


Abstract
IDEAL (Intrinsically Disordered proteins with Extensive Annotations and Literature, http://www.ideal.force.cs.is.nagoya-u.ac.jp/IDEAL/) is a collection of intrinsically disordered proteins (IDPs) that cannot adopt stable globular structures under physiological conditions. Since its previous publication in 2012, the number of entries in IDEAL has almost tripled (120 to 340). In addition to the increase in quantity, the quality of IDEAL has been significantly improved. The new IDEAL incorporates the interactions of IDPs and their binding partners more explicitly, and illustrates the protein–protein interaction (PPI) networks and the structures of protein complexes. Redundant experimental data are arranged based on the clustering of Protein Data Bank entries, and similar sequences with the same binding mode are grouped. As a result, the new IDEAL presents more concise and informative experimental data. Nuclear magnetic resonance (NMR) disorder is annotated in a systematic manner, by identifying the regions with large deviations among the NMR models. The ordered/disordered and new domain predictions by DICHOT are available, as well as the domain assignments by HMMER. Some examples of the PPI networks and the highly deviated regions derived from NMR models will be described, together with other advances. These enhancements will facilitate deeper understanding of IDPs, in terms of their flexibility, plasticity and promiscuity.
INTRODUCTION
Intrinsically disordered or natively unstructured proteins (IDPs) are proteins that do not adopt unique 3D structures under physiological conditions (1–5). They are fully or partially disordered, depending on the amount of intrinsically disordered regions (IDRs). IDPs are abundant among eukaryotic proteins, and are localized preferentially in the nucleus (6–8). They play crucial roles in biological processes, such as signal transduction and transcriptional regulation (1,3,4,9). Naturally, to gain a deep understanding of the nature of IDPs from structural and functional viewpoints, the development of IDP databases is required. For this purpose, Disprot (10), MobiDB (11) and D2P2 (12) were compiled, and each presents original information. By collecting experimentally verified IDPs and annotating them manually, we have developed IDEAL, Intrinsically Disordered proteins with Extensive Annotations and Literature (http://www.ideal.force.cs.is.nagoya-u.ac.jp/IDEAL/) (13). This database provides the ordered and disordered regions, as well as other structural and functional information, for each IDP. In particular, IDEAL pays special attention to the IDRs that adopt 3D structures on binding to their binding partners (14–18), which are described as protean segments (ProS) (13). Although similar concepts of ProS have been proposed as molecular recognition features (MoRFs), eukaryotic linear motifs (ELMs) or ComSins, the details of the definitions are different from each other. ProS is simply defined if both structured and unstructured information are available (13), while MoRF has a length limitation of 70 residues (17), an ELM represents a motif expressed by a regular expression (14) and ComSin requires structures of ligand bound and unbound states (16). In the initial phase of the database construction, IDEAL considered only the human IDPs localized in the nucleus, but currently it encompasses eukaryotic IDPs residing in the nucleus, or in the nucleus and other locations. As a result, the number of entries in IDEAL has tripled (340) in the past 2 years. Although IDEAL compiles the available information for each IDP, we noticed that the relationships between entries were not necessarily described well. One of the significant features of IDPs is their promiscuity (19). In many cases, IDPs function as the hub proteins in protein–protein interaction (PPI) networks (20,21), and thus IDPs are able to interact with many binding partners, and are controlled by posttranslational modifications (PTM) or other mediators. These results indicated the importance of examining the IDPs in the PPI networks and in the context of systems biology. In the upgraded version, IDEAL emphasizes the interactions between IDPs and their binding partners. We regard each IDEAL entry (protein) as a NODE, and the interaction of two entries (PPI) as an EDGE, and thus have prepared both NODE and EDGE pages. The NODE pages are improved versions of the previous pages, and contain detailed information for an IDP. The EDGE pages are new to this version, and show the structural complex of the entry and its binding partner. Since the NODE pages are connected by the EDGE pages, and vice versa, users can easily browse through a PPI network by following these links. The revamped version of IDEAL also features new data and an improved user interface.
NEW DATA
Binding partner
In IDEAL, a protein sequence is divided into regions. A region is considered as a unit sharing the same features (e.g. ordered or disordered), and annotations are provided for each region (13). Although binding partner information for a region was previously described in IDEAL, the new IDEAL presents the binding partners explicitly for each region of the IDPs. The binding partner information is derived from the structures in the Protein Data Bank (PDB) (22). The PDB supplies the ‘biological units’ for each entry, which are a set of subunits that form a protein complex under physiological conditions. Among the biological units, the one defined by the authors was selected, and the shortest Cα distance between any pair of the subunits was calculated. When the distance was <8 Å, the interaction of the subunits was accepted as defining the binary relationship of a PPI. When we considered the author-defined complex to be unsuitable, that is, monomeric structures were deposited for hetero-oligomers, or, a complex had no inter-subunit interactions, the complex in the asymmetrical unit was selected. We did not consider the other types of interactions, for example, those that are measured by large-scale experiments such as a yeast two-hybrid system (23,24).
Clustering results of experimental evidence
Since IDEAL collects all available experimental evidence in the PDB, in which multiple structures for an identical protein or complex have been deposited, some IDEAL entries have redundant information. To extract the unique information from the redundant data, we constructed clusters of almost equivalent PDB entries, by using the biological units described above. In a comparison of two complexes, they are first divided into subunits. When two subunits (a subunit pair) taken from each complex show >70% sequence identity, or the number of gaps in the global alignment is at most six, the subunit pair is considered to be equivalent. Note that the latter condition is applied to compare short segments. When all subunit pairs in two complexes are equivalent, and the interacting-subunit pairs are the same, the complexes are considered to be equivalent, and should be clustered. Based on this rule, we conducted single-linkage clustering, and obtained clusters of protein complexes. Monomers were also clustered in the same manner. IDEAL presently contains >2000 PDB entries, and they are grouped into almost a thousand clusters.
High deviation of nuclear magnetic resonance models
Conventionally, IDRs can be detected as missing residues in X-ray structures or from the descriptions in the literature [see the data collection of the IDR predictors (25,26)], but no standard protocol has been developed to identify the IDRs from structures determined by nuclear magnetic resonance (NMR). NMR-based disorder information (27,28) is useful for understanding IDPs because NMR provides clues about the dynamical nature of proteins. The method has been frequently used to analyze nuclear proteins (29), which contain many IDPs (6–8). Recently, we proposed a computational method to assign IDRs based on NMR structures (29). The missing residues of X-ray structures were compared with the residue-wise Cα root-mean-square deviations (RMSDs) of NMR models [Equation (1) in (29)] for identical proteins, and it was found that the RMSD threshold of 3.2 Å gave the best correlation (30) of the ordered and disordered regions of both structures. This method was applied to the NMR structures for IDEAL entries if the structures satisfied the application conditions of the method (29). To ascertain the significance of the ordered regions, we disregarded the un-deviated segments composed of fewer than four residues.
Methylation and acetylation sites
Methylation and acetylation sites, as well as phosphorylation sites, are cited from the UniProt (31) annotations. Other PTM sites are not considered in this version owing to limited information.
NEW INTERFACE
PPI network
The new version of IDEAL incorporates the binding-partner information explicitly. This enables IDEAL to illustrate the PPI networks. The largest PPI network in IDEAL, composed of 87 entries, is shown in Figure 1, where the yellow-colored protein is the one from which the network was generated. As described in the ‘Introduction’ section, IDEAL highlights ProS (13), which are the functional segments in IDRs (14,15,17) involved in the coupled folding and binding processes (18). The proteins containing at least one ProS are colored green, and the others are blue. The protein name and the PDB code plus chain identifiers appear if the pointer is on the node and the edge (the red box and the red line), respectively. This network is roughly divided into four functional categories: a) proteins related to the cell cycle, centered by the cellular tumor antigen p53 [IDEAL Identification (IID) 00015] (32), such as cyclin-dependent kinase 2 (CDK2, IID00034), cyclin-dependent kinase inhibitor 1B (p27, IID00049) and retinoblastoma-associated protein (Rb, IID00017); b) proteins related to nuclear receptors, such as androgen receptor (ANDR, IID00020), glucocorticoid receptor (GCR, IID00045) and coactivators/repressors of nuclear receptors (NRCA4/NRCR1, IID00074/IID00185) (33); c) proteins related to histones (H4/H3.3, IID00058/IID00239 etc.) (34); and d) proteins binding with importin (IID50009) (35). These classifications are depicted by ellipses and letters in Figure 1. The second largest network consists of 16 entries, and contains proteins involved in the Wnt signaling pathway, such as catenine beta-1 (IID00039), adenomatous polyposis coli protein (IID00035) and Axin-1 (IID00007) (36). As shown in these examples, the interactions among proteins in the same cellular processes are identified and clustered in the PPI networks of IDEAL. From the question mark on the blue header region, a simple instruction of the new interface can be referred. More detailed help document is on the menu bar.
Figure 1.

The largest PPI network in IDEAL. The rectangles indicate proteins (IDEAL entries), and are colored green if they contain at least one ProS. Otherwise, they are colored blue. Users can know the protein name and the PDB entry of the complex by moving the pointer on the node and edge (the red box and the red line), respectively. The networks surrounded by ellipse a involve cell cycle–related proteins (32), those enclosed by ellipse b involve nuclear receptors and their corepressors/activators (33), those surrounded by c involve histone-related proteins (34) and those enclosed by d involve proteins binding with importin (35). Abbreviations for proteins are as follows: p53, cellular tumor antigen p53 (IID00015); CBP, CREB-binding protein (IID50008); p300, histone acetyltransferase p300 (IID00070); CDK2, cyclin-dependent kinase 2 (IID00034); p27, cyclin-dependent kinase inhibitor 1B (IID00049); Rb, retinoblastoma-associated protein (IID00017); ANDR, androgen receptor (IID00020); GLCR, glucocorticoid receptor (IID00014); NRCR1, nuclear receptor corepressor 1 (IID00189); NRCA4, nuclear receptor coactivator 4 (IID00074); H2, histone H2B type 1-K (IID00010); H3.3, histone H3.3 (IID00239); H4, histone H4 (IID00058); SETD8, N-lysine methyltransferase SETD8 (IID00101); BPTF, nucleosome-remodeling factor subunit BPTF (IID00071); and importin α, importin subunit alpha-2 (IID50009).
NODE and EDGE pages
In the PPI networks, each IDEAL entry is presented as a node, and the interaction of two entries is shown as an edge, with their information provided in a NODE page and an EDGE page, respectively (Figure 2). The NODE page is the preexisting IDEAL entry page itself (lower left panel), but it has new links to the EDGE pages (red circle a in Figure 2). The links to the EDGE pages provide the binding partner information explicitly. The EDGE page displays the structural complex of an IDEAL entry and its binding partner (upper left panel), and has two links to two NODE pages (b and c). One can move to the next NODE page by clicking the link to the binding partner (c). When the second NODE page has links to EDGE pages (d), one can move to the next NODE via an EDGE again (d and e). By repeating this step, the user can move around the PPI network.
Figure 2.
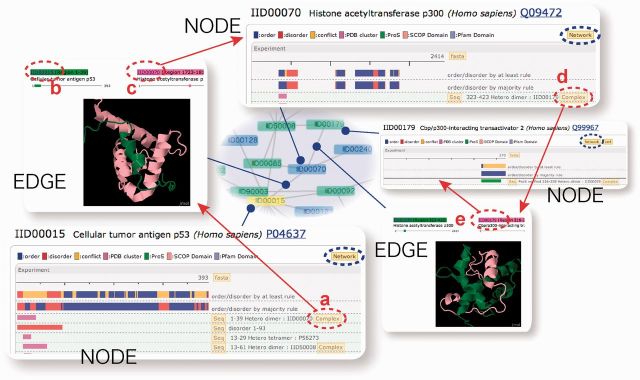
Connection between NODE and EDGE pages. Each entry page in the new version of IDEAL is referred to as a NODE page, and the interaction between two entries is recorded in an EDGE page, which connects two NODE pages. Red circles indicate the link buttons connecting the NODE and EDGE pages. Blue circles are the links to the PPI network.
The link to the PPI networks (Figure 1) was prepared for each of the NODE and EDGE pages (blue circles in Figure 2), and conversely, by clicking the node or the edge of the PPI network, the NODE or EDGE page appears. Therefore, the user can investigate the relationships between IDPs from the perspective of the PPI network, as well as in detail, from the NODE and EDGE pages.
Clusters of experimental evidence and deviation of NMR structures
As described previously (13), the majority-rule bar shows a summary of the experimental evidence based on the majority rule, in which ordered and disordered regions are colored blue and red, respectively. When the bar is clicked, clusters of experimental evidence appear. Each magenta bar represents a cluster of regions contained in equivalent complexes or monomeric proteins. Note that even though the distinct magenta bars indicate an identical region, they represent different clusters if the region is contained in different complexes (e.g. hetero-dimer and hetero-tetramer, see the definition in the ‘New Data’ section). For example, it is common to see a single ProS binding different proteins. In this case, distinct bars spanning the almost identical region appear, to indicate multiple clusters for the different binding partners. In this sense, the clustering is effective not only to avoid redundant PDB information, but also to show the promiscuity of IDPs (3,19).
Under the magenta bar, the experimental evidence in the cluster is provided. The order and disorder annotations for the regions are obtained from the structured (blue bar) and missing (red bar) residues of the X-ray structures as well as the NMR structures. When we found special descriptions for the disordered regions in the literature cited in the PDB, the regions were annotated. For example, a region is disordered if it is involved in the constructed sequence, but the NMR signal for this region cannot be detected or assigned. Disordered regions detected by other methods can also be annotated by literature searches (13). In addition, highly deviated regions found in NMR structures are newly introduced (see the ‘New Data’ section) (29). These regions are shown in red, and labeled with ‘high_rmsd’.
Figure 3 shows part of the IID90003 (early E1A 32 kDa protein) entry page, where two clusters (magenta bars in box a) are depicted. The second cluster (larger magenta bar) contains 2kjeB (PDB ID and chain identifier). Although the 3D coordinates were assigned for its entire region (53–91 residues), the corresponding literature (37) mentioned that residues 81–91 were disordered (the first red bar in box b). When the entry was examined, the terminal region (85–91) was highly deviated (the last red bar in b). This result indicates that the author-defined disorder (81–91) and the high_rmsd region (85–91) coincide well. The Matthews correlation coefficient (MCC) (30) of the author-defined order/disorder and the low/high_rmsd regions is 0.8.
Figure 3.
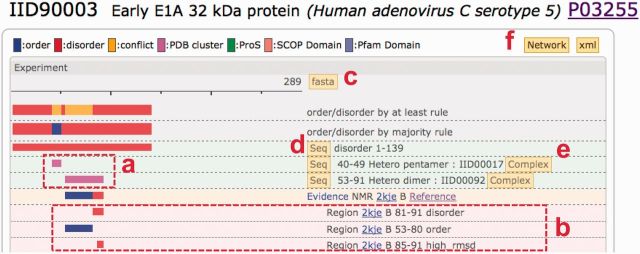
Part of the entry (NODE) page for IID90003. The two magenta bars (box a) indicate that this entry has two clusters of PDB entries. One includes the PDB entry 2kjeB (37). Although the atomic coordinates were provided for the entire region (residues 53–91), the author mentioned that residues 81–91 are disordered (the top red bar in box b). We inferred the others are structured (blue bar). Residues 85–91 are highly deviated (the lowest red bar with ‘high_rmsd’ annotation). The NODE pages also provide several new capabilities, such as the fasta button: sequence in the fasta format, summarizing the order/disorder information (c); the Seq button: sequence of the corresponding bar in the fasta format (d); a link to the EDGE page showing the structural complex with the binding partner (e); and a link to the PPI network (f).
Among the 340 IDEAL entries, 143 include evidence from NMR structures, and 95 have high_rmsd regions. Twenty-five entries comprise disordered regions manually annotated from the literature. For 25 NMR structures, the author-defined disorder and the high_rmsd are both annotated. The MCC, calculated using these 25 samples, is ∼0.8 on average.
Experiment and prediction sections
To differentiate the experimental data and predictions clearly, we divided the bar diagram into two sections, Experiment and Prediction. The prediction section shows domain predictions by reverse PSI-Blast (38) and HMMER (39). In addition, the new version of IDEAL displays ordered/disordered and new domain predictions by DICHOT (6). We anticipate that prediction results by DICHOT are helpful to complement the lack of experimental data, even though IDEAL emphasizes experimentally verified ordered/disordered information (13).
Other improvements
Acetylation and methylation sites, as well as phosphorylation sites, were obtained from UniProt (31), and are shown on the NODE pages. The fasta button (c in Figure 3), located at the top of the bar diagram, provides the ordered/disordered states of the amino acid sequence in the ‘at least rule’ or ‘majority rule’. The ProS regions are also indicated. The Seq button (d) displays the corresponding region in the sequence with color. XML files, showing the PPI network (binary relation), are available.
FUTURE WORK
This revision of IDEAL features the interactions between IDPs and their binding partners, and illustrates the PPI networks. However, the actual PPIs in the cellular systems are more complex, and thus temporally and spatially dynamic. For instance, histone tails have many sites modified by different proteins (34), but these proteins do not bind the tails simultaneously. Beta-catenine partners with different proteins when it is transported from the cytoplasm to the nucleus (36). These examples indicate that the dynamical nature of the PPI network must be integrated in a suitable manner. At first, we will focus on the PTMs that play important roles in the regulation and interactions of IDPs (34,36), using the PTM information from UniProt (31) already available in IDEAL. The relationships between PTMs and alterations of the PPI network will be depicted in more explicit manners.
FUNDING
Grant-in-Aid for Scientific Research on Innovative Areas, ‘Target recognition and expression mechanism of intrinsically disordered proteins’, from Ministry of Education, Culture, Sports, Science and Technology in Japan. Funding for open access charge: Grant-in-Aid for Scientific Research on Innovative Areas, ‘Target recognition and expression mechanism of intrinsically disordered proteins’, from Ministry of Education, Culture, Sports, Science and Technology in Japan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 3.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 4.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 5.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim. Biophys. Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuchi S, Hosoda K, Homma K, Gojobori T, Nishikawa K. Binary classification of protein molecules into intrinsically disordered and ordered segments. BMC Struct. Biol. 2011;11:29. doi: 10.1186/1472-6807-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minezaki Y, Homma K, Kinjo AR, Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006;359:1137–1149. doi: 10.1016/j.jmb.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 10.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Domenico T, Walsh I, Martin AJ, Tosatto SC. MobiDB: a comprehensive database of intrinsic protein disorder annotations. Bioinformatics. 2012;28:2080–2081. doi: 10.1093/bioinformatics/bts327. [DOI] [PubMed] [Google Scholar]
- 12.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, et al. D2P2: database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuchi S, Sakamoto S, Nobe Y, Murakami SD, Amemiya T, Hosoda K, Koike R, Hiroaki H, Ota M. IDEAL: intrinsically disordered proteins with extensive annotations and literature. Nucleic Acids Res. 2012;40:D507–D511. doi: 10.1093/nar/gkr884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, Toedt G, Uyar B, Seiler M, Budd A, et al. ELM-the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–D251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Lobanov MY, Shoemaker BA, Garbuzynskiy SO, Fong JH, Panchenko AR, Galzitskaya OV. ComSin: database of protein structures in bound (complex) and unbound (single) states in relation to their intrinsic disorder. Nucleic Acids Res. 2010;38:D283–D287. doi: 10.1093/nar/gkp963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J. Mol. Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 18.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 19.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and CDK2-bound state: conformational disorder mediates binding diversity. Proc. Natl Acad. Sci. USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Rose PW, Bi C, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada M, et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 25.Jones DT, Ward JJ. Prediction of disordered regions in proteins from position specific score matrices. Proteins. 2003;53(Suppl. 6):573–578. doi: 10.1002/prot.10528. [DOI] [PubMed] [Google Scholar]
- 26.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Martin JA, Witzell J, Blumenstein K, Rozpedowska E, Helander M, Sieber TN, Gil L. Resistance to Dutch ELM disease reduces presence of xylem endophytic fungi in ELMs (Ulmus spp.) PLoS One. 2013;8:e56987. doi: 10.1371/journal.pone.0056987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, et al. BioMagResBank. Nucleic Acids Res. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ota M, Koike R, Amemiya T, Tenno T, Romero PR, Hiroaki H, Dunker AK, Fukuchi S. An assignment of intrinsically disordered regions of proteins based on NMR structures. J. Struct. Biol. 2013;181:29–36. doi: 10.1016/j.jsb.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 31.UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 33.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 34.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 35.Fontes MR, Teh T, Jans D, Brinkworth RI, Kobe B. Structural basis for the specificity of bipartite nuclear localization sequence binding by importin-alpha. J. Biol. Chem. 2003;278:27981–27987. doi: 10.1074/jbc.M303275200. [DOI] [PubMed] [Google Scholar]
- 36.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc. Natl Acad. Sci. USA. 2009;106:13260–13265. doi: 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2013;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]