image for easy visualization. To our knowledge, TISdb is the first public database covering both canonical and alternative TIS positions on a genome-wide scale. The database is a ready resource for researchers looking for alternative translation on individual genes and provides a unique view depicting the richness of genome coding potential.
METHODS
Data generation
Four biological replicates of GTI-seq data from HEK293 and one set of GTI-seq data from a MEF cell line were used to identify putative TIS sites of human and mouse genomes, respectively (16,17). Using Tophat (18), the GTI-seq data were first mapped to the corresponding genome and transcriptome downloaded from UCSC genome browser (hg19 and mm10) (19). From the uniquely mapped reads, the 13th nucleotide (12 nt offset from the 5′ end) was inferred as the ribosome P-site position, which corresponds to the start codon recognized by the initiation Met-tRNA during translation initiation. These uniquely mapped reads were then intersected with the NCBI Refseq gene annotation to quantify the P-site read count for each individual mRNA transcript. Given the fact that many P-sites have a small number of GTI-seq reads and the distribution of the P-site read count is apparently Poisson over-dispersed (unequal mean and variance), we applied the zero-truncated binomial negative (ZTNB) model to determine P-sites with a statistically significant number of read counts. The ZTNB model can handle non-zero digital values of high-throughput sequencing data and has been applied to cross-linking immunoprecipitation sequencing (CLIP-seq) (20). A global ZTBN model was first fit over all the non-empty P-sites in the entire transcriptome. Second, for each individual transcript, a local ZTNB model was trained on the non-zero P-sites of this transcript. The P-site satisfying the P-value cut-offs based on the parameters estimated in the global and local ZTBN model was categorized as a putative TIS codon.
From human GTI-seq data, putative TIS codons discovered in at least three biological replicates (out of four) were compiled into the final database. The final database in the current release of TISdb contains 6991 TIS sites from 4961 human genes and 9973 TIS sites from 5668 mouse genes. Compared with the Refseq gene annotation, ∼30% of the total predicted TIS sites match the annotated TIS (aTIS) codons. Approximately 50% of identified TIS sites belong to the upstream TIS (uTIS) codons and ∼20% are downstream TIS (dTIS) (Table 1). In terms of codon composition, >50% of the identified TIS sites use AUG, whereas ∼30% use near-cognate codons that only differ at one position from AUG (i.e. AUC, AUA, AUU, CUG, GUG, UUG, AUG, AAG and AGG). It is also worth mentioning that the percentages of the AUG and the near-cognate codons are underestimated because a small portion of GTI-seq reads have 1 nt offset to the 13th P-site position. Therefore, <10% of the human TIS sites and 20% of the mouse TIS sites in our database may either use an unconventional TIS codon or represent a false positive identification.
Table 1.
Statistics of translation initiation sites in the TISdb
| Species | Start codon sequence |
TIS category |
Total TIS (gene) | |||||
|---|---|---|---|---|---|---|---|---|
| AUG | Near cognate | Others | Annotated | Upstream | Downstream | Others | ||
| Human | 4090 | 2132 | 769 | 2575 | 3540 | 678 | 198 | 6991 (4961) |
| Mouse | 4400 | 3144 | 2429 | 2729 | 5099 | 2051 | 94 | 9973 (5668) |
Web interface
The web interface is developed using Python Django web framework, which is served by Apache. The TIS data are stored in an SQLite database. All the TIS information, including the codon sequence and predicted ORF, can be downloaded from a separate ‘Download’ page. In addition, the ‘Main page’ and ‘Help’ page contain details about data generation, result interpretation and other relevant information.
Database features
The TISdb database provides a user-friendly web interface for searching putative TIS sites on individual genes from either the human or mouse genome. In addition to the general query, the database allows users to specify a set of filter criteria to focus on the TIS sites of their interests. The query returns a summary table and a carousel containing images illustrating the positional information of TIS sites and the associated ORF in the context of Refseq transcript. In addition, the reading frame information is colour coded for easy visualization. These components of query results offer users a comprehensive and straightforward visualization of TIS codons.
TIS query
On the ‘Search’ page, users can perform a general query for TIS sites or a more specialized search. During general searches, users can input different types of gene identifiers, including official gene symbols, Refseq transcript ID, Ensembl gene ID and Entrez gene ID. By pasting a gene identifier list or uploading a gene identifier file, users can search up to 50 genes during an individual query. Another required field is the choice of species (either human or mouse). To narrow down the search results, users can select several optional search criteria provided by TISdb to address different aspects of TIS information. These search criteria include codon composition (canonical ATG, near-cognate initiation codon or other codons) and the type of TIS (annotated TIS, downstream TIS and upstream TIS). Given the regulatory role of uORF in gene expression, users can select the ‘uORF only’ to limit their search to uTIS and the associated uORFs.
In addition to the TIS information derived from GTI-seq, we also integrated the TIS sites identified by harringtonine-based approach and puromycin-based methods (14,15). Because of the inherent differences of those techniques, variations of TIS resolution and different cell types used in those studies, only ∼30% of TIS sites from TISdb have matching TIS positions reported in other studies. Nonetheless, the general agreement of TIS identification between different studies offers independent clues for experimental biologists to investigate alternative translation events with high confidence. By selecting ‘Supported by other studies’, users can limit their search to those TIS sites commonly identified by different methods within a 3 nt window.
TIS search results
Once the query has matched the records in the database, the ‘Result’ page will display two components: TIS summary table and ORF prediction view.
TIS summary table
The first component of the result section is a TIS summary table from selected species (Figure 1A). This table contains basic TIS information, including the species, gene symbol, transcript ID and genome coordinate of the TIS. To help interpret biological implication of the TIS, it also provides relative positional information compared with the annotated start codon. Given the regulatory role of uORF, any TIS sites associated with uORF are explicitly indicated. As discussed above, we also provide information about whether a TIS was similarly identified in other studies employing different techniques. To demonstrate the conservation feature of TIS site sequences, we calculated the average phyloP score over a window of −3 and +4 nt around the TIS site, according to the UCSC 46-way vertebrate alignment for hg19 and 60-way vertebrate alignment for mm10 (19). In general, a positive average phyloP score reflects ‘conservation’ of a TIS context region, whereas a negative value suggests ‘selection’ of a TIS context. A hyperlink is created for phyloP score in the summary table for users to retrieve the exact sequence alignment and other relevant information from the UCSC genome browser (Figure 1B). The whole summary table is organized on the basis of transcript isoforms instead of the gene. This is because a number of TIS sites are only specific to certain transcript isoforms of the same gene (for example, GAPDH). In this way, we avoid the loss of isoform-specific TIS information and make ORF prediction meaningful within the context of the mRNA transcript. Even though this could cause redundancy of TIS coordinates in the summary table, it provides a more intuitive way for biological interpretation of the TIS codon.
Figure 1.

An example of TIS search results for ACTG1 from both human and mouse genomes. (A) Summary table of TIS information for ACTG1. The coordinates correspond to the first position of the start codon. The genome coordinates are consistent with hg19 (human) or mm10 (mouse). (B) Multiple alignment of TIS context. A window of −3 and +4 nt around an upstream TIS of mouse ACTG1 is displayed. The height in the ‘Placental Cons’ represents the degree of conservation for each base in the window. The orthologous sequences of 10 vertebrate species are shown for multiple alignment.
ORF prediction view
To help users investigate the biological functions of the identified TIS codons in the context of their ORFs, a carousel containing illustrative images of each individual transcript is displayed. Each image corresponds to a queried gene. As explained in the previous section, all the transcript isoforms for that particular gene are displayed. The content of each image contains the coordinate of the TIS codon relative to the transcription start site, codon composition of the identified TIS, colour-coded frame-shift information and the predicted stop codon (TAG, TAA and TGA) position associated with that ORF (Figure 2). The caption of the image is presented as species::gene. Figure 2B is an example of the ORF prediction view of human γ-actin 1 (ACTG1). This gene has two transcript isoforms: NM_001199954 and NM_001614. In the TISdb, there are four TIS sites in both isoforms: two uTIS, one aTIS and one dTIS. However, the biological implications of the two uTIS codons are different for the two transcript isoforms. The upstream CUG uTIS is in the same reading frame as the CDS of NM_001199954, which is expected to result in a protein product with an NH2-terminal extension. In contrast, the same uTIS in NM_001614 only associates with a short uORF overlapping with the CDS. The downstream CUG uTIS is in a different reading frame relative to the annotated CDS and the associated dORFs in both transcript isoforms have distinct sequences.
Figure 2.
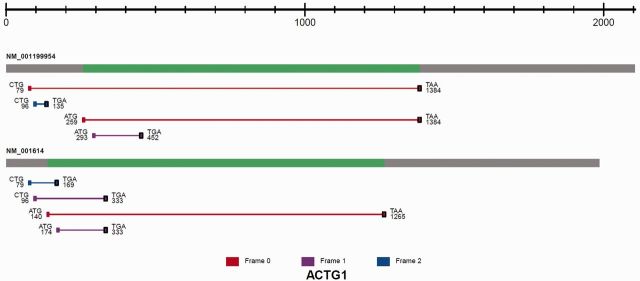
The image of predicted ORFs associated with the identified TIS sites is shown for ACTG1. All isoforms of the transcript are displayed. From left to right, the transcript structure consists of the 5′ UTR (grey), CDS (green) and 3′ UTR (grey). Each ORF is represented as a line between an identified TIS and a predicted stop codon (black square). The ORF is color coded according to reading frames (red, frame 0; purple, frame 1; blue, frame 2). The TIS codon sequence composition is shown together with the coordinates relative to the transcription start site.
FUTURE PROSPECTS
GTI-seq represents a remarkable technological advancement in mapping global mRNA translation initiation. It has the potential to reveal the hidden coding potential of the transcriptome. Comprehensive cataloguing of global TIS sites and the associated ORFs is just the beginning in unveiling the principles governing alternative translation. The enormous biological breadth of translational control has led to an enhanced appreciation of proteome diversity and complexity. Despite the advantages of GTI-Seq on TIS identification, some limitations still exist. First, current ribosome profiling approaches lack quantitative capacity. The sequencing reads density associated with either harringtonine or LTM does not truly reflect the rate of translation initiation. Therefore, the current TISdb provides the position of alternative TIS codons rather than the efficiency of alternation translation initiation. Second, the depth of ribosome profiling is influenced by the abundance of transcripts. TIS information may not be available for transcripts of low abundance. For genes with relatively high expression, GTI-seq could possibly fail to capture some TIS signals that have low initiation efficiency. Such scenarios include transcripts with multiple uTIS sites but no annotated TIS sites. Third, TIS selection is subjected to regulation under different growth conditions. Future GTI-seq experiments will focus on quantitative changes of TIS selection in response to stressors such as nutrient starvation. Fourth, tissue-specific TIS selection remains a formidable task based on technical limitations of the current ribosome profiling protocol. We envision that variants of ribosome profiling will be developed in the future to capture quantitative TIS information from various tissues across a wider range of organismal species. The current TISdb sets the stage for investigation of alternative translation and provides an important platform for studying translational control. There is little doubt that integration of GTI-seq data with other data sets, such as CHIP-seq, RNA-seq, miRNA profiling and proteomics, will present a fresh view of global post-transcriptional and -translational gene regulation. The effort to consolidate the rich biology with detailed understanding of the underlying mechanisms promises an exciting and surprising future.
FUNDING
National Institutes of Health [1 DP2 OD006449-01, 1R01AG042400-01A1]; Ellison Medical Foundation [AG-NS-0605-09] and DOD Exploration-Hypothesis Development Award [W81XWH-11-1-0236 to S.-B.Q.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank members of Qian laboratory for helpful suggestion on the website development and critical comments for the manuscript. We also thank David Smith, Kevin Murphy and Cornell information technologies for providing the virtual hosting space for the TISdb website.
REFERENCES
- 1.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2012;19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 3.Starck SR, Jiang V, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science. 2012;336:1719–1723. doi: 10.1126/science.1220270. [DOI] [PubMed] [Google Scholar]
- 4.Schwab SR, Shugart JA, Horng T, Malarkannan S, Shastri N. Unanticipated antigens: translation initiation at CUG with leucine. PLoS Biol. 2004;2:e366. doi: 10.1371/journal.pbio.0020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mokrejs M, Vopalensky V, Kolenaty O, Masek T, Feketova Z, Sekyrova P, Skaloudova B, Kriz V, Pospisek M. IRESite: the database of experimentally verified IRES structures. Nucleic Acids Res. 2006;34:D125–D130. doi: 10.1093/nar/gkj081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Medenbach J, Seiler M, Hentze MW. Translational control via protein-regulated upstream open reading frames. Cell. 2011;145:902–913. doi: 10.1016/j.cell.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl Acad. Sci. USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science. 2013;341:399–402. doi: 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadershahi A, Fahrenkrug SC, Ellis LB. Comparison of computational methods for identifying translation initiation sites in EST data. BMC Bioinformatics. 2004;5:14. doi: 10.1186/1471-2105-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saeys Y, Abeel T, Degroeve S, Van de Peer Y. Translation initiation site prediction on a genomic scale: beauty in simplicity. Bioinformatics. 2007;23:i418–i423. doi: 10.1093/bioinformatics/btm177. [DOI] [PubMed] [Google Scholar]
- 13.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012;22:2208–2218. doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S, Liu B, Lee S, Huang SX, Shen B, Qian SB. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl Acad. Sci. USA. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Han Y, Qian SB. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol. Cell. 2013;49:453–463. doi: 10.1016/j.molcel.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. The UCSC genome browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LO, Smith AD. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012;28:3013–3020. doi: 10.1093/bioinformatics/bts569. [DOI] [PMC free article] [PubMed] [Google Scholar]