


Abstract
The source of the microbial genomic sequences in the RefSeq collection is the set of primary sequence records submitted to the International Nucleotide Sequence Database public archives. These can be accessed through the Entrez search and retrieval system at http://www.ncbi.nlm.nih.gov/genome. Next-generation sequencing has enabled researchers to perform genomic sequencing at rates that were unimaginable in the past. Microbial genomes can now be sequenced in a matter of hours, which has led to a significant increase in the number of assembled genomes deposited in the public archives. This huge increase in DNA sequence data presents new challenges for the annotation, analysis and visualization bioinformatics tools. New strategies have been developed for the annotation and representation of reference genomes and sequence variations derived from population studies and clinical outbreaks.
INTRODUCTION
From the beginning of microbial genome sequencing, researchers have been interested in representing phylogenetic diversity, and the sequencing of one genome from each prokaryotic division or phylum is still a frequently articulated community goal. However, largely because of interest in human pathogens and advances in sequencing technologies, there is also now a rapidly growing number of closely related genomes representing variations within a species (1). Recent advances in second- and third-generation sequencing technologies and bioinformatics analysis relevant to microbiology and virology are being translated to the needs of public health (2). This is changing the way microbial genome sequences are generated and used. ‘The 100K Genome Project’ (3) aims to sequence genomes of 100 000 strains of important food-borne pathogens, such as Escherichia coli, Listeria and Salmonella, and making them available in the public domain. This will promote developing tests for identification of emerging strains and the sources of outbreaks. To manage the high-level volume of nearly identical genomes and to appropriately represent microbial diversity, National Center for Biotechnology Information (NCBI) is proposing a new approach to RefSeq microbial genome representation and annotation and introducing a new non-redundant protein data model.
REFSEQ MICROBIAL GENOMES
The source of the microbial genomic sequences in the RefSeq collection (4) is the set of primary sequence records submitted to the International Nucleotide Sequence Database public archives. Genomic sequences (nucleotide) in prokaryotic RefSeqs are identical copies of the underlying primary INSDC records (5).
Entrez Genome database at NCBI (6) was launched in 1995 shortly after the first complete microbial genome of Haemophilus influenzae Rd KW20 was released to public (7). Currently (October 2013), public archive contains 24 788 prokaryotic registered genome projects representing 4528 different species; 14 311 of them have assembled genomes either complete (2670) or draft (11 641), and the remainder either do not have submitted sequence data yet or have only raw sequence reads uploaded to Sequence Reads Archive (8).
DATA ACCESS
New genomes are processed for RefSeq, made public in Entrez and added to FTP directories daily. Complete list of prokaryotic genomes is available in Entrez Genome browser.
Link: http://www.ncbi.nlm.nih.gov/genome/browse/
The text version of the table can be downloaded from the FTP site: ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/
Genome sequence data can be downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria
The genomes FTP area supports users who are interested in downloading data for one or a specific subset of organisms and/or in downloading the data that correspond to an annotated genome. Users who are interested in comprehensive downloads can do so via the existing RefSeq release: ftp://ftp.ncbi.nlm.nih.gov/RefSeq/release/microbial/
GENOME REPRESENTATION
Sequenced microbial genomes represent a large collection of strains with different levels of quality and sampling density. They include many important human pathogens and also organisms that are of interest for non-medical reasons, i.e. biodiversity, epidemiology and ecology. These are obligate intracellular parasites, symbionts, free-living microbes, hyperthermophiles, psychrophiles and aquatic and terrestrial microbes, all of which have provided a rich insight into evolution and microbial biology and ecology. Largely because of interest in human pathogens and advances in sequencing technologies (1), there are rapidly growing sets of closely related genomes representing variations within the species. RefSeq is changing the scope of prokaryotic genome collection to include all genomes submitted to public archives to support variation studies and rapid pathogen detection analysis for the disease outbreaks. A single genome is designated to represent a species for comparative analysis. RefSeq prokaryotic genomes are organized in several new categories based on curated attributes and assembly and annotation quality measures.
Reference genome
Manually selected ‘gold standard’ complete genomes with high-quality annotation and the highest level of experimental support for structural and functional annotation. They include community-curated genomes if the annotation quality meets ‘reference genome’ requirements that are manually reviewed by NCBI staff http://www.ncbi.nlm.nih.gov/genome/browse/reference/
Representative genome
Representative genome for an organism (species); for some diverse species can be more than one. Corresponds to Sequence Ontology–[SO:0001505] (9). www.ncbi.nlm.nih.gov/genome/browse/representative/
Variant genome
All other genomes of individual samples representing genome variations within the species. Corresponds to Sequence Ontology- [SO:0001506].
Quality control
RefSeq keeps high standards in representing genome sequence data for each species. The genome assemblies are accepted into RefSeq when they meet the basic validation criteria described later.
Misclassification
Genome submission to GenBank does not require validation of the organism classification other than a check on the organisms’ name for correct spelling and nomenclature. There are several validation checks performed by RefSeq group that includes validation of 16S structural RNA against the reference set, genome alignment to the reference genome for a species and using ribosomal protein reference genes for placement on a phylogenetic tree. Misclassification is reported back to the submitters and usually gets confirmed and corrected or withdrawn in GenBank. Some of the recent examples are as follows:
AJMQ01000000 submitted as Marinilabilia sp. AK2 has been placed into Cyclobacteriaceae family by sequence analysis. The record was removed at the submitter’s request because the source organism cannot be confirmed.
ANNL01000000 submitted as Leptospira kirschneri serovar Valbuzzi str. Duyster has been reclassified using 16S analysis, confirmed by submitters and changed to Leptospira interrogans serovar Valbuzzi str. Duyster.
Genome representation
Only assemblies with full representation of the genome of the organism are taken into RefSeq. A metagenome assembly usually represents not a single organism but rather the composition of a bacterial population (10). Metagenome assemblies are not accepted into RefSeq; however, that policy may change as the technologies and methods evolve. Genome assemblies from mixed cultures, hybrid organisms and chimeras submitted to GenBank are not accepted into RefSeq because they do not represent an organism. They are not clearly marked in GenBank records but usually can be identified by comparing with the reference genome of the species. For example, AP012495 looks like a complete genome representing Bacillus subtilis BEST7613; however, the size of 7.8 Mb is almost double the size of the reference genome (4.2 Mb). A careful user may notice that an article describes this assembly as ‘first chimera genome constructed by cloning the whole genome of Synechocystis strain PCC6803 into the B. subtilis 168 genome’ (11). Another example of misleading genome representation is AKNF01000000—Shigella flexneri 1235-66, whole-genome shotgun sequencing project. There is no publication describing the project, but the size once again looks unusually high compared with the reference, and the submitters did provide an explanation in the comment that reads ‘This is from a mixed culture of S. flexneri 1235-66 and an unknown Shigella species’ (Figure 1).
Figure 1.
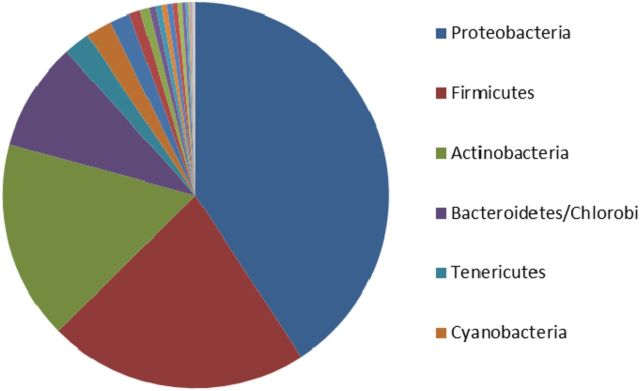
Distribution of bacterial species by phyla. Top four phyla with >100 species sequenced: Proteobacteria–1828, Firmicutes–978, Actinobacteria–747 and Bacteroidetes/Chlorobi group–408.
Many genome assemblies coming from single-cell sequencing technologies give only a partial representation of DNA in a cell, ranging from 10 to 90% (12,13). Genome representation can be validated by comparative analysis if other genomes are available in closely related groups (species or genera). For novel phyla or kingdoms, some indirect criteria are applied (presence of universally conserved genes, total genome size).
Assembly quality
Modern high-throughput sequencing technologies vary in the size of raw sequence reads and the patterns of sequencing errors. Despite many computational advances to genome assembly, complete and accurate assembly from second-generation short-read data remains a major challenge. There are two major approaches that have been used: de novo assembly from raw sequence reads and reference-guided assembly if the closest reference genome is available. The quality of genome assembly can be assessed using a number of different quality metrics. For many years, N50 contig and scaffold lengths have been major measure of assembly quality. More recently, a number of different metrics have been suggested (14,15). Some of the standard global statistic measures and reference-based statistics have been calculated for all RefSeq prokaryotic genomes and used for quality assessment.
Global assembly statistics
Total sequence length
Total assembly gap length
Gaps between scaffolds
Number of scaffolds
Scaffold N50/L50
Number of contigs
Contig N50/L50
Reference-based assembly statistics
Duplication ratio: subject_bases_aligned/query_bases_aligned (14).
Number of mismatches per 100 kb: (mismatches*100k)/query_bases_aligned.
Number of indels per 100 kb: (gap_bases*100k)/query_bases_aligned.
Number of unaligned contigs.
Number of ambiguously mapped contigs: number of contigs with duplication ratio >1.5.
NGx: min contig len to cover x% of reference.
Number of misassemblies: number of hits minus number of aligned contigs (correctly assembled contig should get one hit, more hits indicate misassembly).
Genome assemblies of low quality are filtered out from RefSeq collection. A low-quality criterion is based on the analysis of the genome annotation. Annotation of highly fragmented genomes contains large number of fragmented and frame-shifted genes. The size of the contigs should not be less than five times the average gene length, which in bacteria is known to be 1000 bp.
Minimum assembly quality required for a genome to be included in RefSeq collection:
N50 < 5000 and L50 > 200, and contig# > 1000.
In almost 15 000 genomes currently in the public archive, only 259 do not pass the minimum quality filter. This indicates the majority of genome assemblies submitted to Genbank are of acceptable quality. The reference-based assembly statistics is collected for the highly represented species and used for more detailed comparative analysis.
Annotation quality measure
Evaluation of the genome annotation can assist in making a final decision on assembly quality. One of the warning signs indicating assembly problems is an unusually high number of frame-shifted genes that do not have biological explanation.
For example, two genomes of Mycobacterium tuberculosis strain RGTB327 (CP003233.1) and RGTB423 (CP003234.1) satisfy the minimum assembly quality and average species length criteria. These genomes are finished and circularized; however, they stand apart of other genomes of M. tuberculosis with unusually high number of frame-shifted genes (22% versus 1% in other genomes). When aligned to the reference genome, a significant number of sequence mismatches were found, indicated by vertical red lines in the screenshot (Figure 2). These were primarily indels and were the result of a large number of sequencing and/or assembly errors.
Figure 2.

M. tuberculosis RGTB327 alignments to the reference genome of M. tuberculosis H37Rv. Vertical red lines show sequence mismatches caused by indels, which result in a large number (∼900) of frameshifted genes. These indels are likely caused by sequencing or assembly errors.
Genome assembly and annotation quality are further measured by the presence of complete ribosomal RNAs and essential conserved proteins. Missing or incomplete genes in a genome submitted as ‘complete’ is an indication of a sequencing or assembly error.
Estimation of genome represented in the assembly
Genome coverage is estimated by comparison with a quality reference genome. In the absence of a completely assembled quality reference genome, such a comparison is not possible.
Reannotation project
Historically, RefSeq prokaryotic genomes relied on author-submitted annotation. Curation has been focused primarily on the correction of protein names using protein clusters (first COG, later PRK). Some attempts to correct the start sites were not comprehensive and were subject to manual review that did not scale well when the number of genomes grew to many thousands. The problem of missing genes has not been addressed at all. The result is the inconsistent annotation even in closely related genomes with a good reference such as E. coli K-12 (16).
Researchers recognize that there is a need for high-quality data. However, different annotation procedures, numerous databases and a diminishing percentage of experimentally determined gene functions have resulted in a spectrum of annotation quality. NCBI in collaboration with sequencing centers, archival databases and researchers has developed a set of microbial genome annotation standards (17,18). Over the recent years, NCBI has developed its own annotation pipeline that combines ab initio gene prediction algorithms with homology-based methods (http://www.ncbi.nlm.nih.gov/genome/annotation_prok/). The pipeline has been successfully used for many genomes submitted to GenBank in the past 5 years; it can produce a consistent high-quality automatic annotation that in many cases surpasses the original author-provided annotation. Consensus RefSeq annotation of all prokaryotic genomes will provide a common ground for further analysis of protein clusters, pan-genomes core protein sets and create a single reference system for expert evaluation and experimental validation of functional annotation.
Policy on community-curated genomes and genes
Some organisms of high interest have been manually curated by research community experts.
These genomes will be evaluated by RefSeq curators and will be updated as new information becomes available from community experts. There will be ongoing efforts to establish relationships with the research community to provide accurate and up-to-date annotation for specific organisms or metabolic pathways. Some of the actively community-curated genomes include E. coli str. K-12 substr. MG165 (17), tuberculosis pathogens (18) and Pseudomonas strains (19).
All other genomes, newly or previously submitted, will be annotated for RefSeq with recently re-designed NCBI Prokaryotic Genome Annotation Pipeline (manuscript in preparation).
Autonomous protein
To manage the flood of identical proteins and decrease existing redundancy, particularly from bacterial genomes but soon from viruses and eukaryotes as well, NCBI is introducing a new protein data type in the RefSeq collection signified by a ‘WP’ accession prefix. These new protein records are used to represent a group of identical protein sequences annotated on many genomes from different isolates, strains or species. This new data type is managed independently of the genome sequence record (Figure 3).
Figure 3.

(A) Protein sequence in NP_414555 record annotated on the reference genome of E. coli str. K-12 substr. MG1655 is represented by WP_000516135. (B) This sequence has been annotated on 1285 genomes from 16 Escherichia and Shigella species.
When the NCBI Prokaryotic Genome Annotation Pipeline annotates a bacterial protein that is 100% identical to the existing WP accessioned protein, that protein will be annotated by referencing the existing WP accession, indicating that the genome represents yet another exact example of that known protein sequence. Protein function is automatically assigned by the pipeline. The organism information in the new protein record no longer represents the sample from which the sequence was derived but is calculated as the minimum common taxonomic node of all genomes where the protein is annotated.
In rare cases where identical proteins can be annotated on genomes from different kingdoms (that might happen with bacteria and bacteriophage), protein records will carry taxonomy information for both kingdoms.
Example —
Protein WP_018003289 (30S ribosomal protein S5) crosses kingdoms: http://www.ncbi.nlm.nih.gov/protein/WP_018003289.1
It is identical in some novel species of alpha, delta-Proteobacteria, Verrucomicrobia, Nitrospinae and unclassified Thaumarchaeota. The ASN.1 has two BioSource structures: ‘Bacteria’ and ‘Archaea’; however, the flat file view can show only one.
Reference and representative genomes annotated by community often contain more functional information for the proteins than can be inferred by homology in the automatic process. This information will be preserved in traditional NP-accessioned records, but the protein sequence will refer to the non-redundant WP record.
More details on autonomous proteins and phase implementation plan can be found at ftp://ftp.ncbi.nlm.nih.gov/RefSeq/release/announcements/WP-proteins-06.10.2013.pdf
Microbial genome BLAST
Microbial genomes BLAST is special option of NCBI BLAST that allows the users to search against a subset of sequences from microbial genomes. This option is available from general BLAST home page. Recently, new search options have been added including ‘Representative Genomes’ (now the default database) and ‘All Genomes’ (20).
Representative genomes provide a smaller less-redundant set of records for a given bacterial species. These representatives are selected by the research community and NCBI computational processes and are especially helpful for microbial species that are highly represented by genomes for numerous strains in NCBI databases, such as E. coli. The ‘All Genomes’ option offers the choice of complete genomes, draft genomes or complete plasmids. These sets can be searched individually or in any combination. The microbial BLAST report also has a new ‘Genome’ link to the species page in Entrez Genome in the alignments section of the BLAST report. Microbial protein BLAST has new option to search against ‘Non-redundant RefSeq proteins’ described earlier.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest statement. None declared.
REFERENCES
- 1.Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 2.Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, Holden MT, Dougan G, Bentley SD, Parkhill J, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8:e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Timme RE, Allard MW, Luo Y, Strain E, Pettengill J, Wang C, Li C, Keys CE, Zheng J, Stones R, et al. Draft genome sequences of 21 Salmonella enterica serovar enteritidis strains. J. Bacteriol. 2012;194:5994–5995. doi: 10.1128/JB.01289-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura Y, Cochrane G, Karsch-Mizrachi I. The International nucleotide sequence database collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics. 1999;15:536–543. doi: 10.1093/bioinformatics/15.7.536. [DOI] [PubMed] [Google Scholar]
- 7.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 8.Kodama Y, Shumway M, Leinonen R. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40:D54–D56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6:R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta'omics for microbial community studies. Mol. Syst. Biol. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe S, Shiwa Y, Itaya M, Yoshikawa H. Complete sequence of the first chimera genome constructed by cloning the whole genome of Synechocystis strain PCC6803 into the Bacillus subtilis 168 genome. J. Bacteriol. 2012;194:7007. doi: 10.1128/JB.01798-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blainey PC. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013;37:407–427. doi: 10.1111/1574-6976.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 14.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahman A, Pachter L. CGAL: computing genome assembly likelihoods. Genome Biol. 2013;14:R8. doi: 10.1186/gb-2013-14-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poptsova MS, Gogarten JP. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology. 2010;156:1909–1917. doi: 10.1099/mic.0.033811-0. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Richardson AJ, Rudd KE. EcoGene-RefSeq: EcoGene tools applied to the RefSeq prokaryotic genomes. Bioinformatics. 2013;29:1917–1918. doi: 10.1093/bioinformatics/btt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lew JM, Mao C, Shukla M, Warren A, Will R, Kuznetsov D, Xenarios I, Robertson BD, Gordon SV, Schnappinger D, et al. Database resources for the tuberculosis community. Tuberculosis. 2013;93:12–17. doi: 10.1016/j.tube.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]