


Abstract
JASPAR (http://jaspar.genereg.net) is the largest open-access database of matrix-based nucleotide profiles describing the binding preference of transcription factors from multiple species. The fifth major release greatly expands the heart of JASPAR—the JASPAR CORE subcollection, which contains curated, non-redundant profiles—with 135 new curated profiles (74 in vertebrates, 8 in Drosophila melanogaster, 10 in Caenorhabditis elegans and 43 in Arabidopsis thaliana; a 30% increase in total) and 43 older updated profiles (36 in vertebrates, 3 in D. melanogaster and 4 in A. thaliana; a 9% update in total). The new and updated profiles are mainly derived from published chromatin immunoprecipitation-seq experimental datasets. In addition, the web interface has been enhanced with advanced capabilities in browsing, searching and subsetting. Finally, the new JASPAR release is accompanied by a new BioPython package, a new R tool package and a new R/Bioconductor data package to facilitate access for both manual and automated methods.
INTRODUCTION
Transcription factors (TFs) influence gene expression by binding to specific cis-acting elements in a genomic sequence. Thus, accurate models for describing the binding properties of TFs are essential in modeling transcription. From a set of known transcription factor binding sites (TFBSs) for a given TF, the binding preference is generally represented in the form of a position weight matrix (PWM) (also called position-specific scoring matrix) derived from a position frequency matrix (PFM). A PFM is essentially an occurrence table, summarizing the number of each nucleotide observed at each position of a set of aligned TFBSs (1,2). Compared with simpler models like consensus sequences, PWMs allow for an additive probabilistic description of binding preferences (3).
The JASPAR database holds collections of PFM nucleotide profiles based on published experiments from diverse sources, and has grown gradually from its inception (4–7). The most widely used JASPAR collection is JASPAR CORE, which is a curated non-redundant set of TFBS profiles for multicellular eukaryotes, based on experimental evidence. The JASPAR database aims to provide the best canonical DNA binding profile per TF, as assessed by expert curators. Non-redundancy of TFBS profiles (i.e. one profile per TF) is intended with the exception of cases in which curators observe a clear difference in the sequence (e.g Nkx2-5) or length (e.g. JUND) at the core of a profile. Other JASPAR motif collections, with different characteristics than the CORE database, are available (7).
Over the years, JASPAR has been equipped with functions aimed at casual and power users. The web-based graphical user interface functionality includes browsing, searching, subsetting and downloading, as well as basic sequence searching tools, dynamic clustering of matrices and generation of random PFMs by sampling selected profiles (4–7).
Historically, JASPAR was populated by PFMs generated by in vitro site selection assays or collections of in-depth characterized sites, limiting both the number of TFs with binding profiles and the number of sites contributing to the profiles. With the development of high-throughput techniques that can assess in vitro or in vivo binding (8–10), it is now possible to generate binding models for most regulators, in multiple species. To this end, we have, in this fifth release, expanded the JASPAR CORE collection substantially, as well as updated the profiles of several existing ones with new data from high-throughput experiments.
EXTENSIVE EXPANSION AND IMPROVEMENT OF JASPAR CORE
The JASPAR CORE database has been substantially expanded. In total, 135 new PFMs have been added (a 30% increase), and 43 older PFMs (9% of last release) have been updated with new data, from vertebrate, insect, nematode and plant species (Table 1). These additions are described in more details later.
Table 1.
Summary of content and growth of the JASPAR CORE database
| Subset | Number of non-redundant profiles in JASPAR 4.0 | New non-redundant profiles in JASPAR 5.0 | Updated profiles | Removed profiles | Total profiles (including older versions of profiles) | Total profiles (non-redundant) |
|---|---|---|---|---|---|---|
| Vertebrates | 130 | 74 | 36 | 1 | 260 | 202 |
| Plants | 21 | 43 | 3 | 67 | 64 | |
| Insects | 123 | 8 | 4 | 1 | 136 | 131 |
| Nematodes | 5 | 10 | 15 | 15 | ||
| Fungi | 177 | 177 | 177 | |||
| Urochordata | 1 | 1 | 1 | |||
| Total | 457 | 135 | 43 | 2 | 656 | 590 |
We compiled published sequence-specific DNA binding TF chromatin immunoprecipitation (ChIP)-seq data collections into the PAZAR database (11,12) along with TF ChIP-seq datasets from the ENCODE (13–15) and modENCODE (16,17) consortia for Homo sapiens, Mus musculus, Drosophila melanogaster and Caenorhabditis elegans. From these studies, we extracted the bound regions, identified over-represented motifs close to the ChIP-seq peak max position (corresponding to the region where the maximum number of ChIP-seq reads are mapped) using the MEME suite (18) and constructed PFMs describing the binding preferences of the TFs (see Supplementary Text for details).
As in previous JASPAR CORE additions, we manually curated the profiles. To confirm the putative binding patterns, we identified independent publications with TFBSs or profiles consistent with the candidates, as described in (7). To gain additional profiles, we considered bound regions derived from ChIP-chip experiments from modENCODE and (19) for D. melanogaster. A similar strategy as for ChIP-seq datasets was used to derive PFMs from ChIP-chip data (see Supplementary Text for details). In total, we obtained 45, 28, 8 and 10 high-quality PFMs in H. sapiens, M. musculus, D. melanogaster and C. elegans, respectively, for TFs that have never been described previously in JASPAR (see Supplementary Table S1). It represents a 57, 6 and 200% increase when compared with the previous release for vertebrates, insects and nematodes, respectively. The newly introduced vertebrate profiles are derived from 34 and 40 ChIP-seq experiments collected from PAZAR and ENCODE, respectively. The fact that almost 50% of the new PFMs are from individual studies collected in PAZAR highlights the importance of our manual retrieval of published ChIP-seq data. From ChIP-seq data sets of the vertebrate sequence-specific TFs not previously described in JASPAR, we obtained 71 (∼60%) canonical motifs satisfying our literature-based manual curation (see Supplementary Table S2). The rich data from ChIP experiments allowed replacement of 39 existing profiles for TFs in mammals (36 PFMs updated) and in D. melanogaster (3 PFMs updated).
As part of the curation of ChIP-seq data, and as introduced earlier, we computed a centrality score as described in (20), based on our expectation that the positions where the maximum number of ChIP-seq reads map on the genome of reference will be strongly enriched for binding sites corresponding to the ChIPed TF (21). We provide the centrality plot and log(P-value) for each newly introduced PFMs in vertebrates (see Figure 1), showing the propensity of the motif to be found close to the peak-max position in the corresponding peaks of the ChIP-seq dataset used to generate the profile (see Supplementary Figure S1). The high quality of the vertebrates PFMs and the ChIP-seq datasets used to construct them is reflected by the low centrality log(P-values), which are all below −200, with the exception of the Bach1::Mafk, ESRRA, FOXP1, FOXP2, Hoxa9, Sox6, SP2, SREBF1, SREBF2, and THAP1 binding profiles (see Supplementary Table S1).
Figure 1.

Screenshot of an example TFBS profile in new layout.
Moreover, we expanded the collection of PFMs for Arabidopsis thaliana TFs in JASPAR, with the first targeted JASPAR curation effort for plant TFs. We have included 43 new DNA-binding profiles for A. thaliana TFs, more than tripling the plant content in JASPAR CORE, and we updated three previous PFMs. The profiles are derived from in vitro and in vivo experiments (8 new profiles are constructed from ChIP-seq experiments, 8 from ChIP-chip experiments, 6 from protein binding microarray experiments and 24 from SELEX experiments).
MODELS FOR DUAL BINDING BY THE SAME TF
In this release, in extremely select cases, we introduce multiple binding profiles for a same TF, as motivated by the fact that some TFs display diverse target specificity that cannot be represented using a single PFM model. For instance, JUND has been previously shown to bind the DNA with motifs of flexible lengths (22) with a core composed of either TGACGTCA or TGAC/GTCA, where C/G stands for C or G. The two new profiles introduced for JUND (see Figure 2A) are derived from the same ChIP-seq dataset, confirming the binding to the two subclasses. Similarly, we introduce two profiles for JUN (see Figure 2B), displaying equivalent characteristics to the JUND profiles. A new profile for Nkx2-5 (see Figure 2C) derived from ChIP-seq data has been introduced. It differs substantially from an in vitro SELEX experiment-based profile but has been confirmed to reflect binding properties of Nkx2-5 (23). Finally, we introduce two binding profiles associated to the plant TF RAV1, as it can bind to two unrelated motifs by using two distinct DNA-binding domains (24) (see Figure 2D). The philosophy of maintaining JASPAR as a non-redundant collection remains a driving approach to curation. In these special cases in which we allow unique pairs of profiles for the same TF, the TF presents distinct binding capacities that cannot be captured within a single PFM.
Figure 2.
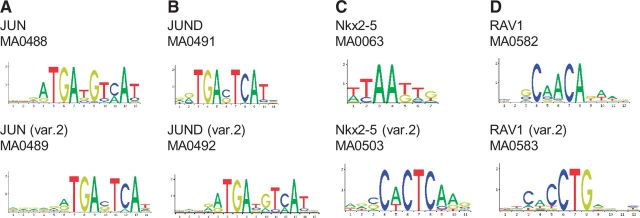
TFBSs with two different profiles. (A) JUN, (B) JUND, (C) Nkx2-5 and (D) RAV1.
ENHANCED WEB INTERFACE AND NEW RESOURCES FOR POWER USERS
For casual users, we have enhanced the web search interface to the JASPAR database. Fuzzy searching is now enabled to search one or multiple profiles by gene name, species official or common name, protein accession ID, DNA-binding domain family or class, experiment type (e.g. ChIP-seq) and any other keyword associated to the profile(s) in the underlying database. This fuzzy searching performs approximate string matching in case-insensitive mode and offers suggestions below the search box while typing. It also includes the gene name aliases from HGNC (PMID:23161694) for searching gene synonyms. Furthermore, for each TF profile, we have now included links to the Transcription Factor Encyclopedia (25) and to the protein structures from the Protein Data Bank when available (26). Each binding profile links to the corresponding TFBSshape profile of DNA structural analysis (27).
For power users, we have developed an open source Python package (freely available at https://github.com/biopython/) within the extensively used tools of the BioPython Project (28). We implemented the jaspar package as part of the ‘motifs’ BioPython package, which provides functions such as reading profiles, writing profiles, scanning sequences for motif instances and more. The specific jaspar ‘motif’ class allows to store all the metadata information related to the profiles in JASPAR, and specific functions allow the user to retrieve profiles from the database. We also developed an R/Bioconductor (PMID: 15461798) software package TFBSTools, available at http://www.bioconductor.org/packages/2.13/bioc/html/TFBSTools.html under the General Public License-2 (GPL-2), to provide developers handy tools to generate, read and convert the JASPAR template, an internal data format to describe each motif instance and its meta information. An R/Bioconductor (29) data package JASPAR2014Data is freely available at http://www.bioconductor.org/packages/devel/data/experiment/html/JASPAR2014.html to provide the users with tools for data analysis using the JASPAR profiles.
In addition, a web-based curator interface was developed for JASPAR, focusing on giving the super-users the ability to edit and update the database: this capacity is released for users wishing to produce custom PFM databases using the JASPAR framework.
CONCLUSIONS AND FUTURE DEVELOPMENTS
In this release of JASPAR, we have focused on the CORE database and expanded it primarily with new ChIP-based data. Although these types of expansions are important and will continue, the increasing availability of rich data sources highlights important questions for the future development of JASPAR, which need to be discussed with its user base. Two such larger questions are as follows.
Non-redundancy versus species-specific matrix models?
JASPAR CORE was originally designed with the clear goal of finding the ‘best’ PFM for a certain TF, unlike other databases that can hold several models for the same factor. Although many users have appreciated the clarity, it is not established how to resolve cases where the same factor has been characterized in-depth in two or more species. While this situation was rare in the early JASPAR versions, new experimental methods allows for probing binding specificity in several species with comparative ease (30). In general, the binding specificity for orthologous TFs rarely changes to a substantial degree, but exceptions exist (31). Thus, future curation of JASPAR will have to resolve whether the non-redundancy approach should be within each species or within larger clades.
New types of models?
Likewise, the sheer amount of sites that the new laboratory methods generate provides sufficient information to produce predictive models that address more aspects than can be readily handled within the classic PWM framework—in particular, dependencies between positions and variable length motifs, which basic PWM models ignore. Here, one will have to consider the trade-off between possible higher specificity in binding predictions [see (32) for a detailed discussion] and the comfort of the community with the simpler PWM models. It is our plan to introduce newly designed Transcription Factor Flexible Models (33) derived from ChIP-seq data within JASPAR in the near future.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank the user community for useful input. They also thank Michiel de Hoon for the help in the development of the jaspar package as part of BioPython.
APPENDIX
During the production process, we analyzed the recently published ChIP-seq data sets from (PMID: 23953112). Three new profiles resulted and have been added to the new release of JASPAR. This late addition is not covered in the manuscript.
FUNDING
The AS lab was supported by grants from the Novo Nordisk Foundation, the Lundbeck Foundation and The European Research Council under the EU 7th Framework Programme [FP7/2007-2013]/ERC grant agreement 204135. The WWW lab was supported by the Canadian Institutes for Health Research [to W.W.W.]; the National Sciences and Engineering Research Council of Canada (to W.W.W. and C.Y.C.); the National Institute of General Medical Sciences [R01GM084875 to W.W.W.]; Michael Smith Foundation for Health Research (C.R.W.H.); GenomeCanada (ABC4DE Project) and Genome British Columbia (ABC4DE and CanEuCre Projects); the Rhônes-Alpes region CMIRA fellowship and CNRS support (to F.P.); Supported by the EU FP7 large-scale integrated project ZF HEALTH [HEALTH-F4-2010-242048 to G.T.];. Supported by the Medical Research Council UK and the Department of Informatics, University of Bergen (to B.L.). Funding for open access charge: Supported by the Novo Nordisk foundation and Lundbeck foundation for the AS lab, the National Institute of General Medical Sciences [R01GM084875] for the W.W.W. lab, and the Medical Research Council (UK) for the B.L. lab.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 2.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 3.Stormo GD. Modeling the specificity of protein-DNA interactions. Quant. Biol. 2013;1:115–130. doi: 10.1007/s40484-013-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vlieghe D, Sandelin A, De Bleser PJ, Vleminckx K, Wasserman WW, van Roy F, Lenhard B. A new generation of JASPAR, the open-access repository for transcription factor binding site profiles. Nucleic Acids Res. 2006;34:D95–D97. doi: 10.1093/nar/gkj115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–D106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger MF, Bulyk ML. Protein binding microarrays (PBMs) for rapid, high-throughput characterization of the sequence specificities of DNA binding proteins. Methods Mol. Biol. 2006;338:245–260. doi: 10.1385/1-59745-097-9:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 10.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev.. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Portales-Casamar E, Kirov S, Lim J, Lithwick S, Swanson MI, Ticoll A, Snoddy J, Wasserman WW. PAZAR: a framework for collection and dissemination of cis-regulatory sequence annotation. Genome Biol. 2007;8:R207. doi: 10.1186/gb-2007-8-10-r207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portales-Casamar E, Arenillas D, Lim J, Swanson MI, Jiang S, McCallum A, Kirov S, Wasserman WW. The PAZAR database of gene regulatory information coupled to the ORCA toolkit for the study of regulatory sequences. Nucleic Acids Res. 2009;37:D54–D60. doi: 10.1093/nar/gkn783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 14. ENCODE Project Consortium, Bernstein, B.E., Birney, E., Dunham, I., Green, E.D., Gunter, C. and Snyder, M. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed]
- 15. Mouse ENCODE Consortium, Stamatoyannopoulos, J.A., Snyder, M., Hardison, R., Ren, B., Gingeras, T., Gilbert, D.M., Groudine, M., Bender, M., Kaul, R. et al. (2012) An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol., 13, 418. [DOI] [PMC free article] [PubMed]
- 16. modENCODE Consortium, Roy, S., Ernst, J., Kharchenko, P.V., Kheradpour, P., Negre, N., Eaton, M.L., Landolin, J.M., Bristow, C.A., Ma, L. et al. (2010) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science, 330, 1787–1797. [DOI] [PMC free article] [PubMed]
- 17.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong EE. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 20.Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CY, Schwartz RJ. Identification of novel DNA binding targets and regulatory domains of a murine tinman homeodomain factor, nkx-2.5. J. Biol. Chem. 1995;270:15628–15633. doi: 10.1074/jbc.270.26.15628. [DOI] [PubMed] [Google Scholar]
- 24.Kagaya Y, Ohmiya K, Hattori T. RAV1, a novel DNA-binding protein, binds to bipartite recognition sequence through two distinct DNA-binding domains uniquely found in higher plants. Nucleic Acids Res. 1999;27:470–478. doi: 10.1093/nar/27.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yusuf D, Butland SL, Swanson MI, Bolotin E, Ticoll A, Cheung WA, Zhang XY, Dickman CT, Fulton DL, Lim JS, et al. The transcription factor encyclopedia. Genome Biol. 2012;13:R24. doi: 10.1186/gb-2012-13-3-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Zhou T, Dror I, Mathelier A, Wasserman WW, Gordan R, Rohs R. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014;42:D148–D155. doi: 10.1093/nar/gkt1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt SF, Jorgensen M, Chen Y, Nielsen R, Sandelin A, Mandrup S. Cross species comparison of C/EBPalpha and PPARgamma profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC Genomics. 2011;12:152. doi: 10.1186/1471-2164-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giese K, Pagel J, Grosschedl R. Distinct DNA-binding properties of the high mobility group domain of murine and human SRY sex-determining factors. Proc. Natl Acad. Sci. USA. 1994;91:3368–3372. doi: 10.1073/pnas.91.8.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benos PV, Bulyk ML, Stormo GD. Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res. 2002;30:4442–4451. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathelier A, Wasserman WW. The Next Generation of Transcription Factor Binding Site Prediction. PLoS Comput. Biol. 2013;9:e1003214. doi: 10.1371/journal.pcbi.1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]