


Abstract
ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) provides a freely available archive of reports of relationships among medically important variants and phenotypes. ClinVar accessions submissions reporting human variation, interpretations of the relationship of that variation to human health and the evidence supporting each interpretation. The database is tightly coupled with dbSNP and dbVar, which maintain information about the location of variation on human assemblies. ClinVar is also based on the phenotypic descriptions maintained in MedGen (http://www.ncbi.nlm.nih.gov/medgen). Each ClinVar record represents the submitter, the variation and the phenotype, i.e. the unit that is assigned an accession of the format SCV000000000.0. The submitter can update the submission at any time, in which case a new version is assigned. To facilitate evaluation of the medical importance of each variant, ClinVar aggregates submissions with the same variation/phenotype combination, adds value from other NCBI databases, assigns a distinct accession of the format RCV000000000.0 and reports if there are conflicting clinical interpretations. Data in ClinVar are available in multiple formats, including html, download as XML, VCF or tab-delimited subsets. Data from ClinVar are provided as annotation tracks on genomic RefSeqs and are used in tools such as Variation Reporter (http://www.ncbi.nlm.nih.gov/variation/tools/reporter), which reports what is known about variation based on user-supplied locations.
INTRODUCTION
Interactive and programmatic access to the interpretation of medically important human variation is critical to realizing the promise of genomic medicine. ClinVar was developed to meet that need. Building from the foundation of the variants submitted with minimal phenotypic descriptions to dbSNP (1) and dbVar (2), ClinVar now accepts direct submissions with rich, structured details of phenotype, interpretation of functional and clinical significance, methodology used to capture variant calls and supporting evidence. Submissions are categorized by type of data capture (e.g. clinical testing, literature evaluation and research) and level of review status (single submission, expert panel and practice guideline). ClinVar thus provides all users access to a broader set of clinical interpretations than they may have collected on their own and the promise of a comprehensive site for obtaining current and historical data. ClinVar is available to individual users and organizations that want to incorporate the data into local applications and workflows. ClinVar was released as a prototype in November 2012, with the official launch in April 2013.
CONTENT
ClinVar’s content can be divided into five major categories: submitter, variation, phenotype, interpretation and evidence. The unique combination of submitter, variation and phenotype determines a record unit and is assigned an accession in the format SCV000000000.0 (SCV). Whenever possible, the content is highly structured and harmonized to controlled vocabularies or other data standards. These standards are discussed in more detail in the following sections. Controlled terms used by ClinVar and the NIH Genetic Testing Registry (GTR) (3) are reported on GTR’s File Transfer Protocol (FTP) site (ftp://ftp.ncbi.nlm.nih.gov/pub/GTR/standard_terms/).
Scope
ClinVar accepts submissions for variations identified through clinical testing, research and literature curation. At this time, ClinVar does not include unreviewed data from GWAS studies, although variants that were determined to be of interest through GWAS and have been curated to provide an interpretation of clinical significance are in scope.
Submitter
ClinVar represents submitters as both organizations and individuals. The infrastructure supporting this content is shared with the GTR, dbSNP and dbVar. Submitters have the right to request anonymity, although to date no submitter has elected this option. Summary data about submissions are provided on the web (http://www.ncbi.nlm.nih.gov/clinvar/submitters/).
Variation
Variation is a key component of ClinVar’s data model, especially to be able to represent the relationship of variation to phenotype. Variation is thus reported as the sequence at one location or as a combination of sequence changes at multiple locations. In other words, ClinVar can represent the interpretation of a single allele, compound heterozygotes, haplotypes and combinations of alleles in different genes. Variation is modeled in the database as a set of distinct sequences, but currently most sets have only one member (Table 1). The goal is to represent each variation relative to at least one reference sequence, but the data flow from some submitters is not amenable to establishing this immediately. Thus, free text describing a variant is accepted if that text is connected to a public record.
Table 1.
Overview of elements of a ClinVar record
| Content | XML | Comment |
|---|---|---|
| Variation | ||
| Set of variants | MeasureSet | Categorized by type, e.g. haplotype |
| Single variant | Measure | @ID identifies the sequence at that location |
| Descriptors | Name, Symbol, AttributeSet | Open-ended structure to capture HGVS expressions and other descriptors; structured as content, source, citation and comment. |
| Location on an assembly | SequenceLocation | Will report on both GRCh37 and GRCh38 |
| Phenotype | ||
| Set of phenotypes | TraitSet | |
| Single phenotype | Trait | Categorized by type, e.g. disease, pharmacologic response, finding |
| Descriptors | Name, Symbol, AttributeSet | Open-ended structure to capture names, identifiers and other descriptors; structured as content, source, citation and comment. |
| Evidence | ||
| Effect of sequence change based on NCBI annotation | MolecularConsequence | Cross-referenced to Sequence Ontology |
| Observations | ObservedData, Co-OccurrenceSet | Types of evidence to request and maintain for interpretation are in development |
| Experimental results | FunctionalConsequence | From published literature |
Variations submitted to ClinVar are compared with variant locations accessioned by dbSNP or dbVar. If variation at that location is known, ClinVar adds the rs# or variant call identifier to the Reference ClinVar (RCV) record. If the variation location is novel, the variant is submitted to dbSNP or dbVar to be accessioned and the identifiers from those databases are then added to the ClinVar RCV record. In other words, ClinVar does not create its own identifiers for locations of variation. Nonetheless, to support internal data flows and some public reports (Maintenance and Access sections), ClinVar does assign an internal unique identifier to the specific allele at each location, which is reported in the XML and tab-delimited exports as an integer identifier (Table 1).
ClinVar reports multiple types of attributes for each variant. Human Genome Variation Society (HGVS) (4) expressions are reported based on the current reference assembly, RefSeqGenes (5), cDNAs and proteins as appropriate. When there are multiple transcripts for a gene, ClinVar selects one HGVS expression to construct a preferred name. By default, this selection is based on the first reference standard transcript identified by the RefSeqGene/LRG (Locus Reference Genomic) collaboration (6), but can be overridden by stakeholder request.
Some of the data ClinVar reports related to variation are added by NCBI. These data are reported only as part of the aggregate record (accession starting with RCV), and can include alternate HGVS expressions, allele frequencies from the 1000 Genomes project (7) or GO-ESP (8), identifiers from dbSNP or dbVar, molecular consequences (e.g. nonsense/missense/frameshift) and location data (splice site, untranslated regions, cytogenetic band, gene symbols and names). Values for molecular consequence, type of variation and location relative to a gene are standardized by reference to identifiers from the Sequence Ontology (9).
Phenotype
Similar to variation, ClinVar represents phenotype either as a single concept or a set of concepts. Sets are used primarily to report a combination of clinical features; single values are used to represent diagnostic terms or indications for genetic testing. Submitters are encouraged to submit phenotypic information via identifier, e.g. MIM number, MeSH term, or identifier from the Human Phenotype Ontology (HPO) (10). Free text is accepted, however, and ClinVar staff will work with submitters to determine if that text can be mapped to standardized concepts. When such mapping is possible, ClinVar connects the phenotype to a concept in MedGen (11) and adds MedGen’s identifier to the RCV record.
Although ClinVar is structured to represent detailed descriptions of phenotype, to date the majority of submissions provide only a single, often broad, diagnostic term. An exception is the submission from ISCA (https://www.iscaconsortium.org, e.g. http://www.ncbi.nlm.nih.gov/clinvar/?term=isca[submitter]), which included more phenotypic information.
Interpretation
All content in this category is submitter-driven. ClinVar represents interpretations of clinical significance and when that significance was last interpreted by the submitter, mode of inheritance of a variation relative to a disorder, qualification of severity of phenotype, etc. Terms for clinical significance are those recommend by the American College of Medical Genetics and Genomics (ACMG; 12). If submitters disagree on the interpretation of the clinical significance of any variation, the aggregate record is marked as having conflicts. If one submitter does not provide this information and another does, that is not marked as conflicting.
Evidence
Evidence that supports an interpretation of the variation–phenotype relationship can be either highly structured or a free-text summary discussing how the evidence was evaluated. When structured, content includes the description of how the variants were called and in what context (genetic testing, family studies, comparison of tumor/normal tissue, animal models, etc.) Based on that context, the results can be represented as number of observations per person or chromosome, number of segregations observed, the number of times other rare variations were identified in the same gene or other genes, etc. At present, most structured data are reports of number of individuals, in which non-somatic variation was observed, sometimes with indication of number of families.
MAINTENANCE
Data in ClinVar are currently being released weekly to the web site for access by interactive browsing or programmatically via E-Utilities (http://www.ncbi.nlm.nih.gov/books/NBK25500). The first week of every month, there are comprehensive extractions for ftp access, coordinated with dbSNP. This section will summarize how submissions are managed and how content is processed for retrieval.
Submissions and SCV accessions
There are several streams by which data flow into ClinVar. There are three current semi-automated data streams, namely from OMIM (13), GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/), dbSNP and one planned from the GTR. For OMIM, ClinVar automatically processes the allelic variant portions of any gene record and converts them to ClinVar’s submission XML, using the title of each allelic variant to establish the phenotype, the germline or somatic source and the description of the allele set. The full text is stored as a comment. As the reference sequence is rarely provided in the title or text, after the automatic submission or update the variant may remain uncharacterized. Scripts are used to determine if there is a unique nucleotide solution for any missense or nonsense record, and if so, that variant is mapped to reference sequences. Otherwise, as resources allow, curators review the primary literature for the OMIM record not mapped to sequence to define the variant.
The second semi-automatic flow is from ‘GeneReviews’. Data in tables are converted to ClinVar submission XML. In this case, standard transcript-based HGVS expressions are usually provided, so the variant is defined in nucleotide coordinates, but the text does have to be checked interactively to identify the name of the disorder.
Third, groups that submitted data to dbSNP associated with phenotype before April 2013 are being asked if their current data can be accessioned in ClinVar and if they want to revise or add content. If they do not opt out, or if they submitted to dbSNP with the understanding that the data would also be included in ClinVar, the data in dbSNP are converted to ClinVar submission XML and processed. Submissions to the GTR have been made with the understanding they be accessioned by ClinVar. Conversion of submissions to dbSNP or dbVar into records in ClinVar should be complete early in 2014.
ClinVar also accepts direct submissions. Information about how to submit to ClinVar is posted on the web site (http://www.ncbi.nlm.nih.gov/clinvar/docs/submit/). If submitted as text or spreadsheet, ClinVar converts to the standard submission XML. This XML is archived, but some content is extracted to relational tables to facilitate standardization of data values and aggregation of comparable submissions into an RCV accession.
The submission process involves several validation steps and consultation with the submitter if questions arise. Once the format of the submission is valid, the data are loaded to a test database to compare content to other submissions for the same variation. If there are conflicting data, these are reported to the submitter for review. Views of how the data will look on the website may also be provided. When preprocessing is complete, the submission XML is submitted to the production database. XML is updated to include the SCV accession that was assigned and converted from submission format (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/clinvar_submission.xsd) to public (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/clinvar_public.xsd). The XML corresponding to a submission (as accession.version) is not changed again unless there was a minor typographical error that was identified after the accessioning was complete. Even then, the history of the XML changes are retained, but the version is not incremented. A new version is assigned to an SCV accession only when the submitter resubmits to alter content.
Reviewed submissions and RCV accessions
In addition to records from individual submitters, ClinVar also accepts reviewed submissions of variations that have been curated by an expert group or are included in practice guidelines from a professional society. Expert curation groups may download a set of ClinVar SCV records to review or submit variations to the database as novel records. For reviewed submissions, the unique combination of submitter, variation and phenotype is accessioned in the format RCV000000000.0. If there are SCV records for the same variation and phenotype as the reviewed record, the interpretation on the reviewed record takes precedence over the interpretations from individual submitters.
Aggregation and RCV accessions
RCV accessions also represent aggregation of data from multiple submitters for the same variation/phenotype combination as well as content added by NCBI. A new version is assigned to the RCV accession only if the content of SCV accessions is changed (addition to the group of SCV accessions or update of a previous record), but not if content from NCBI is changed. For example, if NCBI defines new transcripts for a gene and a variation that was previously reported as intronic is now discovered to cause a missense change as well, that data will be added to the record but the version will not change. The alteration in the record is archived, however, and will be able to be retrieved from the web site as record histories soon.
Validation and standardization
As part of the test submission described in the previous section, ClinVar has established a set of checks to validate new content. For example, HGVS expressions are tested and submitters are informed if the reference sequence has been suppressed or the expression is not valid. Any term used to report clinical significance or phenotype that differs from standard is verified with the submitter.
Updates
A submitter can provide an update at any time. With a new submission, ClinVar assigns a new version to the SCV accession based on the new content, updates the RCV accession based on the submission and assigns the RCV a new version as well. To facilitate accurate management of updates, submitters should provide the SCV accession assigned to the record.
ACCESS
Web
ClinVar maintains a web site, namely, http://www.ncbi.nlm.nih.gov/clinvar. The database is part of NCBI’s Entrez system and is searchable with the standard query interface and ‘Advanced query’ options. ClinVar supports retrieval by variation (HGVS expression, rs, nsv, nssv, OMIM allelic variant identifier and identifier used in an LSDB), genes (symbol or full name), disease (names and identifiers), submitters, etc. If a query is detected to be a gene symbol (which may also match content in records not specific to that gene), an option is displayed to restrict results to records for that gene. The default result set is a table of 20 rows, but that can be altered using ‘Display’ settings (Figure 1). When multiple results are returned from a query, filters are provided at the left that reflect the content of the retrieval set (values and counts of each). Clicking on one of those options removes all but that set from the display, a restriction that can be reversed by using the ‘Clear’ option.
Figure 1.
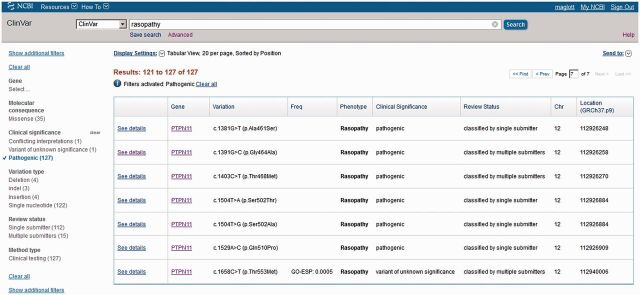
Tabular display of query results with filters applied. In this example http://www.ncbi.nlm.nih.gov/clinvar?term=rasopathy&cmd=DetailsSearch), the query ‘rasopathy’ returned more than 225 results, but when the selection of ‘Pathogenic’ was applied, the number of results was reduced to 127. The number of records in each category is reported. The check mark to the left of the filter name as well as the report at the top of the page are used to remind the user about the filters that have been applied. These filters can be removed using the ‘Clear’ option. To see the details of any record that was found, click on ‘See details’. To alter the display and change the sorting order, open the ‘Display Settings’ menu above the table.
To see a full record, click on ‘See details’. At present (September 2013), this display corresponds to content of an RCV accession. The ‘Clinical significance’, ‘Allele description’ and ‘Condition(s)’ sections and the Genome view report aggregate data; the Clinical Assertions are submitter-specific and the Evidence is provided both in aggregate and submitter-specific sections (Figure 2). Before the end of 2013, a new display will be provided via ‘See details’, quite similar to the RCV report but aggregated per single variation rather than variation–phenotype combination. This new display allows users to see all data for a variation even when submitters’ representation of phenotype differs.
Figure 2.

Representative display of an RCV accession RCV000033555.3 generated by aggregating information in two submissions (SCV000057460 and SCV000058286). The information above the tabbed section includes information from the submitters and values added by NCBI (e.g. GeneIDs, Mendelian Inheritance in Man (MIM) numbers, rs numbers and links to MedGen). The information in the ‘Clinical Assertions’ and ‘Evidence’ tabs are organized by what each submitter contributed and are extracted from the SCV records.
Data in ClinVar can also be accessed via other NCBI databases, based on the links that are built when content is shared. Examples include dbSNP, dbVar, Gene, MedGen, Nucleotide and PubMed. Locations of variation represented in ClinVar are annotated on RefSeqs and visible in the graphical sequence displays (e.g. http://www.ncbi.nlm.nih.gov/nuccore/125662814?report=graph) and browsers such as 1000 Genomes (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). ClinVar also provides specialized pages for certain types of access. One is the list of genes and disorders for which ACMG recommends that incidental findings should be reported (14) (http://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/); another would be the listings of submitters and all their submissions (http://www.ncbi.nlm.nih.gov/clinvar/submitters/).
FTP
Data from ClinVar are reported from several directories at NCBI. The README file (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/README.txt) provides a comprehensive list, pointing, for example, to the file converting MIM numbers, GeneIDs and MedGen concepts ids on Gene’s FTP site (mim2gene_medgen), the standard terms at GTR’s FTP site and the tab-delimited, XML and VCF files from ClinVar. The latter is available from dbSNP (with the symbolic link from ClinVar).
E-utilities
ClinVar currently supports programmatic access via E-Utilities. The esearch, esummary and elink options (but not efetch) are enabled. Please note that esearch [e.g. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=clinvar&term=brca1(gene)&retmax=1000] returns the unique identifiers for an RCV record, which does not correspond 1:1 with an accession version. The unique identifiers represent an instance of that record, which may change without a version change if NCBI adds data to the record such as an rs# or a ConceptUID from MedGen. A record retrieved by an outdated ID provides a link to the current record.
FUTURE DIRECTIONS
Tracking changes to variant interpretation over time is an important function of ClinVar, and all past versions of a record are maintained. ClinVar will provide web access to that history, similar to revision history in other NCBI’s sequence databases. Additionally, three new web views are in development: (i) the allele-specific display mentioned in the Access section, (ii) a full report for each SCV record that will display all details for each submission and (iii) a variation browser that is a significant upgrade of Variation Viewer (http://www.ncbi.nlm.nih.gov/sites/varvu). This browser will provide a sequence-based view similar to what is available under the Genome View tab (Figure 2), but also facilitate importing local data, exon–exon navigation and downloads. ClinVar will also start calculating and displaying HGVS expressions on LRG sequences for all records, in addition to the HGVS expressions on RefSeqGenes that are already provided. And as noted above, variation data included as part of GTR submissions will be converted to ClinVar records in the near future to improve connections to GTR for tested variants.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41:D936–D941. doi: 10.1093/nar/gks1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubinstein WS, Maglott DR, Lee JM, Kattman BL, Malheiro AJ, Ovetsky M, Hem V, Gorelenkov V, Song G, Wallin C, et al. The NIH genetic testing registry: a new, centralized database of genetic tests to enable access to comprehensive information and improve transparency. Nucleic Acids Res. 2013;41:D925–D935. doi: 10.1093/nar/gks1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutation. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell C, Hart J, Landrum MJ, McGarvey KM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacArthur JAL, Morales J, Tully RE, Astashyn A, Gil L, Bruford EA, Dalgleish R, Larsson P, Flicek P, Maglott DR, et al. Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence. Nucleic Acids Res. 2014;42:D873–D878. doi: 10.1093/nar/gkt1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, NHLBI GO Exome Sequencing Project—ESP Lung Project Team. Christiani DC, Wurfel MM, Lin X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mungall CJ, Batchelor C, Eilbeck K. Evolution of the Sequence Ontology terms and relationships. J. Biomed. Inform. 2011;44:87–93. doi: 10.1016/j.jbi.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson PN, Kohler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014;42:D7–D17. doi: 10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 13.Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 14.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]