


Abstract
In the hierarchy of data, information and knowledge, computational methods play a major role in the initial processing of data to extract information, but they alone become less effective to compile knowledge from information. The Kyoto Encyclopedia of Genes and Genomes (KEGG) resource (http://www.kegg.jp/ or http://www.genome.jp/kegg/) has been developed as a reference knowledge base to assist this latter process. In particular, the KEGG pathway maps are widely used for biological interpretation of genome sequences and other high-throughput data. The link from genomes to pathways is made through the KEGG Orthology system, a collection of manually defined ortholog groups identified by K numbers. To better automate this interpretation process the KEGG modules defined by Boolean expressions of K numbers have been expanded and improved. Once genes in a genome are annotated with K numbers, the KEGG modules can be computationally evaluated revealing metabolic capacities and other phenotypic features. The reaction modules, which represent chemical units of reactions, have been used to analyze design principles of metabolic networks and also to improve the definition of K numbers and associated annotations. For translational bioinformatics, the KEGG MEDICUS resource has been developed by integrating drug labels (package inserts) used in society.
INTRODUCTION
Bioinformatics has been a data-driven discipline. In the era of high-throughput biology it enables extraction of meaningful information from large amounts of data generated by systematic experiments. Such information may be of practical use, but it does not necessarily represent biological knowledge. A more focused approach in traditional biology, which is often hypothesis-driven or model-driven, is necessary to analyze low-throughput but high-quality data and to acquire knowledge. For example, a disease-associated gene identified by a genome-wide association study may be just information, but a disease mechanism involving a perturbed signaling pathway uncovered by a more detailed analysis may be called knowledge. In the hierarchy of data, information and knowledge, computations with elaborate algorithms play a major role in the initial processing of data to information, but computations with good reference databases become more important in the following processing to compile knowledge.
In 1995, we started the Kyoto Encyclopedia of Genes and Genomes (KEGG) database project foreseeing the need for a reference resource that can be used for biological interpretation of genome sequence data. In particular, we started developing a reference pathway database by capturing and organizing experimental knowledge from published literature, first focusing on metabolism but soon followed by other cellular processes. The KEGG resource has been expanded significantly over the years to meet the needs for integrating and interpreting various types of high-throughput data, as well as for supporting translational bioinformatics (1). We have recently returned to metabolism with a new attempt, which is a synthesis of biological knowledge toward understanding basic principles of metabolic networks (2). The concept of data, information, knowledge and principle is now used to improve the architecture and content of the KEGG database. We expect that this will eventually lead to logic-driven bioinformatics and first principle-based computations.
LINKING GENOMES TO PHENOTYPES
Overview of KEGG
KEGG is an integrated database resource consisting of 15 main databases maintained in the internal Oracle database. As shown in Table 1 they are categorized into systems information (PATHWAY, BRITE and MODULE), genomic information (ORTHOLOGY, GENOME and GENES), chemical information (COMPOUND, GLYCAN, REACTION, RPAIR, RCLASS and ENZYME) and health information (DISEASE, DRUG and ENVIRON). The chemical and health information categories are collectively called KEGG LIGAND and KEGG MEDICUS, respectively. KEGG MEDICUS contains two outside databases for drug labels (package inserts) of all drugs marketed in Japan and the USA, which are maintained as part of the KEGG Oracle database. There are additional databases not listed in Table 1, which are computationally generated and maintained outside of the KEGG Oracle database. They include the sequence similarity database SSDB for genome annotation and auxiliary gene catalog databases DGENES, MGENES and VGENES for draft genomes, metagenomes and viral genomes, respectively.
Table 1.
The KEGG resource including drug labels
| Category | Category name | Database name | Content |
|---|---|---|---|
| Systems Information | KEGG PATHWAY | KEGG pathway maps | |
| KEGG BRITE | BRITE functional hierarchies | ||
| KEGG MODULE | KEGG modules | ||
| Genomic Information | KEGG ORTHOLOGY | KEGG Orthology (KO) groups | |
| KEGG GENOME | KEGG organisms with complete genomes | ||
| KEGG GENES | Gene catalogs in complete genomes | ||
| Chemical Information | KEGG LIGAND | KEGG COMPOUND | Metabolites and other small molecules |
| KEGG GLYCAN | Glycans | ||
| KEGG REACTION | Biochemical reactions | ||
| KEGG RPAIR | Reactant pairs | ||
| KEGG RCLASS | Reaction class | ||
| KEGG ENZYME | Enzyme nomenclature | ||
| Health Information | KEGG MEDICUS | KEGG DISEASE | Human diseases |
| KEGG DRUG | Drugs | ||
| KEGG ENVIRON | Crude drugs and health-related substances | ||
| JAPICa | Drug labels in Japan | ||
| DailyMedb | Drug labels in the USA (linked through NDCc) |
Reference knowledge base
The original concept of KEGG was to create a reference knowledge base of metabolism and other cellular processes, so that it can be used for inferring higher-level functions from genome sequence data. This concept is unchanged after significant expansion of the knowledge base, now containing organismal systems, human diseases and drugs and the variety of data, now including metagenomes, transcriptomes, metabolomes and other high-throughput data. The reference knowledge base consists of KEGG PATHWAY, BRITE and MODULE databases (systems information category in Table 1). During the past 2 years we implemented improvements of the KEGG MODULE and PATHWAY databases to automate interpretation of phenotypic features, especially metabolic capacities, from genome and metagenome sequences.
Genes to K numbers
The KEGG pathway maps, BRITE functional hierarchies and KEGG modules are represented in a generic way to be applicable to all organisms using the KEGG Orthology (KO) system. For example, when a pathway map is drawn based on experimental evidence in specific organisms, additional work is performed for generalizing genes and proteins in those specific organisms to other organisms by converting to KO entries (ortholog groups), and if necessary by creating new KO entries (Figure 1). Each KO entry (identified by K number) is defined as a sequence similarity group, although the degree of similarity is context (pathway) dependent. This allows manually created reference pathways to be computationally expanded to organism-specific pathways, once genes in the genome are annotated with K numbers based on sequence similarity.
Figure 1.

A schematic diagram of genome annotation in KEGG. It consists of two parts: defining KO entries represented by K numbers (right) and assigning K numbers to genes in complete genomes (left). The KO definition is manually done, but the K number assignment is highly computerized (see text).
The genome annotation in KEGG is highly computerized as illustrated in Figure 1. SSDB is a huge graph of genes, whose edges are weighted by sequence similarity scores and directed by best-hit relations. It is continuously updated from the KEGG GENES database by pairwise genome comparisons using the SSEARCH program. The sequence similarity group of each KO entry corresponds to a clique-like subgraph in the SSDB graph, and the genome annotation involves extending and modifying this subgraph. This is accomplished by the KOALA (KEGG Orthology And Links Annotation) program, which evaluates sequence similarity scores, best-hit relations, protein domains and taxonomy groups for each gene in each genome using the GFIT (Gene Function Identification Tool) table created from SSDB. KOALA’s computational assignments (currently performed three times a week) for a clean (clearly defined) set of K numbers (currently 73%) are used to automatically annotate genes in a newly determined genome and also in the existing genomes that meet certain criteria. Discrepancies between KOALA’s assignments and current annotations are examined by annotators with the manual version of KOALA and GFIT tools (read-only copies are made available from KO and GENES web pages), which are linked to additional tools including gene cluster, ortholog table and phylogenetic analysis tools. The manual tools are also extensively used for grouping and regrouping of KO entries to increase the clean set of K numbers (Figure 1).
K numbers to M numbers
The KEGG pathway map is drawn to present an overall picture of the molecular interaction and reaction network, often combining experimental evidence from multiple organisms. In contrast, the KEGG module (http://www.kegg.jp/kegg/module.html) is a tighter functional unit of molecules that generally corresponds to a conserved subpathway in the KEGG pathway map. Each KEGG module (identified by M number) is manually defined as a combination of K numbers by examining gene conservation patterns among organism groups and positional correlations of genes (operon-like structures). The definition is described by a simple Boolean expression of K numbers allowing computerized evaluation of whether the gene set is complete, i.e. the functional unit is present, based on the annotation (K number assignment) of genes in the genome. For example, M00010, which is named ‘Citrate cycle, first carbon oxidation, oxaloacetate => 2-oxoglutarate’ and one of the most conserved modules, is defined as:
where a comma sign represents OR and a space or a plus sign (for a molecular complex) represents AND when evaluating this expression. The computerized evaluation, which is implemented in both the internal annotation procedure and the KEGG module web page, also allows detecting incomplete but almost complete modules. In many cases these modules can become complete after a few more genes are correctly annotated, but in some cases they remain incomplete due to pseudogenes, fragmented genes and apparently missing genes in the genome, which may indicate alternative genes and pathways that are still unknown.
M numbers to phenotypes
The KEGG module collection is being expanded to allow hierarchical definition of M numbers, where M numbers are part of the Boolean expression to define another M number. This will be used mostly in what we call signature modules to describe phenotypes. For example, the modules for methanogenesis M00567, M00357, M00356 and M00563 are separately defined by different K number sets representing different reaction pathways. Then, the phenotypic feature of methanogen M00617 is defined as a combination:
The genome based annotation of phenotypes, mostly metabolic capacities, is being added to the Keyword field of the Genome entry page. Experimental evidence taken from literature is added to its Comment field as part of the metadata annotation of complete genomes. Now that the number of KEGG organisms (complete genomes) is reaching 3000, new tools will be developed to examine relationships between organism groups and metabolic capacities by using the organism level annotation of signature modules.
Reaction modules
The KEGG modules are defined by K numbers meaning that they represent features of genes and genomes. In contrast, the reaction modules that have been introduced recently for metabolic pathways are defined by RC (Reaction Class) numbers describing conserved sequences of chemical structure transformation patterns of small molecules. Figure 2 shows how reaction data are processed in KEGG. The reaction class is like an ortholog group of reactions representing functionally important local structural changes and accommodating global structural differences. The reaction modules are extracted from purely chemical properties without using any information about enzymes and enzyme genes (3). Nevertheless, reaction modules tend to correspond to KEGG modules. For example, the KEGG module M00010 from oxaloacetate (four-carbon or C4) to 2-oxoglutarate (C5) matches the reaction module RM001 for ‘2-Oxocarboxylic acid chain extension by tricarboxylic acid pathway’. Furthermore, as illustrated in Figure 3, RM001 is found in other pathways in combination with other reaction modules for synthesizing basic amino acids, branched-chain amino acids, coenzyme B and glucosinolates. There are four different KEGG modules currently defined for the same reaction module RM001, and the constituent genes for the corresponding reaction steps are similar or paralogous in these cases. The reaction modules have thus been used to improve the K number grouping and associated annotations.
Figure 2.

Reaction data processing in KEGG. The reaction formula is decomposed into a set of reactant pairs, one-to-one relationships of substrate–product pairs. Each reactant pair is characterized by the local structure transformation pattern, called RDM pattern of KEGG atom type changes. Among the reactant pairs that appear in the KEGG pathway maps, distinct RDM patterns are used to define reaction class entries identified by RC numbers. The reaction module is a conserved sequence of RC numbers observed in different pathways. This example shows the RDM pattern (R for reaction center atoms in red, D for difference region atoms in green and M for matched region atoms in blue) of RC00067, which appears in the reaction module RM001 variant 01 (see details in http://www.kegg.jp/kegg/reaction/rmodule.html).
Figure 3.
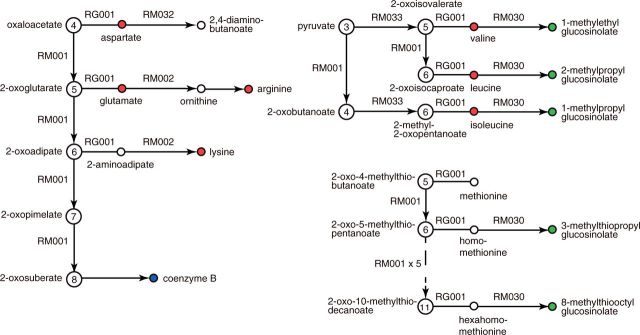
An example of the modular architecture of the metabolic network. The reaction module RM001 for chain extension of 2-oxocarboxylic acids (large circles with the number of carbons) is used in combination with other reaction modules to generate amino acids (red circles), glucosinolates (green circles) and coenzyme B (blue circle) (see details in http://www.kegg.jp/pathway/map01210).
The reaction modules contain interesting features, possibly design principles of a series of organic reactions (2,3), including how to achieve an activated transition state (e.g. phosphorylation), how to introduce a protective group (e.g. N-acetylation), how to increase specificity (e.g. using a carrier protein) and how to increase efficiency (e.g. switching a carbon source from acetyl-CoA to malonyl-CoA). An example is found in the two pathways for fatty acid biosynthesis: the minor pathway in mitochondria (RM020) utilizing acetyl-CoA as a carbon source, which is a reversal of beta-oxidation (RM018), and the major pathway (RM021) utilizing acyl carrier protein and malonyl-CoA as a carbon source. RM021 appears to be more advanced than RM020 in the possible evolution of reaction modules.
Metabolism overview maps
The pathway maps similar to Figure 3 are now made available under the category of metabolism overview maps (map numbers 01200s). In contrast to the previously developed global maps (map numbers 01100s), which omit intermediate reaction steps, the overview maps contain all the reaction steps as in the regular KEGG metabolic pathway maps. In addition, they are manually annotated with KEGG modules and reaction modules. These maps represent our efforts to present design principles of the metabolic network rather than traditional views of individual pathways. The modular architecture of the metabolic network is apparent in 2-oxocarboxylic acid metabolism (map01210), fatty acid metabolism (map01212) and degradation of aromatic compounds (map01220), but the central carbon metabolism (map01200) seems to contain a different design principle. It is an extensive use of the same paths with minor modifications (2), such as reductive pentose phosphate pathway that contains two key reaction steps catalyzed by RuBisCO and PRK. The overview maps, together with KEGG modules and reaction modules, will be expanded toward understanding basic principles of metabolic networks.
Note that for these new overview maps, the EC number reference pathways (map numbers prefixed with ec) are no longer supported. The EC numbers are given to biochemically well-characterized enzymes and enzymatic reactions in Enzyme Nomenclature (4), but there are many reactions that do not qualify for EC numbers, such as those identified by genetic experiments or inferred from metabolic pathways. Less than one half of the reactions in the KEGG REACTION database are associated with EC numbers, and this ratio becomes smaller for those reactions that appear in the KEGG pathway maps (3). There are also many EC numbers whose sequence information is unknown (5). Since the EC numbers are given as attributes of KO entries (K numbers) and reaction entries (R numbers), their use as identifiers are highly discouraged in KEGG Mapper and other applications.
LINKING GENOMES TO SOCIETY
KEGG MEDICUS
KEGG MEDICUS is an integrated information resource of diseases, drugs and health-related substances, aiming to bring the genomic revolution to society. Specifically, the drug labels (package inserts) are now part of KEGG MEDICUS (Table 1). The Japanese drug labels are fully integrated in the KEGG Oracle database using the XML data provided monthly from the Japan Pharmaceutical Information Center (JAPIC). For the drug labels in the USA only the information taken from the FDA’s National Drug Code (NDC) database is in Oracle, and the actual contents are referred to the DailyMed website. The KEGG DRUG database was originally developed as a unified resource for drugs in Japan, the USA and Europe, where active ingredients and their mixtures are distinguished by chemical structures and/or chemical components and represented by KEGG DRUG entries (identified by D numbers). The D numbers have been linked, for example, to targets in the KEGG pathway maps and drug ontologies in the BRITE hierarchies. Now that the drug labels data in Japan and the USA are linked to D numbers, both scientific data used in the scientific community and practical data used in society are integrated through the KEGG DRUG database. Figure 4 illustrates a drug hierarchy showing that slightly different chemical structures (D numbers) of warfarin are linked to WHO’s Anatomical Therapeutic Chemical (ATC) classification, and also to drug labels and drug products in the USA and Japan (not shown).
Figure 4.

The KEGG DRUG D numbers play a role of integrating different types of data and information, now including drug labels and drug products. Here, D numbers are used to integrate the ATC classification and drug products, effectively classifying all drug products in the USA (see http://www.kegg.jp/brite/br08303_ndc).
Drug interaction database
The full text information of the entire set of Japanese drug labels is a great source for developing scientific databases. In particular, a drug interaction database has been developed (6). Adverse drug interactions associated with contraindications and precautions are listed in a tabular form in the Japanese drug labels. Drug names and drug group names are extracted and converted to D numbers and D number groups using internally developed dictionaries for standardizing variations and synonyms. The resulting D number pairs are annotated with possible causes, such as CYP enzyme or drug target involvement, using the information stored in the KEGG DRUG database. Drug interactions can be searched for a given set of D numbers at the KEGG DRUG web page and also through the drug–drug interaction (DDI) search button in each KEGG DRUG entry. In the Japanese version of KEGG MEDICUS, drug interactions can be searched at the product level, giving more detailed information extracted from the drug labels.
Chemical ingredients of drugs include not only active ingredients but also different types of pharmaceutical additives, which may also cause adverse effects. All additives in each drug product are extracted from the Japanese drug labels and converted to KEGG DRUG D numbers, KEGG COMPOUND C numbers and KEGG ENVIRON E numbers. The search interface is available for Japanese drug products in the Japanese version of KEGG MEDICUS.
Translational bioinformatics
Translational bioinformatics usually means bioinformatics resources and technologies that help bring research results into practical applications. Here it also means bioinformatics resources directly targeted to society that help to understand the scientific basis of diseases and drugs of personal interest. The latter aspect of translational bioinformatics is successfully implemented in the Japanese version of KEGG MEDICUS, which has a big impact on the web access statistics. The number of visitors (unique IP addresses) per month increased from 200 000 to 500 000 in the past 2 years, and many of them now come directly from web search engines. The drug information is universal at the level of D numbers representing active ingredients, but actual drug products vary in different countries. We will be unable to directly provide a service of the same quality as in Japan, but the KEGG MEDICUS resource can be used to develop a similar service in many countries. The Japanese and US drug labels currently maintained in KEGG are like KEGG organisms for genome analysis. As shown in Table 2 new genome sequences and other user-defined data can be integrated with KEGG, once the IDs are properly converted to KEGG IDs. Similarly, once drug labels in any country are linked to the KEGG DRUG D number identifiers, the whole array of KEGG resources including the drug interaction database, drug targets, drug metabolism and drug classifications become available.
Table 2.
Examples of queries against KEGG
| Query data | Procedure |
|---|---|
| Genome or metagenome |
|
| Transcriptome |
|
| Metabolome |
|
| Drug labels |
|
Accessing KEGG
KEGG is made available at both the KEGG website (http://www.kegg.jp/) and the GenomeNet website (http://www.genome.jp/kegg/). The internal KEGG Oracle database is copied daily to a public version as a read-only PostgreSQL database, which is available only at the KEGG website. LinkDB and various analysis tools including KAAS and SIMCOMP are available only at the GenomeNet website. However, these differences may not be noticeable by the users because mutual links are made as if they are a single site.
FUNDING
Japan Science and Technology Agency (in part). Funding for open access charge: Japan Science and Technology Agency.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Tomoko Komeno and Yuriko Matsuura for developing new overview maps of KEGG metabolic pathways. The computational resource was provided by the Bioinformatics Center, Institute for Chemical Research, Kyoto University.
REFERENCES
- 1.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanehisa M. Chemical and genomic evolution of enzyme-catalyzed reaction networks. FEBS Lett. 2013;587:2731–2737. doi: 10.1016/j.febslet.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 3.Muto A, Kotera M, Tokimatsu T, Nakagawa Z, Goto S, Kanehisa M. Modular architecture of metabolic pathways revealed by conserved sequences of reactions. J. Chem. Inf. Model. 2013;53:613–622. doi: 10.1021/ci3005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald AG, Boyce S, Tipton KF. ExplorEnz: the primary source of the IUBMB enzyme list. Nucleic Acids Res. 2009;37:D593–D597. doi: 10.1093/nar/gkn582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lespinet O, Labedan B. ORENZA: a web resource for studying ORphan ENZyme activities. BMC Bioinformatics. 2006;7:436. doi: 10.1186/1471-2105-7-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takarabe M, Shigemizu D, Kotera M, Goto S, Kanehisa M. Network-based analysis and characterization of adverse drug-drug interactions. J. Chem. Inf. Model. 2011;51:2977–2985. doi: 10.1021/ci200367w. [DOI] [PubMed] [Google Scholar]