


Abstract
Diversified histone modifications (HMs) are essential epigenetic features. They play important roles in fundamental biological processes including transcription, DNA repair and DNA replication. Chromatin regulators (CRs), which are indispensable in epigenetics, can mediate HMs to adjust chromatin structures and functions. With the development of ChIP-Seq technology, there is an opportunity to study CR and HM profiles at the whole-genome scale. However, no specific resource for the integration of CR ChIP-Seq data or CR-HM ChIP-Seq linkage pairs is currently available. Therefore, we constructed the CR Cistrome database, available online at http://compbio.tongji.edu.cn/cr and http://cistrome.org/cr/, to further elucidate CR functions and CR-HM linkages. Within this database, we collected all publicly available ChIP-Seq data on CRs in human and mouse and categorized the data into four cohorts: the reader, writer, eraser and remodeler cohorts, together with curated introductions and ChIP-Seq data analysis results. For the HM readers, writers and erasers, we provided further ChIP-Seq analysis data for the targeted HMs and schematized the relationships between them. We believe CR Cistrome is a valuable resource for the epigenetics community.
INTRODUCTION
Nucleosome function and modification represent important epigenetic features. In eukaryotes, the nucleosome is composed of an octamer of core histones (two copies of H2A, H2B, H3 and H4) and 146 DNA base pairs of DNA wrapped around the histone octamer (1). Histone modifications (HMs), such as methylation and acetylation, two typical types of nucleosome modifications, play essential roles in modulating chromatin structures and functions, making them indispensable in epigenetic regulation (2–5).
Histone marks tend to occur in an observable pattern known as the histone code, which is coded and decoded by chromatin regulators (CRs) including readers, writers and erasers (2,4,6–19). Readers usually contain specific domains that can recognize specific modified histone residues, and they determine the modification type (e.g. methylation or acetylation) and state (e.g. mono-, di- or tri- for lysine methylation) (20). Writers and erasers can post-translationally modify and de-modify chromatin, adding and removing certain modifications, such as methylation and acetylation, to and from some specific histone sites, thus altering chromatin structure and recruiting regulatory factors (20,21). In addition to the factors that are directly related to HMs, chromatin remodelers are also regarded as a type of CR (22–24). Chromatin remodelers can make nucleosomal DNA easier to access or allow nucleosomes to move to a different position along the DNA, remove or exchange nucleosomes using energy from ATP hydrolysis (20,21,25). CRs display vital functions in many common cellular processes, such as transcription, replication, recombination, apoptosis, differentiation and development, as well as in some pathologic processes, especially in cancer (21,26–43).
With increasing attention being paid to CRs and the development of ChIP-Seq technology, there are abundant available CR ChIP-Seq data and CR-related HM ChIP-Seq data that have been obtained under the same conditions (i.e. in the same cell line/type). Analysis of the linkages between CR and HM ChIP-Seq data has proven to be an effective method for revealing new CR functions. EZH2, a subunit of the PRC2 complex, has been acknowledged as a transcriptional repressor that mediates the generation of H3K27me3. A recent study by Xu et al. shows a new role of EZH2 in metastatic prostate cancer (44). In this research, by comparing and analyzing EZH2 and H3K27me3 ChIP-Seq data, they found that a subset of EZH2 peaks is irrelevant to H3K27me3. Further study confirms that those irrelevant peaks perform as a transcriptional activator of androgen receptor, which is independent of other PRC2 subunits and its known product H3K27me3.
The integration and presentation of CR ChIP-Seq data and related HM ChIP-Seq data obtained under the same conditions can contribute greatly to the study of epigenetics. However, among the relevant publicly available databases, such as Histome and Factorbook, there is no specific resource providing linkage analysis of CR and HM ChIP-Seq data. Histome is a knowledge base that integrates detailed information about all human HM sites and their related writers and erasers; however, it has not associated CRs and HMs with ChIP-Seq data (45). Factorbook is a wiki-based database collecting all of the TF ChIP-Seq data from human generated by ENCODE, together with additional downstream analysis, which does not specifically focus on linkage pairs between CR and HM (46). This situation has driven us to develop CR Cistrome, a unique knowledgebase integrating curated information of 36 CRs, 194 qualified CR ChIP-Seq data sets and 177 qualified HM ChIP-Seq data sets, and analysis of the relationship between 458 pairs of CRs and HMs in human and mouse. The CRs with related HMs are restricted to chromatin readers, writers and erasers, as remodelers possess no related HMs. We believe this database represents a valuable resource for systematically examining the genome-wide functions of CRs and that it may motivate investigators who are interested in epigenetics.
CONSTRUCTION AND CONTENTS
Data sources
CR information was derived from different sources. The list of readers was acquired from Yun et al. (20), and the reader-recognized HMs were summarized through manual literature mining. Writers, erasers and their related HMs were obtained from the Histome database (45). The list of remodelers was obtained from Bao et al. (47). The names of all of these CRs are consistent with those in the Cistrome Map (23), a database we previously constructed, containing all the articles involving human and mouse ChIP-Seq data. Furthermore, for each CR, CR Cistrome provides its aliases, which are based on the NCBI Gene database. In addition, we manually collected some detailed and curated information, including summaries, functions and interactions and known disease associations. The information of ChIP-Seq data on CRs and related HMs, including the species, cell line/population, cell type, tissue origin and GSE and GSM accession numbers were derived from the Cistrome Map, and the raw ChIP-Seq data were downloaded in the fastq format from GEO at NCBI, EBI and ENCODE from the UCSC Genome Browser.
Database contents
For each collected CR, CR Cistrome provides three layers of contents, as shown in Figure 1. The first layer provides information including the CR’s introduction in other public databases (NCBI, UniProt, Wikipedia and GeneCards are included), its full name and aliases, type (writer, eraser, reader or remodeler), manually curated function and known associated diseases as well as a CR summary.
Figure 1.

Content of the CR Cistrome database. For each CR collected, we provide basic information and ChIP-Seq data analysis results. For each reader, writer and eraser (if there are available ChIP-Seq data for the related HMs from the same cell line/pop, or the cell type, when the same cell line is not available), we also provide the ChIP-Seq analysis results for these HMs, including the same results as provided for the CR data, except that HMs don’t have motif scan results. Furthermore, the resultant Venn diagram, the genomic distribution of overlap peaks between CR and HM, average CR and HM profile in CR’s binding sites and reads density plot of CR and HM in CR’s peaks are shown to illustrate the relationship between the CRs and related HMs.
In this database, the publically available ChIP-Seq data on each CR from human and mouse were collected and processed, and the results are shown as the second layer of content. In this layer, the peak file (.bed) generated through MACS (48) and the reads density file (.bw) obtained from bedGraphToBigWig (49) were provided for free download. In addition, some annotation results were also displayed and can be freely downloaded, such as the binding DNA sequence (motif) acquired through MDseqpos, average conservation profile across CR’s peaks, the average profile near the transcription start site (TSS), the transcription terminal site (TTS), through the gene body, and genome-wide enrichment as indicated by CEAS (50).
The third layer is specifically aimed at CR-HM linkage pairs. For the CR-HM linkages presented here, the Reader-HM linkage was defined as the reader and its recognized HM obtained from the literature and the writer- and eraser-related HMs referred to the Histome database. For each CR (readers, writers and erasers), if there were available ChIP-Seq data for the related HMs from the same cell line/type (or, if data from the same cell line/type were not available, the species and tissue origin were considered), the results of the analysis of the linkage pair between CR and the related HM were presented, including the Venn diagram between them, the distribution of their overlap peaks, the average CR and HM profile in CR’s binding sites and the reads density plot of the CR and HM in CR’s binding sites. Furthermore, the results of the analysis of the related HM ChIP-Seq data, including the profile near the TSS, the TTS, and through the gene body as well as the observed genome-wide enrichment and conservation and the freely downloaded peak file and reads density file, are contained in this layer.
To guarantee the quality of the ChIP-Seq data in the database, we set criteria (total sequencing reads >5 M and detected peaks >500), and only those data sets that met these criteria could be added. As a result, 36 CRs associated with 194 ChIP-Seq data sets from human and mouse were collected in CR Cistrome, and the detailed statistics of these data are shown in Table 1. The detailed statistics of HM ChIP-seq data are listed in Table 2. The database includes 13 pairs and 165 data sets for Writer-HM linkages, 12 pairs and 171 data sets for Eraser-HM linkages and 5 pairs and 122 data sets for Reader-HM linkages (Table 3). All of the analysis results and information noted earlier in text are stored and managed through the MySQL relational database management system on a Linux server.
Table 1.
The statistics of CR ChIP-Seq data set
| CR type | CR number | CR name | ChIP-seq dataset number (human/mouse) |
|---|---|---|---|
| Reader | 5 | CHD1, RAG2, TAF3, PHF8, WDR5 | 18/7 |
| Writer | 11 | CREBBP, Ep300, EZH1, EZH2, KAT7, PCAF, SETDB1, WHSC1, WDR5, RAC3, KAT2A | 71/21 |
| Eraser | 14 | PHF8, KDM5B, KDM5A, KDM2A, KDM1A, HDAC6, HDAC3, HDAC2, HDAC1, SIRT6, SIRT1, KDM5C, KDM4A, KDM6B | 49/10 |
| Remodeler | 9 | CHD1, CHD2, CHD4, CHD7, MTA3, SMARCA4, SMARCB1, SMARCC1, SMARCC2 | 26/14 |
| Total | 36 | 146/48 |
After the quality control, we finally got 5 readers with 18 ChIP-Seq data sets in human and 7 ChIP-Seq data sets in mouse, 11 writers with 71 ChIP-Seq data sets in human and 21 ChIP-Seq data sets in mouse, 14 erasers with 49 ChIP-Seq data sets in human and 10 ChIP-Seq data sets in mouse, 9 remodelers with 26 ChIP-Seq data sets in human and 14 ChIP-Seq data sets in mouse, in total, that is 146 ChIP-Seq data sets in human and 48 ChIP-Seq data sets in mouse.
Table 2.
The statistics of HM ChIP-Seq data set
| HM type | HM number | HM name | ChIP-seq data set number (human/ mouse) |
|---|---|---|---|
| Methylation | 7 | H3K4me1, H3K4me2, H3K4me3, H4K20me1, H3K9me3, H3K27me3, H3K36me3 | 96/25 |
| Acetylation | 6 | H3K9ac, H3K56ac, H3K27ac, H3K18ac, H4K8ac, H4K5ac | 45/11 |
| Total | 13 | 141/36 |
After the quality control, we finally got 7 kinds of histone methylation with 96 ChIP-Seq data sets in human and 25 ChIP-Seq data sets in mouse and 6 kinds of acetylation with 45 ChIP-Seq data sets in human and 11 ChIP-Seq data sets in mouse.
Table 3.
The statistics of CR-histone linkage data set pair
| CR-HM link type | CR-HM number | ChIP-seq data set number |
|---|---|---|
| Writer-HM | 13 | 165 |
| Eraser-HM | 12 | 171 |
| Reader-HM | 5 | 122 |
| Total | 30 | 458 |
There are 13 pairs and 165 data sets of Writer-HM linkage, 12 pairs and 171 data sets of Eraser-HM linkage, 5 pairs and 122 data sets of Reader-HM linkage.
CR Cistrome is a part of the Cistrome Project (51). And for users’ convenience to further analyze our CR and HM data (.bed and.w), we have now added an interface between CR Cistrome and Galaxy/Cistrome (http://cistrome.org/ap/). The users can either import CR Cistrome data (.bed and.w) from the ‘Import Data’ drop-down menu in the CISTROME TOOLBOX or send the data to Galaxy/Cistrome by choosing the SEND TO CISTROME function in CR Cistrome. Then they can use the powerful CISTROME TOOLS and GALAXY TOOLBOX of the Galaxy/Cistrome webserver to explore the data.
UTILITY AND DISSCUSSION
Interface and visualization
Our database provides two different methods for users to survey the ChIP-Seq data sets. One is through the regulator atlas and the other is based on the advanced search menu. The ‘regulator atlas’ can display all of the CR ChIP-Seq data sets from each cohort, and users can survey one data set in one cell type at a time, whereas using the advanced search, they can examine a single CR in different cell lines, cell types, tissues and species at the same time. To make the data presentation more intuitive, the statistics on the cell lines and cell types from which the ChIP-Seq data were obtained are also shown in the ‘collection stats’ menu.
Case exploration
If the user is interested in a specific CR, he can select the name and cell lines (or cell types, tissues) and species of the CR ChIP-Seq data (if the cell lines, cell types, tissues and species are not set, the database will return all the ChIP-Seq data for this CR) in the advanced search menu. In this article, we use PHF8, an eraser of H3K9me2/3 and H4K20me1 and a reader of H3K4me3, as an example of the exploration procedure.
Step 1
Assuming the user is interested in the ChIP-Seq data on PHF8 in human fibroblast, they can select PHF8 in human fibroblast in the search menu, as shown in the first step of Figure 2. The database will then return a page containing the manually curated information for PHF8 and its ChIP-Seq data information and the related HM ChIP-Seq data information for human fibroblast. In human fibroblast, two PHF8 ChIP-Seq data sets are generated (GSE20753, GSM520383 and GSE20753, GSM520384), and the peak file (.bed), the read density file (.bw) and the analysis results are freely downloaded on this page.
Figure 2.
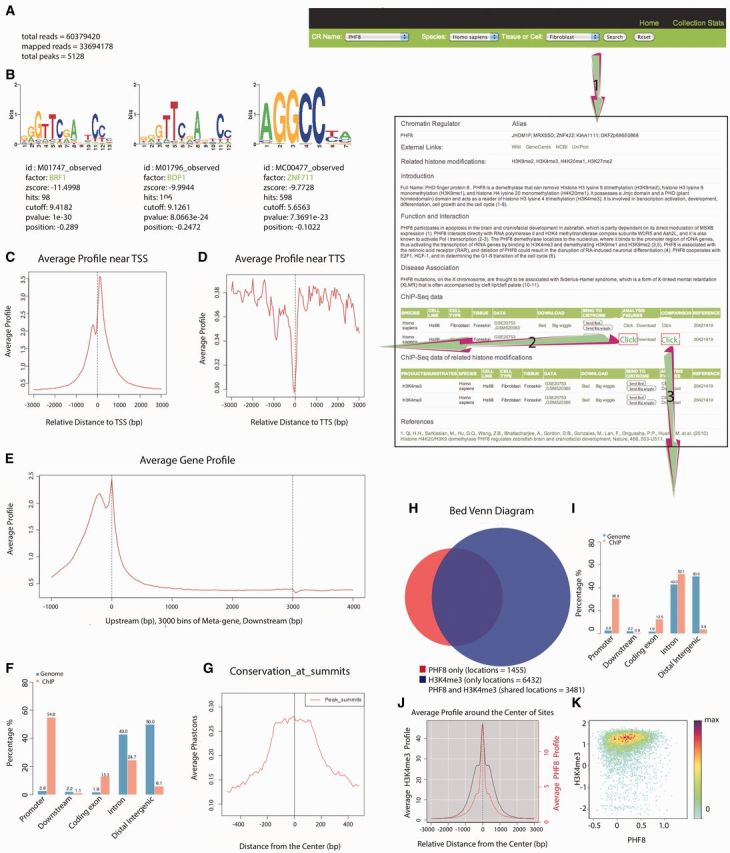
Screenshot depicting an example run of the CR Cistrome database. Assuming the user is interested in PHF8 in human fibroblasts, step 1 will return a page containing the basic information on PHF8, including its alias, introduction, functions and disease associations, which are manually generated from the literature and also external links to NCBI, UniProt, Wikipedia and GeneCards are provided, as well as ChIP-Seq data information and ChIP-Seq data information for the related HMs in human fibroblasts. Step 2 will provide a page containing the top three motifs for PHF8 in human fibroblasts and the average profile near the TSS and TTS as well as the average gene profile, genome enrichment, average conservation profile across PHF8 ChIP-Seq peaks and a brief data summary, which could help indicate the general quality of this data set. Step 3 will generate PHF8 data set id and H3K4me3 data set id for each comparison as well as the Venn diagram, distribution of their overlap peaks, the average PHF8 and H3K4me3 ChIP-Seq signal profile in PHF8’s binding sites and the reads density plot of PHF8 and H3K4me3 in PHF8’s binding sites.
Step 2
If the user wishes to acquire a detailed analysis of the result from the second ChIP-Seq data set (GSE20753, GSM520384), they can follow the second step shown in Figure 2. This action will provide a page containing the following analysis results: (i) a brief summary of this PHF8 data set (Figure 2A); (ii) the top three enriched DNA binding motifs in the genome region of PHF8 in fibroblasts (Figure 2B); (iii) the average ChIP-Seq signal profile near the TSS (Figure 2C), the TTS (Figure 2D) and across the gene body (Figure 2E); (iv) the genomic distribution of PHF8 ChIP-Seq peaks (Figure 2F); and (v) the average conservation profile across PHF8 ChIP-Seq peaks (Figure 2G).
(1) Modern high-throughput sequencers can generate tens of millions of sequences in a single run. A summary of these raw ChIP-Seq data is presented in Figure 2A. Bowtie (52) was used to align short DNA sequence reads to the genomes. Here, ‘total reads’ means all of reads sequenced in a single ChIP-Seq experiment, which indicates the resolution, whereas ‘mappable reads’ means reads that align to the genomes with two mismatches allowed at most. Next, the mappable reads are used to find the peaks using MACS. ‘Total peaks’ means the number of regions in which the factor is enriched under the cutoff Q-value (0.01). Here, this PHF8 data set includes 60 379 420 total reads, 33 694 178 mappable reads and 5128 total peaks, suggesting that this data set is of good quality.
(2) Sequence motifs are often defined as sequence-specific binding sites for proteins such as nucleases and transcription factors (TFs). They are usually short, recurring DNAs and are believed to have biological functions. MDSeqPos is an internal laboratory software platform used for de novo motif detection and known motif detection, with the top 1000 peaks being sorted by the Q-value. Figure 2B shows the top three motif detection results, including the sequence logo, the z-score and the factor name and position. In this case, additional factor (BRF1, BDP1 and ZNF711) motifs were enriched in the top 1000 peaks obtained for PHF8, suggesting that there may be co-binding between them. The information including the expression of these factors in transcription factor encyclopedia (TFe) (http://www.cisreg.ca/cgi-bin/tfe/home.pl) could be acquired through the hyperlink.
(3) Biologists are capable of visualizing the average ChIP signal profile over specific genomic features through CEAS (50), such as the TSS (Figure 2C), the TTS (Figure 2D) and across the gene body (Figure 2E). The profile near the TSS (TTS) focuses on the 3000 bp upstream and downstream of the TSS (TTS), whereas the average profile across the gene body focuses on the 1000 bp upstream of the TSS and 1000 bp downstream of the TTS (all meta genes were divided into 3000 bins). Here, there is a strong peak that can be viewed near the TSS, indicating that PHF8 is enriched and exhibits functions in fibroblast promoter regions.
(4) The orange bar in Figure 2F represents the genomic distribution of PHF8 ChIP-Seq peaks, including the following five regions: the promoter, downstream, coding exon, intron and distal intergenic regions. Based on comparison with the genomic distribution generated by chance (the blue bar), we can see that PHF8 is clearly enriched in the promoter region, which is consistent with the average ChIP profile near the TSS (Figure 2C).
(5) The average conservation profile across the PHF8 ChIP-Seq peaks (Figure 2G) is also presented. Conserved sequences indicate similar or identical sequences across different species, and highly conserved sequences tend to be biologically functional. Here, we focused on the 500 bp (for broad HM peaks, we set it to 2000 bp) upstream and downstream of the summit of each PHF8 peak, and the peak height was much higher in the middle than the surroundings, indicating that the middle part is more conserved and functional than the surroundings.
Step 3
As PHF8 is a reader of H3K4me3, and there are also ChIP-Seq data for H3K4me3 in human fibroblasts, figures representing a detailed analysis between PHF8 and H3K4me3 can be obtained through the third step. Here, we list the PHF8 data set ID and H3K4me3 data set ID for each compared pair as well as providing (i) the Venn diagram between PHF8 and H3K4me3 (Figure 2H) and the distribution of their overlap peaks (Figure 2I), (ii) the average PHF8 and H3K4me3 ChIP-Seq signal profile in PHF8’s binding sites (Figure 2J) and (iii) the reads density plot of PHF8 and H3K4me3 in PHF8’s binding sites (Figure 2K).
(1) The Venn diagram (Figure 2H) shows the overlap of the PHF8 and H3K4me3 peaks; the red circle represents all PHF8 peaks; the blue circle represents all H3K4me3 peaks; and the overlap represents the shared peaks between PHF8 and H3K4me3. Here, PHF8 and H3K4me3 overlapped greatly, indicating that they are functionally related. The orange bar in Figure 2I shows where the overlap peaks in Figure 2H are enriched throughout the genome, including the promoter region, downstream region, coding exon region, intron region and distal intergenic region, whereas the blue bar shows the distribution generated by chance. Here, the overlap peaks for PHF8 and H3K4me3 are enriched in the promoter region compared with the by-chance distribution.
(2) The average PHF8 and H3K4me3 ChIP-Seq signal profile observed within the PHF8 peaks (Figure 2J) provides the reads density of PHF8 and H3K4me3 in the PHF8 peaks. Here, we focus on the 1000 bp upstream and downstream sections of each PHF8 peak summit, calculate the PHF8 and H3K4me3 read density every 50 bp and obtain 40 reads density values and line them. The red line and the right red y-axis represent the reads density of PHF8 and the black line and the left black y-axis represent the reads density of H3K4me3. Additionally, H3K4me3 is enriched within the PHF8 peak summits.
(3) The reads density plot for PHF8 and H3K4me3 within PHF peaks (Figure 2K) also provides the H3K4me3 reads density among PHF8 peak regions. It is generated to reflect the reads density of a given HM among the binding sites for a given CR. Each dot refers to one CR binding site, which has been trimmed to 150 bp upstream and downstream of the peak center. The value of X-axis (Y-axis) of each dot stands for CR’s (HM’s) read density in this CR binding site, that is the CR (HM) ChIP-Seq reads number in this binding site normalized by the binding lenghth (300 bp) and then transformed by using log10. We produced an image scatter plot of two data sets in which the colors indicate the density of the points in the scatter plot. Here, H3K4me3 is enriched in the PHF8 peak regions.
CR CISTROME SUMMARY
CR Cistrome is a ChIP-Seq database containing information on CRs and CR-HM linkages in human and mouse, and it comprises all qualified CRs with available public ChIP-Seq data, manually curated information on these CRs, including their full names, aliases, introductions, functions, known disease associations and CR type as well as the ChIP-Seq data analysis results. This database also provides related HMs’ ChIP-Seq data analysis and CR-HM linkage analysis results for readers, writers and erasers in cases where there are available HM ChIP-Seq data collected under the same condition as the associated CRs. Each CR could be linked to NCBI, UniProt, Wikipedia and GeneCards to provide user alternative information. CRs can be surveyed through either the advanced search options or the regulator atlas menu. This database will be useful for different users, for individuals who are interested in epigenetic mechanisms, it is easy to acquire the features of the CR ChIP-Seq data and associations between CR and HM ChIP-Seq data. For advanced users, it is convenient to download the processed ChIP-Seq data, and if the users generate CR ChIP-Seq data themselves, they can achieve a better comparison and integration with the public ChIP-Seq data through our database.
FUTURE DEVELOPMENTS
We will pay close attention to any updated ChIP-Seq data for our collected CRs and HMs, and we will process them and add the results to the database as quickly as possible.
AVAILABILITY AND REQUIREMENTS
CR Cistrome is available at http://compbio.tongji.edu.cn/cr and http://cistrome.org/cr/. Although we recommend Safari as the default web browser, this database also supports other standard web browsers.
TOOLS FOR ANALYZING CHIP-SEQ DATA
We have provided all the tools and parameters we used for analyzing data in the FAQ part of our database.
Bowtie
Bowtie is an ultrafast memory-efficient tool for quickly mapping large numbers of short DNA sequences (reads) to large genomes (52). Our query input files were FASTQ files, and the alignments were conducted in the SAM format. If >1 reportable alignment was mapped to a particular read, we only retained one alignment and suppressed all the others.
Samtools
Samtools is a set of tools that processes alignments in the BAM format (53). Here, we used Samtools to convert SAM files into compressed BAM files and to merge samples that are replicates.
MACS
Calling peaks is the main function of MACS (model-based analysis of ChIP-Seq), which is used for identifying TF binding sites (48). It is a powerful analysis method for ChIP-Seq data. We used 0.01 as the Q-value cutoff, as this represents a stringent standard and can generate peaks with a higher confidence level. In the case of duplicate tags at the same location, we retained duplicate tags up to 1 because such results can improve prediction accuracy given the same complexity of the ChIP-Seq library. To conveniently and horizontally compare all of the ChIP-Seq data in this database, we processed all of the ChIP-Seq data without building a shifting model and used 73 bp as the shift size. When the experimental design included two biological replicates, we only generated the merged peak file (.bed) and read density file (.bw). If the data set possessed control data files (‘Input DNA’ or ‘IgG control’), the binding site prediction preferentially uses the control data files as the background; otherwise, MACS will randomly sample the genome as a control. When there were >10 000 peaks for a data set, we only used the top 10 000 for further analysis.
BEDTools
BEDTools is a set of utilities for addressing common genomics tasks (54). We used its intersectBed function to find the overlap regions of bdg files generated by MACS and the chromosome length bed file, in case that the MACS peak calling crossed the boundary of chromosome length.
bedGraphToBigWig
bedGraphToBigWig is a UCSC tool (49). Here, we used it to convert a bdg file into a bw file to reduce the storage burden.
CEAS
CEAS (cis-regulatory Element Annotation System) is a tool for providing statistics on ChIP enrichment at important genome features, such as for specific chromosomes and promoters. Here, we used it to generate average ChIP enrichment signals over specific genomic features, including the TSS and TTS, as well as gene profiles and genome enrichment. For each factor, we used the top 5000 peaks to analyze the distribution of cis-regulatory elements.
FUNDING
The National Basic Research Program of China [973 Program; 2010CB944904 and 2011CB965104]; National Natural Science Foundation of China [31071114, 31371288 and 31329003]; New Century Excellent Talents in the University of China [NCET-11-0389]; Innovative Research Team Program Ministry of Education of China [IRT1168]; National Institutes of Health [HG4069]. Funding for open access charge: The National Basic Research Program of China [2010CB944904].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Chengyang Wang, Xueqiu Lin and Chenfei Wang for their helpful discussions about the project.
REFERENCES
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8.Brinkman AB, Roelofsen T, Pennings SWC, Martens JHA, Jenuwein T, Stunnenberg HG. Histone modification patterns associated with the human X chromosome. EMBO Rep. 2006;7:628–634. doi: 10.1038/sj.embor.7400686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein BE, Mikkelsen TS, Xie XH, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 12.Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 13.Garcia BA, Barber CM, Hake SB, Ptak C, Turner FB, Busby SA, Shabanowitz J, Moran RG, Allis CD, Hunt DF. Modifications of human histone H3 variants during mitosis. Biochemistry. 2005;44:13202–13213. doi: 10.1021/bi050906n. [DOI] [PubMed] [Google Scholar]
- 14.Barratt MJ, Hazzalin CA, Cano E, Mahadevan LC. Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction. Proc. Natl Acad. Sci. USA. 1994;91:4781–4785. doi: 10.1073/pnas.91.11.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 16.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 17.Taverna SD, Ueberheide BM, Liu YF, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD. Long-distance combinatorial linkage between methylation and acetylation on histone H3N termini. Proc. Natl Acad. Sci. USA. 2007;104:2086–2091. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sims JK, Houston SI, Magazinnik T, Rice JC. A trans-tail histone code defined by monomethylated H4 Lys-20 and H3 Lys-9 demarcates distinct regions of silent chromatin. J. Biol. Chem. 2006;281:12760–12766. doi: 10.1074/jbc.M513462200. [DOI] [PubMed] [Google Scholar]
- 19.Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell. 2006;24:309–316. doi: 10.1016/j.molcel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 20.Yun MY, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin B, Zhou M, Ge Y, Taing L, Liu T, Wang Q, Wang S, Chen JS, Shen LL, Duan XK, et al. CistromeMap: a knowledgebase and web server for ChIP-Seq and DNase-Seq studies in mouse and human. Bioinformatics. 2012;28:1411–1412. doi: 10.1093/bioinformatics/bts157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–2489. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Bigas N, Kisiel TA, DeWaal DC, Holmes KB, Volkert TL, Gupta S, Love J, Murray HL, Young RA, Benevolenskaya EV. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol. Cell. 2008;31:520–530. doi: 10.1016/j.molcel.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, Schreiber SL. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl Acad. Sci. USA. 1998;95:3519–3524. doi: 10.1073/pnas.95.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. J. Intern. Med. 2008;263:128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl Acad. Sci. USA. 2011;108:13130–13134. doi: 10.1073/pnas.1110081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi XX, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Gene Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Pugh BF. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dodge JE, Kang YK, Beppu H, Lei H, Li E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell Biol. 2004;24:2478–2486. doi: 10.1128/MCB.24.6.2478-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tell R, Rivera CA, Eskra J, Taglia LN, Blunier A, Wang QT, Benya RV. Gastrin-releasing peptide signaling alters colon cancer invasiveness via heterochromatin protein 1(Hs beta) Am. J. Pathol. 2011;178:672–678. doi: 10.1016/j.ajpath.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalal I, Tasher D, Somech R, Etzioni A, Garti BZ, Lev D, Cohen S, Somekh E, Leshinsky-Silver E. Novel mutations in RAG1/2 and ADA genes in Israeli patients presenting with T-B- SCID or Omenn syndrome. Clin. Immunol. 2011;140:284–290. doi: 10.1016/j.clim.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Fathi AT, Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv. Hematol. 2012;2012:469592. doi: 10.1155/2012/469592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 39.Saramaki OR, Tammela TLJ, Martikainen PM, Vessella RL, Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Gene Chromosome Cancer. 2006;45:639–645. doi: 10.1002/gcc.20327. [DOI] [PubMed] [Google Scholar]
- 40.Collett K, Eide GE, Arnes J, Stefansson IM, Eide J, Braaten A, Aas T, Otte AP, Akslen LA. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin. Cancer Res. 2006;12:1168–1174. doi: 10.1158/1078-0432.CCR-05-1533. [DOI] [PubMed] [Google Scholar]
- 41.Kleer CG, Cao Q, Varambally S, Shen RL, Ota L, Tomlins SA, Ghosh D, Sewalt RGAB, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl Acad. Sci. USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte SD, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127–1133. doi: 10.18632/oncotarget.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elsasser SJ, Allis CD, Lewis PW. New epigenetic drivers of cancers. Science. 2011;331:1145–1146. doi: 10.1126/science.1203280. [DOI] [PubMed] [Google Scholar]
- 44.Xu KX, Wu ZJ, Groner AC, He HS, Cai CM, Lis RT, Wu XQ, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. HIstome-a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2012;40:D337–D342. doi: 10.1093/nar/gkr1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Zhuang JL, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong XJ, Virgil D, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41:D171–D176. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao Y, Shen X. SnapShot: chromatin remodeling complexes. Cell. 2007;129:632. doi: 10.1016/j.cell.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr. Protoc. Bioinform. 2007 doi: 10.1002/0471250953.bi0104s17. Chapter 1, Unit 1 4. [DOI] [PubMed] [Google Scholar]
- 50.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25:2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 51.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]