


Abstract
MycoCosm is a fungal genomics portal (http://jgi.doe.gov/fungi), developed by the US Department of Energy Joint Genome Institute to support integration, analysis and dissemination of fungal genome sequences and other ‘omics’ data by providing interactive web-based tools. MycoCosm also promotes and facilitates user community participation through the nomination of new species of fungi for sequencing, and the annotation and analysis of resulting data. By efficiently filling gaps in the Fungal Tree of Life, MycoCosm will help address important problems associated with energy and the environment, taking advantage of growing fungal genomics resources.
INTRODUCTION
Genomics is a powerful tool for understanding the genomic basis of biological processes in individual organisms and their communities. The Fungal Genomics Program (http://jgi.doe.gov/fungi) of the US Department of Energy (DOE) Joint Genome Institute (JGI) has partnered with the international scientific community to support genomic exploration of the kingdom Fungi and to help address problems related to energy and the environment. The exponential growth of data resulting from several large-scale genomics initiatives like the 1000 Fungal Genomes Project requires new approaches for data integration and analysis (1). We designed MycoCosm, an integrated fungal genomics resource, to allow researchers to easily access large amounts of genomics data and analyze them using web-based analytical tools. The MycoCosm portal enables in-depth multidimensional analysis of individual genomes and efficient comparative genomics of fungi, which may be applied to phylogenetically related fungi, or to those sharing the same lifestyle, ecological niche or phenotypic trait.
The first JGI fungal portal was created for the white rot fungus Phanerochaete chrysosporium in 2004 (2). To enable comparative genome analysis within the same analytical framework, in 2008 JGI integrated data from six genomes of Dothideomycete plant pathogens (three sequenced by JGI and three others provided by the Dothideomycetes Genome Consortium). This served as a prototype for MycoCosm, the first official version of which was released in March 2010 in response to a call for a pan-fungal genomic resource from the community of fungal biologists brought together by the Burroughs Wellcome Fund (Alexandria, VA). By 2011, MycoCosm included over 100 fungal genomes (3), and continues to grow. Here we provide an update on the latest developments of MycoCosm, which includes flexible tools for the analysis of exponentially growing genomics and other ‘omics’ data sets, and extends user community participation to the entire genome project cycle, from nomination of species for sequencing through genome annotation and comparative genomics.
Significantly larger and better-integrated genomics data
Since our last report (3), MycoCosm has grown to host >250 publicly available fungal genomes (Figure 1), 80% of which were sequenced by JGI and 20% by other groups added for comparative purposes. Individual genomes, or biologically relevant groups of them, may be searched using keywords or sequence, investigated using MycoCosm analytical tools, and downloaded for custom analyses.
Figure 1.

Growth of annotated genomes in MycoCosm. The genomes sequenced by JGI are shown in blue and those sequenced by others are shown in red.
Also in the past two years, all MycoCosm nodes have been populated with newly sequenced and annotated genomes including the recently activated nodes for Glomeromycota, Entomophthoromycotina, Kickxellomycotina, Blastocladiomycota, Neocallimastigomycota and others. Several new branches such as Xylonomycetes, Zoopagomycotina and Cryptomycota have been added for a more complete picture of the Fungal Tree of Life (Figure 2). Several model fungi have been added to MycoCosm, for which genes are linked to the corresponding records in curated databases such as the Saccharomyces Genome Database (4), the Aspergillus Genome Database (5) and others. GenBank records for JGI-sequenced fungal genomes are linked to MycoCosm using “JGIDB” qualifiers (http://www.ncbi.nlm.nih.gov/genbank/collab/db_xref/).
Figure 2.

MycoCosm front page. The MycoCosm tree illustrates relationships between major groups of fungi. Each node of the tree is linked to the corresponding group page and portals for individual genomes within the group.
Each genome in MycoCosm has its own portal where genomics and other ‘omics’ data can be explored in depth, in a single-genome context (Figure 3). Genomes in MycoCosm are also grouped for comparative genomics analyses (Figure 4), into two kinds of groups: phylogenetic and ecological. Phylogenetic groups are represented (and linked to) by the nodes of the MycoCosm tree, and facilitate comparative analyses of organisms with a shared evolutionary history. Ecological groups represent a curation effort in which fungi of similar lifestyles (e.g. endophytes, mycorrhizal symbionts, plant pathgens, etc.) have been assembled into groups that are linked to the chapters of the Genomics Encyclopedia of Fungi (6).
Figure 3.
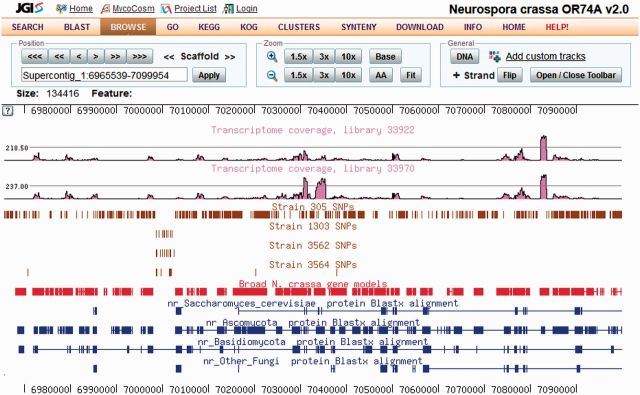
Genome Browser for Neurospora crassa. Horizontal tracks represent genomic features of a given assembly scaffold. Red bars indicate N. crassa genes predicted by the Broad Institute and uploaded into MycoCosm. The four tracks with brown bars illustrate SNPs derived from resequencing of mutant strains of N. crassa (7). The two pink curves represent transcriptome profiling using RNA-Seq during N. crassa growth under two different conditions. Additional tracks can be displayed and configured using the toolbar.
Figure 4.
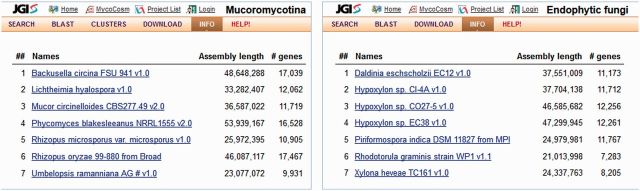
Examples of MycoCosm group pages. A PhyloGroup containing seven annotated genomes of fungi from Mucoromycotina is shown on the left. An EcoGroup with seven annotated genomes of phylogenetically diverse endophytes is shown on the right.
To encourage the research community to participate in the further growth of MycoCosm, users can now nominate diverse fungal species for sequencing (Figure 5) in the context of the 1000 Fungal Genomes Project (http://1000.fungalgenomes.org). Nominated species will be peer reviewed, and, if approved, will be sequenced, annotated and fully integrated into MycoCosm’s comparative genomics framework.
Figure 5.
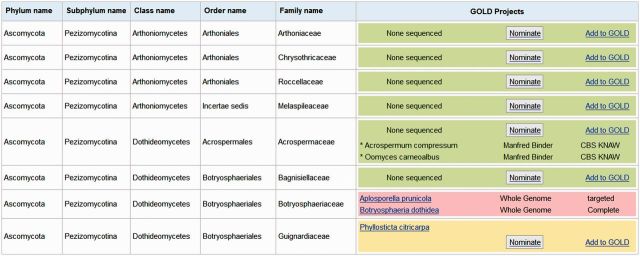
1000 Fungal Genomes nomination page. Over 500 families of fungi and associated genome projects are displayed. Green and yellow rows highlight families with no, or only one, sequenced genome, where additional species can be nominated for sequencing via the use of the ‘Nominate’ button. Nomination requires user registration and commitment to provide JGI with DNA and RNA samples for sequencing.
Uniform genome annotation
For genome comparison to produce meaningful results, application of the same set of functional annotation databases to all genomes is required. For genomes sequenced and annotated outside of JGI, the existing gene models are fed into the functional annotation part of the pipeline, using the same databases used for annotation of JGI genomes, to ensure consistent references for genome comparisons.
All fungi sequenced at JGI are annotated using the JGI Annotation Pipeline, which integrates an array of tools for gene prediction, annotation and analysis (8). Using repeat-masked genome assemblies as input, protein-coding gene models are predicted using a combination of gene predictors of three general kinds: ab initio, homology-based and transcriptome-based. The ab initio Fgenesh (9) and GeneMark (10) are specifically trained for the given genome. Homology-based Fgenesh+ (9) and Genewise (11) are seeded by BLASTx (12) alignments of fungal sequences in NCBI’s nonredundant protein database. Transcriptome-based models are predicted from assembled RNA-sequencing data derived from the cultured fungus. For each genomic locus, the best representative gene model is selected based on a combination of protein homology and transcriptome support; these make up a ‘gene catalog’. In addition to protein-coding genes, tRNAs are predicted using tRNAscan-SE (13).
All predicted proteins are then functionally annotated. SignalP (14) is used to detect the sequence motifs responsible for protein localization, TMHMM (15) identifies possible transmembrane domains and InterProScan (16) predicts functional domains from Pfam (17) and other databases. Protein alignments to the NCBI’s nonredundant (http://www.ncbi.nlm.nih.gov), Swiss-Prot (18), KEGG (19) and KOG (20) databases additionally facilitate functional interpretation. Interpro, KEGG and Swiss-Prot hits are used to map gene ontology (GO) terms (21). Detailed annotation reports from these methods are provided on every coding gene’s protein page. Additionally, summaries listing numbers of genes by category in the GO, KEGG and KOG classifications are accessible from the portal menu, and can be compared with other selected genomes to explore gene family expansions and contractions across genomes.
Predicted gene models, along with different lines of supporting evidence including transcriptomics and similarity to proteins in other genomes, are displayed on a genome browser (Figure 3). The browser, which is based on the UCSC Genome Browser (22), displays genomic features as horizontal tracks, which can be customized to the needs of each user. Individual elements from these tracks are linked to the corresponding pages for additional details. For example, predicted gene models are linked to annotation reports, while genome conservation tracks are linked to VISTA Point (23). Two new types of data, single-nucleotide polymorphisms (SNPs) and gene expression profiles, have been added to the browser, to enable the visualization of resequencing and transcriptomics data, respectively (Figure 3). These are important additions to the resources developed, for example, for model fungi such as the ascomycete Neurospora crassa (7) or the basidiomycete Schizophyllum commune (24).
Predicted gene models, and their annotations, can be improved through the process of manual curation, which is open to all authorized users and coordinated by the principal investigator of each project. Such collective efforts were critical for the analysis of several fungal genomes (24–31). Additionally, users can upload custom tracks to the browser and share them, either with specified collaborators or globally. This capability to upload data for individual genes or entire genomes, makes MycoCosm a powerful and customizable data exchange resource for the community.
Grouping genomes for comparative analyses
The wealth of genomes in MycoCosm creates an opportunity to compare groups of genomes, and the need for efficient tools to enable these comparisons. Mycocosm therefore includes tools that integrate single genomes into a comparative context, such as the ability to visualize variation in gene counts in different GO, KEGG and KOG categories across a user-selected assortment of genomes. Additionally, MycoCosm includes predefined groups of fungi to enable users to ask questions for fungal genomes in a group context. Two types of genome groups are available in MycoCosm and shown in Figure 4: (i) PhyloGroups, consisting of phylogenetically related species (corresponding to the nodes of the MycoCosm tree), and (ii) EcoGroups, containing fungi of similar lifestyle or ecology (but which may be phylogenetically distant).
Both kinds of group offer consistent interfaces for comparative analyses. Genome sizes and gene counts may be compared on the Info page (Figure 4), giving a coarse-grained overview. Group-specific BLAST, search and download of genomes and annotations are available. Genes shared between genomes within a group can be explored using precomputed Markov clustering (32) of the full set of protein sequences in the group. Derived from all-versus-all BLAST alignments of the proteins, these gene clusters can be thought of as protein ‘families’. The clusters can be filtered to show genes shared between all, or some, organisms in the group, or to find genes unique to them as described in details in help pages. Each cluster is linked to a page listing all the genes of the cluster with graphical display of protein domains, intron–exon gene structure and local synteny of cluster members. A larger genome-scale synteny calculated by VISTA (23) is available from the Synteny menu of individual genome portals, where dot-plots, for example, can illustrate the full spectrum of micro-, meso- and macro-synteny between different pairs of Dothideomycete species (31). This phenomenon is limited to this class of fungi and illustrates the utility of the group-centric approach for comparative genome analysis.
User nomination of new species for sequencing
Even though all major nodes of the MycoCosm tree are represented by at least one sequenced genome, many fungal clades remain unrepresented. The 250+ annotated genomes in MycoCosm are a small fraction of the total number of fungal species, estimates of which range from 1.5 million to as high as 5.1 million (33,34). Thus, sampling of the Fungal Tree of Life by genomics is sparse, and likely has left many biologically interesting clades unexplored. This undersampling will surely limit our attempts to understand functional diversity and evolution through fungal genomes, and will hamper metagenomics studies of fungal communities, where a broad array of reference genomes is required to interpret complex sequence data.
To encourage the scientific community to participate in determining what gaps in the Fungal Tree of Life will be filled in next, every node of MycoCosm is now equipped with a ‘Nominate’ link to enable users to recommend new genomes for sequencing via the 1000 Fungal Genomes Project (http://1000.fungalgenomes.org). Following this link from a given node, families of fungi with no, or only one, sequenced genomes (Figure 5, green and yellow rows) display a ‘Nominate’ button, which invites users to describe the species, access JGI User Guidelines and provide DNA and RNA samples to JGI for sequencing (if approved by peer review). The 1000 Fungal Genomes Project aims to have at least two sequenced genomes for every family of fungi. Through this and other JGI Community Sequencing Projects (35), we aim to fill the remaining gaps in the Fungal Tree of Life.
To enable visibility of ongoing JGI genome projects, the Projects menu of MycoCosm (http://jgi.doe.gov/projects) lists each project’s status, principal investigator and other pertinent information. Combined with other genome projects listed in the GOLD database (36) and other sources, they form a ‘master’ list of fungal genome sequencing projects worldwide (http://1.usa.gov/JGI-1000-Fungi). This ‘master’ list helps us better focus our sequencing efforts on unexplored regions of the Fungal Tree of Life. Thus, we integrate a full genome project cycle (from sequencing target selection through assembly and annotation, to biology-driven data analysis) into MycoCosm enabling users to be active contributors to this process.
FUTURE PLANS
Since the release of its first fungal genome 10 years ago, the number of fungal genomes sequenced by JGI has jumped to >200. Collecting, integrating and visualizing these genomes, and those in sequencing pipeline, for efficient comparative analysis requires new approaches and tools. Especially, functional genomics will quickly reveal putative functions of a large number of genes identified only through genome sequencing (as opposed to smaller-scale biochemical and genetic studies). New interactive tools for transcriptomics, epigenomics, population genomics and metagenomics will therefore be developed and integrated into MycoCosm, in support of data analysis and utilization, and hypothesis development.
FUNDING
Director, Office of Science, Office of Biological and Environmental Research, Life Sciences Division, US Department of Energy [DE-AC02-05CH11231]. Funding for open access charge: US Department of Energy Contract [No. DE-AC02-05CH11231].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank MycoCosm users for valuable feedback, members of JGI portal group for development of software modules used in MycoCosm and Byoungnam Min for technical assistance.
REFERENCES
- 1.Grigoriev IV. A changing landscape of fungal genomics. In: Martin F, editor. The Ecological Genomics of Fungi. Hoboken, NJ: John Wiley & Sons, Inc; 2013. pp. 1–20. [Google Scholar]
- 2.Martinez D, Larrondo LF, Putnam N, Gelpke MD, Huang K, Chapman J, Helfenbein KG, Ramaiya P, Detter JC, Larimer F, et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004;22:695–700. doi: 10.1038/nbt967. [DOI] [PubMed] [Google Scholar]
- 3.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, et al. The genome portal of the department of energy joint genome institute. Nucleic Acids Res. 2012;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnaud MB, Cerqueira GC, Inglis DO, Skrzypek MS, Binkley J, Chibucos MC, Crabtree J, Howarth C, Orvis J, Shah P, et al. The Aspergillus genome database (AspGD): recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res. 2012;40:D653–D659. doi: 10.1093/nar/gkr875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigoriev IV, Cullen D, Goodwin SB, Hibbett D, Jeffries TW, Kubicek CP, Kuske C, Magnuson JK, Martin F, Spatafora JW, et al. Fueling the future with fungal genomics. Mycology. 2011;2:192–209. [Google Scholar]
- 7.McCluskey K, Wiest AE, Grigoriev IV, Lipzen A, Martin J, Schackwitz W, Baker SE. Rediscovery by whole genome sequencing: classical mutations and genome polymorphisms in Neurospora crassa. G3 (Bethesda) 2011;1:303–316. doi: 10.1534/g3.111.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grigoriev IV, Martinez DA, Salamov AA. Fungal genomic annotation. In: Arora DK, Berke RA, Singh GB, editors. Applied Mycology and Biotechnology. Vol. 6. Amsterdam: Elsevier; 2006. pp. 123–142. [Google Scholar]
- 9.Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, Madden TL, Matten WT, McGinnis SD, Merezhuk Y, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 15.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 16.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;33:W116–W120. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The UniProt Consortium. Update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blake JA, Dolan M, Drabkin H, Hill DP, Li N, Sitnikov D, Bridges S, Burgess S, Buza T, McCarthy F, et al. Gene ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. The UCSC genome browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE, de Vries RP, Record E, Levasseur A, Baker SE, et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010;28:957–963. doi: 10.1038/nbt.1643. [DOI] [PubMed] [Google Scholar]
- 25.Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, Ishmael N, John T, Darmond C, Moisan MC, et al. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat. Biotechnol. 2011;29:922–927. doi: 10.1038/nbt.1976. [DOI] [PubMed] [Google Scholar]
- 26.Duplessis S, Cuomo CA, Lin YC, Aerts A, Tisserant E, Veneault-Fourrey C, Joly DL, Hacquard S, Amselem J, Cantarel BL, et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl Acad. Sci. USA. 2011;108:9166–9171. doi: 10.1073/pnas.1019315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eastwood DC, Floudas D, Binder M, Majcherczyk A, Schneider P, Aerts A, Asiegbu FO, Baker SE, Barry K, Bendiksby M, et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science. 2011;333:762–765. doi: 10.1126/science.1205411. [DOI] [PubMed] [Google Scholar]
- 28.Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martinez AT, Otillar R, Spatafora JW, Yadav JS, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 29.Goodwin SB, M'Barek SB, Dhillon B, Wittenberg AH, Crane CF, Hane JK, Foster AJ, Van der Lee TA, Grimwood J, Aerts A, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011;7:e1002070. doi: 10.1371/journal.pgen.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin F, Aerts A, Ahren D, Brun A, Danchin EG, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V, et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88–92. doi: 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- 31.Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012;8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawksworth DL. The fungal dimension of biodiversity-magnitude, significance, and conservation. Mycol. Res. 1991;95:641–655. [Google Scholar]
- 34.Blackwell M. The Fungi: 1, 2, 3.. 5.1 million species? Am. J. Bot. 2011;98:426–438. doi: 10.3732/ajb.1000298. [DOI] [PubMed] [Google Scholar]
- 35.Martin F, Cullen D, Hibbett D, Pisabarro A, Spatafora JW, Baker SE, Grigoriev IV. Sequencing the fungal tree of life. New Phytologist. 2011;190:818–821. doi: 10.1111/j.1469-8137.2011.03688.x. [DOI] [PubMed] [Google Scholar]
- 36.Pagani I, Liolios K, Jansson J, Chen IMA, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]