

Abstract
Radiosensitivity can be influenced both by factors intrinsic and extrinsic to the cancer cell. One of the factors in the tumor microenvironment (TME) extrinsic to the cancer cell that can affect radiosensitivity is oxygenation. Severely hypoxic cells require a 2–3 fold higher dose of radiation to achieve the same level of cell killing as do well-oxygenated cells. Other elements in the microenvironment that may influence tumor radiosensitivity are the response of stromal cells to radiation and the expression of factors such as vascular endothelial growth factor (VEGF) and hypoxia inducible factor-1 (HIF-1). There are currently several classes of agents that may increase tumor radiosensitivity by modulating the TME. Pre-clinical evidence indicates that inhibition of VEGF may increase local control after radiation. Several mechanisms have been postulated to explain this including radiosensitization of tumor endothelial cells, prevention of the establishment of new vasculature post-radiation, and increased oxygenation secondary to vascular normalization. Agents targeting HIF-1 also increase local control after radiation in pre-clinical models. This may occur via indirect inhibition of VEGF, which is a downstream target of HIF-1, or by VEGF-independent means. When combined with radiation, the EGFR inhibitor cetuximab improves local control and survival in patients with head and neck cancer. Pre-clinical data indicate that EGFR inhibitors can increase the intrinsic radiosensitivity of cancer cells. They can also improve tumor blood flow and oxygenation, which may increase extrinsic radiosensitivity. One of the pathways downstream of EGFR that may contribute to this effect is the PI3K/Akt pathway. Agents that directly inhibit this pathway improve blood flow and increase tumor oxygenation in pre-clinical models. The challenge remains to obtain clinical data from patients showing that modulation of the TME is an important mechanism by which biological agents can radiosensitize tumors and then to utilize this information to optimize therapy.
Keywords: radiation, radiosensitization, vascular normalization, EGFR, VEGF, HIF, PI3 kinase, Akt, tumor microenvironment
Introduction
Radiation therapy has been used for over a hundred years to treat patients with cancer; however, in spite of this long track record, it is difficult to achieve local control in many patients. To improve the efficacy of radiation therapy, it is important to understand mechanisms of radioresistance. Radiosensitivity can be influenced by both factors intrinsic and extrinsic to the cancer cell. Some investigators feel that the sensitivity of the cancer cell itself is the predominant factor in determining the radiation response of a tumor.1 As will be discussed later in this review, the activation of certain oncogenes may be associated with increased resistance to radiation. Factors extrinsic to a tumor cell can also contribute significantly to a tumor’s response to radiation. There has been a great deal of recent interest in the role that host cells that reside in the stroma may play in the radiation response. Studies have shown that when identical tumors are implanted into mice with variable genetic backgrounds, there is an increased radiation response in tumors in mice with sensitive genetic backgrounds.2,3 These studies suggest that the host component plays an important role in tumor response and is thus an important target in cancer therapy. In particular, some investigators feel that the vascular endothelium may play a major role in the radiation response.3–5 Kolesnick and colleagues performed experiments suggesting that radiation-induced apoptosis in the vascular endothelium of the microvasculature supplying the gastrointestinal (GI) tract is responsible for radiation-related GI toxicity rather than direct damage to the stem cells lining the GI tract.4 However, Ogawa et al. used isogenic tumor cell lines that were either competent or defective in DNA double-strand break repair and reached the conclusion that tumor cell radiosensitivity was the major determinant of tumor response in nude mice.6 However, in severe combined immunodeficiency (SCID) mice, both tumor cell sensitivity and radiation-induced stromal damage played a role.6 Therefore, the question of the relative importance of stromal versus tumor cells in the radiation response is unresolved.
Another factor extrinsic to the tumor cell that can influence radiosensitivity is hypoxia, which is commonly seen in human cancers.7 Cancer cells also display increased glycolysis and CO2 production, resulting in acidification of the tumor microenvironment.8 This acidic and hypoxic microenvironment can make cells resistant to both radiation and chemotherapy.9 Hypoxic cells require higher doses of radiation compared to oxic cells to achieve the same level of killing due to the requirement that oxygen be present during irradiation to elicit maximal DNA damage through the creation of free radicals.10 The oxygen enhancement ratio (OER), the ratio of radiation doses that confer the same level of cell death without and with oxygen is 2.5–3 at a high fractional dose (10–20 Gy) and approximately 2 at a fractional dose of ≤2 Gy.10,11 Clinical data indicate that hypoxia is associated with a worse outcome following radiation in many tumors including head and neck cancers.12–14
There are currently several classes of agents that might alter the tumor microenvironment TME in a way that could increase tumor radiosensitivity. In this review, we discuss the preclinical and clinical data that exist for using these agents, focusing on their ability to alter the TME. The biological agents that are currently being tested with radiation may alter both the cancer cell’s intrinsic radiosensitivity, and, by altering the TME, their extrinsic radiosensitivity. In this review we will briefly discuss the former but focus on the latter.
Targeting the Vasculature to Improve Radiation Response
The idea of targeting the tumor vasculature as cancer therapy goes back to Dr. Judah Folkman, who in 1971 first proposed the idea that this might be an effective cancer treatment.15 The rationale for this idea was the finding that tumors required the development of a new network of blood vessels to sustain growth beyond 1 mm3.16,17 The idea of targeting the vasculature as an adjunct to radiation therapy has also been proposed.
There have been two main strategies used to target tumor vessels, vascular disruptive agents (VDAs) and anti-angiogenic agents. VDAs target pre-existing tumor vessels17 whereas the latter target the angiogenic process itself by inhibiting the action of factors that stimulate new blood vessel development.18 ZD6126, an example of a VDA, disrupts the tubulin cytoskeleton of endothelial cells, leading to their death. In preclinical studies, this agent has been shown to enhance the anti-tumor effects of radiation in large tumors (>1 g) but not small ones (<0.3 g).19 Another VDA, OXi4503, was shown to reduce the radiation dose required to control 50% of the tumors (TCD50) from 52 Gy without the drug to 41 Gy with the drug.20
The other strategy to target tumor vasculature is to inhibit angiogenesis. As tumors grow, they outstrip their blood supply, leading to regions that are underperfused and poorly oxygenated. This stimulates the expression of the α subunit of hypoxia-inducible factor-1 (HIF-1), a key transcription factor that plays an important role in cellular and systemic responses to hypoxia. HIF-1α is upregulated across a broad range of cancers and regulates key features of tumor biology such as angiogenesis and glucose metabolism.21,22 One of the genes upregulated by HIF-1 is vascular endothelial growth factor (VEGF), a potent mediator of angiogenesis that enhances endothelial cell survival, induces vasodilatation and regulates pericyte coverage.23 Both HIF-1α and VEGF have been tested as potential targets for improving the radiation response.
VEGF expression leads to hyperproliferation of blood vessels, which should improve oxygenation within tumors. However, VEGF also has another important property, that of increasing vascular permeability.24 In fact, VEGF was originally discovered as a vascular permeability factor and termed VPF.25 Perhaps because the supraphysiologic secretion of VEGF in tumors causes excessive leakiness or perhaps because there is not a coordinate expression of other key angiogenic factors, the vessels within a tumor appear dilated, saccular, tortuous and are poorly functional with sluggish blood flow. Therefore, in spite of high levels of VEGF, which should act as a compensatory mechanism, tumors continue to have regions of hypoxia.
Pre-clinical work has shown that VEGF can be induced in response to radiation and that inhibition of VEGF can increase tumor control after radiation. Gorski et al. showed that VEGF was induced after irradiation both in vitro and in vivo in several different cell lines.26 Treatment of mice bearing xenografts with a neutralizing antibody to VEGF prior to irradiation led to a synergistic effect on the inhibition of tumor regrowth. The authors postulated that this was mediated via an effect on the endothelial cell rather than the tumor cells because in vitro anti-VEGF treatment potentiated radiation-induced lethality of human umbilical vein endothelial cells (HUVECs) but not tumor cells. Lee et al. also used an anti-VEGF antibody in a mouse xenograft model to show that VEGF inhibition in combination with radiation led to a delay in tumor regrowth, which was additive for LS174T colon carcinoma xenografts and greater than additive for U87 glioblastoma xenografts.27
Another strategy to inhibit VEGF signaling is to block its function downstream at the level of the endothelial cell. Hess et al. used PTK787/ZK222584, an inhibitor of the VEGF receptor, and found that when this drug was given concurrently with radiation there was a greater delay in the regrowth of SW480 colon adenocarcinoma xenografts compared with either treatment by itself.28 The same agent was also tested with radiation by Zips et al. in head and neck squamous cell carcinoma xenograft models.29 Using different scheduling regimens, they found that if PTK787 was given before or during the course of fractionated radiation therapy, there was no beneficial effect; however, when given following the completion of the course of radiotherapy, there was a marked improvement in growth delay. Similar to these results, Williams et al. found that although the VEGFR inhibitor ZD6474 delayed tumor regrowth when given concurrently with radiation, a much more marked inhibition was noted if the drug was given after the radiotherapy had been completed (sequential treatment).30
More recently, the combination of radiation and cediranib (RECENTIN; AZD2171), a highly potent orally available inhibitor of VEGF receptor tyrosine kinase activity, has been investigated. In mouse xenograft models of human lung cancer and colon cancer cells, the combination resulted in greater tumor growth delay than either treatment alone.31–33 One of these studies examined two different schedules of the drug, either once daily 2 hours prior to each fraction of radiation and daily thereafter or started immediately after the course of radiotherapy was completed.32 Both regimens were effective in delaying tumor regrowth.
In theory, targeting the vasculature might lead to increased hypoxia, which would hinder the ability of radiation to kill cells; however, in reality the effect is much harder to predict. For VDAs that obliterate the vasculature, this is likely the case, so if one were to use radiation after administration of these agents, this might be counterproductive. The study using the VEGFR inhibitor ZD6474 that was discussed above found that this treatment was associated with decreased tumor perfusion.30 The same group found that cediranib by itself resulted in a decrease in total vessel density and an increase in hypoxic fraction.31 However, a review of the literature on the effects of anti-angiogenic agents on tumor oxygenation shows this to be far more complicated, with some drugs increasing hypoxia and others decreasing it.34
What is the explanation for such varied effects on tumor oxygenation by anti-angiogenesic agents? The concept of “vascular normalization”, proposed by Rakesh Jain35 may provide an explanation. According to this model, high levels of VEGF within tumors actually impede tumor oxygenation because the vessels function poorly. Paradoxically, inhibition of VEGF expression in these tumors can cause the vessels to remodel to become less permeable and less aberrant, resulting in decreased interstitial fluid pressure (IPF), improved blood flow and increased tumor oxygenation. Consistent with this model, Jain’s group showed that blocking VEGF signaling in mice with xenografts using DC101, a VEGF-receptor-2 antibody, decreased IFP by producing vessels that were more “normal” in appearance and in function with greater coverage by pericytes.36 Furthermore, DC101 induced a hydrostatic pressure gradient across the vascular wall, which led to deeper penetration of the drugs into tumors. In a different study, the same group showed that this “normalization window” was accompanied by an improvement in oxygenation which was transient.37 Lee et al. found that anti-VEGF antibody resulted in a significant decrease in vascular density and IPF in U87 xenografts accompanied by an improvement in tumor oxygenation. In contrast, in LS174 xenografts, the drug did not change tumor oxygenation although it did decrease IFP.27
The evidence discussed above supporting the concept of vascular normalization is pre-clinical. There are some data indirectly supporting vascular normalization in human patients. The most commonly used anti-VEGF agent in the clinic is bevacizumab (Avastin), an antibody with a half-life of 2–3 weeks that binds to VEGF to block VEGFR activation. Willett and colleagues reported on a clinical trial in which six patients with locally advanced adenocarcinoma of the rectum received bevacizumab alone, followed after 2 weeks by concurrent bevacizumab, 5FU and external beam radiation, then surgery.38 Twelve days after the initial bevacizumab injection, IFP was reduced in four of four patients who had endoscopic measurements. The decrease in IFP was postulated to be due to vascular “normalization”, and this idea was supported by the fact that the fraction of vessels positive for α-smooth muscle actin increased in four of five patients by day 12.
In a study of patients with recurrent glioblastomas treated with the VEGFR inhibitor cetiranib, Batchelor et al. used MRI techniques to show that the drug induced a decrease in relative vessel size as early as one day after the start of treatment which persisted to day 28.39 By day 56 vessel size had started to increase, suggesting a closure of the structural vascular normalization window. Dynamic contrast-enhanced MRI (DCE-MRI) showed a decrease in Ktrans at days 1 and 28 that persisted to day 112. Ktrans measures the rate of contrast agent between plasma and the extravascular extracellular space and is dependent upon both vascular permeability and vessel surface area. Therefore, this result is consistent with the idea that vascular permeability is decreased with cetiranib, although effects on vessel surface area confound these results.
In summary there are a great deal of pre-clinical data suggesting that anti-angiogenic agents in combination with radiation may improve outcome. Several mechanisms have been proposed as to how this might happen. Vascular normalization is one means by which the efficacy of radiation therapy might be increased after anti-angiogenic therapy, via increased oxygenation. It remains to be seen whether this happens in patients and what the kinetics of this effect might be. If this occurs, then the optimal time to start anti-VEGF therapy would be prior to the start of radiation. Another potential mechanism by which anti-VEGF therapy might work is via radiosensitization of the endothelial cells. Alternatively, VEGF may be induced by stromal cells following radiation. This may allow for establishment of new vasculature, which anti-angiogenic therapy might prevent. Pre-clinical data indicating that delivering anti-angiogenic therapy after the completion of radiation therapy results in better control support this idea.29,30 Whether these agents will work with radiation therapy in the clinic, and if so, which of these explanations is correct remains unknown. Currently, there are numerous trials listed with the National Cancer Institute (NCI) that are investigating the use of bevacizumab or other agents with anti-angiogenic properties in combination with radiation with or without standard chemotherapeutic agents. Therefore, the answers to these questions will be forthcoming.
Anti-HIF Therapy
Another potential target that could influence radiosensitivity is HIF-1, which as previously discussed, is a master transcription factor activated in response to hypoxia. It consists of two subunits, termed α and β.21,22 The α subunit is encoded by three different genes, HIF-1α, which is expressed ubiquitously and HIF-2α and HIF-3α, which are tissue specific. The β subunit is constitutively expressed regardless of the level of oxygenation. Under normoxia there is continual degradation of the α subunit, but this is inhibited under hypoxia. This allows the α subunit to accumulate rapidly and then bind to the β subunit to form the active HIF-1 factor. This transactivates dozens of target genes including VEGF, glucose transporter 1 (glut1) and various glycolytic enzymes that help cells adapt to hypoxia. Moeller et al. have reported that following radiotherapy, HIF-1α accumulates and upregulates the expression of cytokines that enhance endothelial cell radioresistance.40 This group used YC-1,41 an agent originally developed for circulatory disorders that inhibits platelet aggregation and vascular contraction and also targets HIF-1. In a subsequent study Moeller et al. found the net effect of HIF-1 blockade with YC-1 on tumor radioresponsiveness was highly dependent on treatment sequencing, with regimens using radiation first being significantly more effective than the alternative.42
More recently another group showed that treatment with YC-1 followed by radiation mitigated the effect of radiation therapy, while YC-1 treatment following radiation suppressed the post-irradiation upregulation of HIF-1 activity and subsequently resulted in delayed tumor growth.43 This study indicated that sequencing determines whether a HIF1 inhibitor enhances or inhibits the therapeutic effect of radiation. YC-1 significantly suppressed HIF-1 activity, leading to a decrease in microvessel density and an increase in tumor hypoxia. This led the authors to suggest that the sequence of YC-1 followed by radiation was associated with a poorer outcome because it resulted in an increase in the radioresistant hypoxic fraction.
Two other agents that are HIF-1α inhibitors that have been tested as radiosensitizers are TAS106 and S-2-amino-3-[4′-N,N,-bis(2-chloroethyl)amino]phenyl propionic acid N-oxide dihydrochloride (PX-478). TAS106 has been reported to be helpful against radiation-resistant hypoxic cells in solid tumors.44 PX-478 was found to enhance radiosensitivity in prostate cancer cell lines in both normoxic and hypoxic conditions.45 Further studies demonstrated that the effect of PX-478 as a radiosensitizer was even more potent in vivo than in vitro, likely due to blunting of the tumor stromal response to radiation.46 Specifically PX-478 blunted HIF-1 dependent angiogenic signaling, which was induced by ischemia caused by vascular damage due to radiation. Currently, PX-478 is being investigated in phase I clinical trials.
Epidermal Growth Factor Receptor (EGFR) Inhibitors
The EGFR receptors (erb1/EGFR, erb2/HER2, erb3/HER3 and erb4/HER4) are members of a family of receptor tyrosine kinases implicated in the development of some types of cancer, especially non-small cell lung cancers and head and neck cancers. EGFR activation leads to an intracellular signaling cascade via Ras activation, which stimulates the extracellular signal pathway regulated kinase (ERK)/mitogen-activated protein kinase (MAP) kinase pathway (Fig. 1). A number of EGFR inhibitors including the monoclonal antibodies cetuximab (Erbitux) and panitumumab (Vectoibix) and small molecule tyrosine kinase inhibitors gefitinib (Iressa) and erlotinib (Tarceva) have shown potential in the treatment of several types of human cancers and have been reviewed by us previously.47 Radiation can increase the expression of EGFR, which has been implicated in increasing the radiation resistance of cancer cells. In a landmark randomized phase III trial, the combination of cetuximab and radiation was found to be superior to radiation only in the treatment of locally advanced head and neck squamous cell carcinoma with clear improvement in local control and survival favoring the combined modality group.48
Figure 1.

Signaling pathways that can be inhibited to increase the radiation response of tumors. Activation of growth factor receptors including EGFR leads to upregulation of numerous downstream pathways. One of these, the PI3K/Akt pathway, has been implicated in radioresistance and in the regulation of hypoxia-inducible factor 1α (HIF-1α) and vascular endothelial growth factor (VEGF) expression. Inhibition of the PI3K/Akt pathway has been shown to increase the intrinsic radiosensitivity of cancer cells. However, recent evidence suggests that inhibition of the EGFR/PI3K/Akt pathway can also increase the extrinsic radiosensitivity of tumors by altering the tumor microenvironment, perhaps by improving blood flow and tumor oxygenation.
Harari and colleagues found that the monoclonal antibody C225 enhanced the radiosensitivity of head and neck cancer cells as measured by clonogenic survival.49 C225 also augmented radiation killing in human xenografts implanted in nude mice.50,51 Early studies with gefitinib found that it inhibited growth factor production and angiogenesis in various cancer cells and enhanced the antitumor activity of ionizing radiation.52,53 Investigators found that gefitinib radiosensitized cells in vitro perhaps by suppressing cellular DNA repair capacity.54,55 Gefitinib not only inhibited radiation-induced activation of DNA-dependent protein kinase56 but it also inhibited homology-directed recombination repair in human breast cancer cells.57 Chinnayan et al. showed that erlotinib radiosensitized cells in vitro, an effect that might have occurred through a combination of increased apoptosis, cell cycle arrest and changes in DNA damage repair.58
We have investigated the effects of EGFR inhibition on the tumor microenvironment to see whether there might be effects that could affect subsequent therapy. Our previous work demonstrated that the small molecule EGFR inhibitors erlotinib and gefitinib decreased VEGF mRNA expression, decreased secretion of VEGF protein, and blunted HIF-1α induction in response to hypoxia in SQ20B head and neck squamous cell carcinoma cells.59 More recently we have shown that erlotinib treatment altered vessel morphology and decreased vessel permeability within human xenografts grown in nude mice.60 Furthermore, we have found that erlotinib increased tumor blood flow measured by power Doppler ultrasound and decreased hypoxia and tumor O2 saturation.60 Others have shown that EGFR inhibition with C225,61 or gefitinib62,63 can lead to improved oxygenation in tumors. Solomon et al. used 18F-FAZA PET scanning with the nitroimidazole FAZA to show this,63 and Warburton et al. used EF5 flow cytometry.62 Our results offer a mechanism for this phenomenon. We hypothesize that these changes in tumor physiology are an indirect effect of EGFR inhibition, which causes decreased VEGF secretion by the tumor cells, leading to vascular normalization, improved blood flow, and improved oxygenation. We speculate that this may be part of the mechanism by which the drug may increase in vivo radiosensitivity, in addition to the other effects it has been noted to have in vitro.
The Phosphatidylinositol 3-Kinase (PI3K)/Akt/mTOR Pathway
The PI3K/Akt pathway is commonly activated in human cancers64,65 (Fig. 1). Phosphatidylinositol 3-kinases are a family of enzymes that phosphorylates the 3′-OH of the inositol ring of phosphatidylinositol. Phosphatidylinositol 3,4,5-trisphosphate (PIP3) is an important lipid second messenger generated by PI3K that plays a vital role in several signal transduction pathways.66,67 PIP3 activates the serine/threonine kinase AKT by physically binding to it via a pleckstrin homology domain. This leads to translocation of Akt to the inner cell membrane, allowing it be phosphorylated on 2 specific residues, Ser473 and Thr308. Phosphatase and tensin homologue gene (PTEN) is a phosphatase that opposes the action of PI3K, thereby reducing the level of activated (phosphorylated) AKT. AKT controls protein synthesis and cell growth by phosphorylation of mammalian target of rapamycin (mTOR). The PI3K pathway, which is known to play a key role in controlling cell proliferation, growth and survival, is activated in many cancers.68
There are three classes of PI3K: I, II and III. Class I PI3Ks are heterodimers composed of a catalytic and a regulatory subunit and are further subdivided into two subclasses: IA and IB. Class IA PI3K contains the p85 regulatory subunit that binds to receptor tyrosine kinases (RTK) and either an alpha, beta or delta p110 catalytic subunit (p110α, p110β or p110δ). Additionally p110 is also a direct effector of Ras. Class IB PI3K is composed of the p110γ catalytic subunit complexed with the p101 regulatory protein. Class II consists of three members, PI3KC2α, PI3KC2β and PI3KC2γ, while Class III contains only one member encoded by the gene VPS34.67,69,70
PI3K activation can occur via loss of PTEN, by Ras mutation or by increased expression of a growth factor receptor such as EGFR. There appears to be a relationship between PI3K/Akt pathway activation and radiosensitivity. The radioresistance of cells can be increased by the overexpression or activation of oncogenes such as epidermal growth factor receptor (EGFR)71 and Ras72 or loss of the tumor suppressor gene PTEN.73 These mutations share in common the fact that they activate the PI3K-Akt pathway. Conversely, many groups have shown that blocking the Ras and/or PI3K pathway enhances the radiation response in vitro and in vivo.74–77
PI3K inhibitors
LY294002 and wortmannin are the first generation PI3K inhibitors that inhibit most of the PI3Ks at low doses. Wortmannin, a fungal secondary metabolite, is a noncompetitive, irreversible inhibitor of PI3K with an IC50 of 2–4 nM.78 Wortmannin radiosensitizes cancer cells, however, it is biologically unstable and toxic to the liver.79 Quercetin, a naturally occurring bioflavinoid, with an IC50 of 3.8 μM was shown to inhibit PI3K.80 LY294002 the first synthetic PI3K inhibitor was based on quercetin.81,82 LY294002 has been found to radiosensitize both in vitro77,79 as well as in vivo.76 However, LY294002 is unsuitable for human use due to toxicity.83
SF1126, a pan-PI3K inhibitor developed by Semafore Pharmaceuticals is a novel RGDS-conjugated LY294002 prod-rug. It was conjugated to RGDS purposely in order to have it bind to specific integrins within the tumor compartment, resulting in enhanced delivery of the active compound to the tumor vasculature and tumor. Indeed, it was shown to have potent anti-tumor activity with a better pharmacokinetic and dynamic effect compared to its parent drug.84 SF1126 also has radiosensitizing effect in glioma tumor models.85
Of a series of isoform-selective inhibitors of PI3K,86 the pyridinylfuranopyrimidine inhibitor PI-103 was found to selectively inhibit p110α and mTOR in malignant gliomas.87 Chen et al. found that this inhibitor produced additive cytotoxic effects in combination with radiation therapy.88 Their in vivo studies indicated a significant reduction in tumor growth rate induced by the combined treatment compared with each treatment modality alone. Prevo et al. showed that PI-103 enhanced the radiation sensitivity of tumor cells in vitro and resulted in the persistence of DNA damage as evidenced by elevated levels of γH2AX foci.89 This same group showed that the drug caused changes in the vessel morphology with increased pericyte coverage, consistent with vascular normalization.90 This was accompanied by increased blood flow and decreased tumor hypoxia.
Akt inhibitors
The Akt inhibitor 1L-6-hydroxymethyl-chiro-inositol 2(R)-2-O-methyl-3-O-octadecylcarbonate, has been tested against, HL60AR, an HL60 leukemia subclone with a constitutively activated PI3K/Akt pathway, which is resistant to multiple chemotherapeutic drugs and ionizing radiation.91 At 20 μM concentration the drug was able to inhibit Akt without affecting the PI3K activity and markedly reduced resistance of HL60AR cells to chemotherapeutic agents and ionizing radiation. Another novel Akt inhibitor 8-(1-Hydroxy-ethyl)-2-methoxy-3-(4-methoxy-benzyloxy)-benzo[c]chromen-6-one (Palomid 529 or P529) has been shown to have anti-cancer and radiosensitizing effects on PC3 prostate carcinoma cells and xenografts.92
HIV Protease inhibitors
Our group and others have demonstrated that a class of drugs in common clinical use, the HIV protease inhibitors (HPIs), interferes with PI3K-Akt signaling and radiosensitizes a variety of tumor types.93–95 These drugs have been widely used in HIV patients with well-characterized pharmacokinetics and documented toxicity profile.96 A prominent side effect of HPI treatment is insulin resistance.97 Akt plays a key role in the coordinated regulation of growth and metabolism by the insulin/IGF-signaling pathway.98 Therefore, it was hypothesized that HPIs might block the PI3K-Akt signaling axis in tumor cells and hence might be used clinically as radiation sensitizers.93 In this study, three HPIs, nelfinavir, amprenavir and saquinavir inhibited Akt phosphorylation at Ser473 and radiosensitized a variety of tumors.
The effect of nelfinavir often appeared greater in vivo than in vitro, leading us to hypothesize that the drug may have effects on the TME that increase radiosensitivity. We have shown that nelfinavir decreases HIF-1α and VEGF expression both in vitro and in vivo95,99 and that it improves tumor oxygenation in A549 lung carcinoma xenografts.95 We have recently found that nelfinavir can increase blood flow in tumors (Fig. 2). Thus, our results with nelfinavir are very similar to our results with erlotinib.60 We hypothesize that the drug leads to decreased VEGF secretion by tumor cells, resulting in vascular normalization. Consistent with this idea, Qayum et al. found that nelfinavir treatment led to vessels that were more regular with increased interbranch length and reduced tortuosity, consistent with normalization.90
Figure 2.
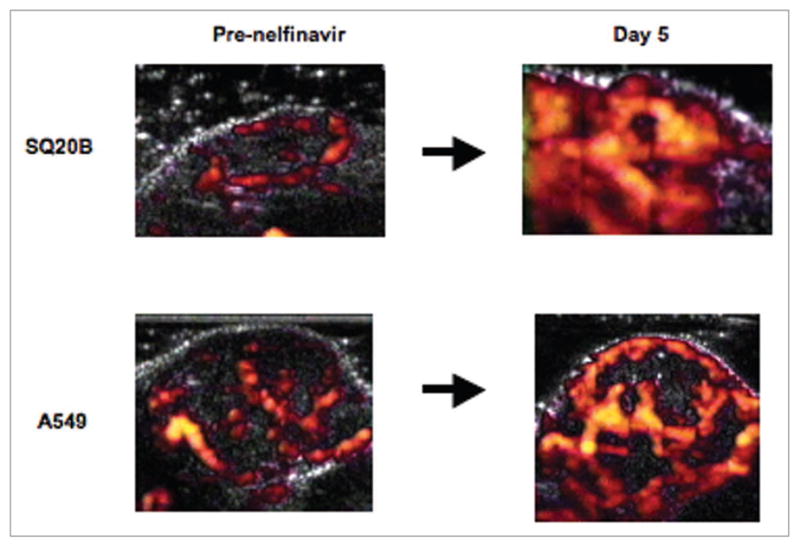
Xenograft blood flow response to nelfinavir treatment. Nude mouse was injected subcutaneously in the flank with SQ20B head and neck cancer cells or A549 lung cancer cells to form xenograft. When tumor reached a size of ~1 cm in diameter, mouse was anesthetized and injected with microbubble contrast agent, then power Doppler ultrasound was performed. Mouse was then started on nelfinavir-containing diet (80 mg/kg/day). Repeat Doppler measurement was performed at day 5. Doppler flow images are shown pre-nelfinavir (A) and at day 5 (B). Details regarding xenograft production and Doppler imaging can be found in Cerniglia et al.60 and Pore et al.95
mTOR inhibitors
The mTOR pathway is downstream of the Akt pathway (Fig. 1) and is an attractive target for cancer therapy. Rapamycin was the first mTOR inhibitor to be discovered. The other mTOR inhibitors currently in clinical development as anticancer agents include temsirolimus (CCI779), everolimus (RAD001) and deforolimus (AP23573).100,101 Rapamycin has been reported to radiosensitize both in vitro and in vivo soft tissue sarcoma cell lines and xenografts.102 However, in another study, rapamycin and RAD001 radiosensitized vascular endothelial cells but failed to sensitize glioma cells.103 Still another study demonstrated no direct radiosensitizing effects in glial cells treated with rapamycin in vitro, but a strong radiosensitizing effect in vivo.104 Hence, the role of mTOR inhibitors in radiosensitization is still an open one.
Conclusion
It is apparent that we now how have access to a variety of agents with radiosensitizing abilities that can be used in the clinic. These include agents that inhibit either growth factors or growth factor receptors such as EGFR and VEGFR. These factors are often overexpressed in tumors or are released in response to radiation. Inhibitors are also available that can inhibit pathways downstream of the receptor. Inhibition of a single factor may affect multiple downstream events. These agents may affect the intrinsic radiosensitivity of tumor cells, but there is growing evidence that they can influence the microenvironment of the tumor to increase response to radiation. For example, inhibition of EGFR can lead to decreased VEGF secretion by tumor cells and alteration of tumor oxygenation in preclinical models. However, clinical data from patients showing that modulation of the tumor microenvironment is an important mechanism by which these agents radiosensitize are sparse. A major challenge remains to obtain this information non-invasively in patients and then to determine whether the sequencing of these agents with respect to radiation therapy can be optimized.
Acknowledgments
This work was supported by NIH RO1 CA093638 (A.M.)
References
- 1.Gerweck LE, Vijayappa S, Kurimasa A, Ogawa K, Chen DJ. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006;66:8352–5. doi: 10.1158/0008-5472.CAN-06-0533. [DOI] [PubMed] [Google Scholar]
- 2.Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, et al. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005;65:4987–92. doi: 10.1158/0008-5472.CAN-04-4250. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 4.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 5.Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89–91. doi: 10.1016/j.ccr.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Ogawa K, Boucher Y, Kashiwagi S, Fukumura D, Chen D, Gerweck LE. Influence of tumor cell and stroma sensitivity on tumor response to radiation. Cancer Res. 2007;67:4016–21. doi: 10.1158/0008-5472.CAN-06-4498. [DOI] [PubMed] [Google Scholar]
- 7.Vaupel P, Hoeckel M. Predictive power of the tumor oxygenation status. Adv Exp Med Biol. 1999;471:533–9. doi: 10.1007/978-1-4615-4717-4_63. [DOI] [PubMed] [Google Scholar]
- 8.Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol. 2004;14:198–206. doi: 10.1016/j.semradonc.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Cairns R, Papandreou I, Denko N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol Cancer Res. 2006;4:61–70. doi: 10.1158/1541-7786.MCR-06-0002. [DOI] [PubMed] [Google Scholar]
- 10.Koch CJ, Kruuv J, Frey HE. Variation in radiation response of mammalian cells as a function of oxygen tension. Radiat Res. 1973;53:33–42. [PubMed] [Google Scholar]
- 11.Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26:638–48. doi: 10.1259/0007-1285-26-312-638. [DOI] [PubMed] [Google Scholar]
- 12.Vaupel P. Prognostic potential of the pre-therapeutic tumor oxygenation status. Adv Exp Med Biol. 2009;645:241–6. doi: 10.1007/978-0-387-85998-9_36. [DOI] [PubMed] [Google Scholar]
- 13.Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 1997;38:285–9. doi: 10.1016/s0360-3016(97)00101-6. [DOI] [PubMed] [Google Scholar]
- 14.Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941–3. [PubMed] [Google Scholar]
- 15.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 16.Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 17.Denekamp J. Review article: angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol. 1993;66:181–96. doi: 10.1259/0007-1285-66-783-181. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–63. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- 19.Siemann DW, Rojiani AM. The vascular disrupting agent ZD6126 shows increased antitumor efficacy and enhanced radiation response in large, advanced tumors. Int J Radiat Oncol Biol Phys. 2005;62:846–53. doi: 10.1016/j.ijrobp.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 20.Hokland SL, Horsman MR. The new vascular disrupting agent combretastatin-A1-disodium-phosphate (OXi4503) enhances tumour response to mild hyperthermia and thermoradiosensitization. Int J Hyperthermia. 2007;23:599–606. doi: 10.1080/02656730701739554. [DOI] [PubMed] [Google Scholar]
- 21.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–6. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 23.Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006;12:5018–22. doi: 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 24.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–19. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 26.Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM, et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999;59:3374–8. [PubMed] [Google Scholar]
- 27.Lee CG, Heijn M, di TE, Griffon-Etienne G, Ancukiewicz M, Koike C, et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60:5565–70. [PubMed] [Google Scholar]
- 28.Hess C, Vuong V, Hegyi I, Riesterer O, Wood J, Fabbro D, et al. Effect of VEGF receptor inhibitor PTK787/ZK222584 [correction of ZK222548] combined with ionizing radiation on endothelial cells and tumour growth. Br J Cancer. 2001;85:2010–6. doi: 10.1054/bjoc.2001.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zips D, Krause M, Hessel F, Westphal J, Bruchner K, Eicheler W, et al. Experimental study on different combination schedules of VEGF-receptor inhibitor PTK787/ZK222584 and fractionated irradiation. Anticancer Res. 2003;23:3869–76. [PubMed] [Google Scholar]
- 30.Williams KJ, Telfer BA, Brave S, Kendrew J, Whittaker L, Stratford IJ, et al. ZD6474, a potent inhibitor of vascular endothelial growth factor signaling, combined with radiotherapy: schedule-dependent enhancement of antitumor activity. Clin Cancer Res. 2004;10:8587–93. doi: 10.1158/1078-0432.CCR-04-1147. [DOI] [PubMed] [Google Scholar]
- 31.Williams KJ, Telfer BA, Shannon AM, Babur M, Stratford IJ, Wedge SR. Inhibition of vascular endothelial growth factor signalling using cediranib (RECENTIN; AZD2171) enhances radiation response and causes substantial physiological changes in lung tumour xenografts. Br J Radiol. 2008;81:21–7. doi: 10.1259/bjr/59853976. [DOI] [PubMed] [Google Scholar]
- 32.Williams KJ, Telfer BA, Shannon AM, Babur M, Stratford IJ, Wedge SR. Combining radiotherapy with AZD2171, a potent inhibitor of vascular endothelial growth factor signaling: pathophysiologic effects and therapeutic benefit. Mol Cancer Ther. 2007;6:599–606. doi: 10.1158/1535-7163.MCT-06-0508. [DOI] [PubMed] [Google Scholar]
- 33.Cao C, Albert JM, Geng L, Ivy PS, Sandler A, Johnson DH, et al. Vascular endothelial growth factor tyrosine kinase inhibitor AZD2171 and fractionated radiotherapy in mouse models of lung cancer. Cancer Res. 2006;66:11409–15. doi: 10.1158/0008-5472.CAN-06-2414. [DOI] [PubMed] [Google Scholar]
- 34.Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006;66:11520–39. doi: 10.1158/0008-5472.CAN-06-2848. [DOI] [PubMed] [Google Scholar]
- 35.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 36.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 37.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1 and matrix metalloproteinases. Cancer Cell. 2004;6:553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Willett CG, Boucher Y, di TE, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batchelor TT, Sorensen AG, di TE, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals and stress granules. Cancer Cell. 2004;5:429–41. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 41.Yeo EJ, Chun YS, Cho YS, Kim J, Lee JC, Kim MS, et al. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003;95:516–25. doi: 10.1093/jnci/95.7.516. [DOI] [PubMed] [Google Scholar]
- 42.Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 43.Harada H, Itasaka S, Zhu Y, Zeng L, Xie X, Morinibu A, et al. Treatment regimen determines whether an HIF-1 inhibitor enhances or inhibits the effect of radiation therapy. Br J Cancer. 2009;100:747–57. doi: 10.1038/sj.bjc.6604939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasui H, Ogura A, Asanuma T, Matsuda A, Kashiwakura I, Kuwabara M, et al. Inhibition of HIF-1alpha by the anticancer drug TAS106 enhances X-ray-induced apoptosis in vitro and in vivo. Br J Cancer. 2008;99:1442–52. doi: 10.1038/sj.bjc.6604720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radio-sensitivity of prostate carcinoma cells. Int J Cancer. 2008;123:2430–7. doi: 10.1002/ijc.23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz DL, Powis G, Thitai-Kumar A, He Y, Bankson J, Williams R, et al. The selective hypoxia inducible factor-1 inhibitor PX-478 provides in vivo radiosensitization through tumor stromal effects. Mol Cancer Ther. 2009;8:947–58. doi: 10.1158/1535-7163.MCT-08-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutta PR, Maity A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007;254:165–77. doi: 10.1016/j.canlet.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 49.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–40. [PubMed] [Google Scholar]
- 50.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics and tumor angiogenesis. Clin Cancer Res. 2000;6:2166–74. [PubMed] [Google Scholar]
- 51.Milas L, Mason K, Hunter N, Petersen S, Yamakawa M, Ang K, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6:701–8. [PubMed] [Google Scholar]
- 52.Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001;7:1459–65. [PubMed] [Google Scholar]
- 53.Bianco C, Tortora G, Bianco R, Caputo R, Veneziani BM, Caputo R, et al. Enhancement of antitumor activity of ionizing radiation by combined treatment with the selective epidermal growth factor receptor-tyrosine kinase inhibitor ZD1839 (Iressa) Clin Cancer Res. 2002;8:3250–8. [PubMed] [Google Scholar]
- 54.Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res. 2008;14:1266–73. doi: 10.1158/1078-0432.CCR-07-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang SM, Li J, Armstrong EA, Harari PM. Modulation of radiation response and tumor-induced angiogenesis after epidermal growth factor receptor inhibition by ZD1839 (Iressa) Cancer Res. 2002;62:4300–6. [PubMed] [Google Scholar]
- 56.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–9. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 57.Li L, Wang H, Yang ES, Arteaga CL, Xia F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008;68:9141–6. doi: 10.1158/0008-5472.CAN-08-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chinnaiyan P, Huang S, Vallabhaneni G, Armstrong E, Varambally S, Tomlins SA, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Res. 2005;65:3328–35. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 59.Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 60.Cerniglia GJ, Pore N, Tsai JH, Schultz S, Mick R, Choe R, et al. Epidermal growth factor receptor inhibition modulates the microenvironment by vascular normalization to improve chemotherapy and radiotherapy efficacy. PLoS ONE. 2009;4:6539. doi: 10.1371/journal.pone.0006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krause M, Ostermann G, Petersen C, Yaromina A, Hessel F, Harstrick A, et al. Decreased repopulation as well as increased reoxygenation contribute to the improvement in local control after targeting of the EGFR by C225 during fractionated irradiation. Radiother Oncol. 2005;76:162–7. doi: 10.1016/j.radonc.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 62.Warburton C, Dragowska WH, Gelmon K, Chia S, Yan H, Masin D, et al. Treatment of HER-2/neu overexpressing breast cancer xenograft models with trastuzumab (Herceptin) and gefitinib (ZD1839): drug combination effects on tumor growth, HER-2/neu and epidermal growth factor receptor expression, and viable hypoxic cell fraction. Clin Cancer Res. 2004;10:2512–24. doi: 10.1158/1078-0432.ccr-03-0244. [DOI] [PubMed] [Google Scholar]
- 63.Solomon B, Binns D, Roselt P, Weibe LI, McArthur GA, Cullinane C, et al. Modulation of intratumoral hypoxia by the epidermal growth factor receptor inhibitor gefitinib detected using small animal PET imaging. Mol Cancer Ther. 2005;4:1417–22. doi: 10.1158/1535-7163.MCT-05-0066. [DOI] [PubMed] [Google Scholar]
- 64.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ihle NT, Powis G. Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther. 2009;8:1–9. doi: 10.1158/1535-7163.MCT-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–6. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 67.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–6. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 68.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–9. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 70.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855–65. doi: 10.1038/sj.onc.1206698. [DOI] [PubMed] [Google Scholar]
- 72.Gupta AK, Bakanauskas VJ, Cerniglia GJ, Cheng Y, Bernhard EJ, Muschel RJ, et al. The Ras radiation resistance pathway. Cancer Res. 2001;61:4278–82. [PubMed] [Google Scholar]
- 73.Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007;282:21206–12. doi: 10.1074/jbc.M703042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakamura JL, Karlsson A, Arvold ND, Gottschalk AR, Pieper RO, Stokoe D, et al. PKB/Akt mediates radiosensitization by the signaling inhibitor LY294002 in human malignant gliomas. J Neurooncol. 2005;71:215–22. doi: 10.1007/s11060-004-1718-y. [DOI] [PubMed] [Google Scholar]
- 75.Grana TM, Rusyn EV, Zhou H, Sartor CI, Cox AD. Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res. 2002;62:4142–50. [PubMed] [Google Scholar]
- 76.Gupta AK, Cerniglia GJ, Mick R, Ahmed MS, Bakanauskas VJ, Muschel RJ, et al. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int J Radiat Oncol Biol Phys. 2003;56:846–53. doi: 10.1016/s0360-3016(03)00214-1. [DOI] [PubMed] [Google Scholar]
- 77.Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, et al. Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clin Cancer Res. 2006;12:250–6. doi: 10.1158/1078-0432.CCR-05-1084. [DOI] [PubMed] [Google Scholar]
- 78.Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–23. [PubMed] [Google Scholar]
- 79.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149–56. [PubMed] [Google Scholar]
- 80.Matter WF, Brown RF, Vlahos CJ. The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem Biophys Res Commun. 1992;186:624–31. doi: 10.1016/0006-291x(92)90792-j. [DOI] [PubMed] [Google Scholar]
- 81.Srivastava AK. Inhibition of phosphorylase kinase, and tyrosine protein kinase activities by quercetin. Biochem Biophys Res Commun. 1985;131:1–5. doi: 10.1016/0006-291x(85)91761-9. [DOI] [PubMed] [Google Scholar]
- 82.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–8. [PubMed] [Google Scholar]
- 83.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–92. [PubMed] [Google Scholar]
- 84.Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and anti-angiogenic activity. Cancer Res. 2008;68:206–15. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 85.Shu HG, Gao H, Chang C, Durden DL. SF1126, a Novel Integrin-Targeting Phosphatidylinositol-3 Kinase Inhibitor, Has Radiosensitizing and Anti-Tumor Effects in Glioma Model Systems. Int J Radiat Oncol Biol Phys. 2007;69:98. [Google Scholar]
- 86.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110al-pha in insulin signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen JS, Zhou LJ, Entin-Meer M, Yang X, Donker M, Knight ZA, et al. Characterization of structurally distinct, isoform-selective phosphoinositide 3′-kinase inhibitors in combination with radiation in the treatment of glioblastoma. Mol Cancer Ther. 2008;7:841–50. doi: 10.1158/1535-7163.MCT-07-0393. [DOI] [PubMed] [Google Scholar]
- 89.Prevo R, Deutsch E, Sampson O, Diplexcito J, Cengel K, Harper J, et al. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI-103 enhances tumor radiosensitivity. Cancer Res. 2008;68:5915–23. doi: 10.1158/0008-5472.CAN-08-0757. [DOI] [PubMed] [Google Scholar]
- 90.Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, et al. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martelli AM, Tazzari PL, Tabellini G, Bortul R, Billi AM, Manzoli L, et al. A new selective AKT pharmacological inhibitor reduces resistance to chemotherapeutic drugs, TRAIL, all-trans-retinoic acid, and ionizing radiation of human leukemia cells. Leukemia. 2003;17:1794–805. doi: 10.1038/sj.leu.2403044. [DOI] [PubMed] [Google Scholar]
- 92.Diaz R, Nguewa PA, Diaz-Gonzalez JA, Hamel E, Gonzalez-Moreno O, Catena R, et al. The novel Akt inhibitor Palomid 529 (P529) enhances the effect of radiotherapy in prostate cancer. Br J Cancer. 2009;100:932–40. doi: 10.1038/sj.bjc.6604938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005;65:8256–65. doi: 10.1158/0008-5472.CAN-05-1220. [DOI] [PubMed] [Google Scholar]
- 94.Jiang Z, Pore N, Cerniglia GJ, Mick R, Georgescu MM, Bernhard EJ, et al. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007;67:4467–73. doi: 10.1158/0008-5472.CAN-06-3398. [DOI] [PubMed] [Google Scholar]
- 95.Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, et al. Nelfinavir downregulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006;66:9252–9. doi: 10.1158/0008-5472.CAN-06-1239. [DOI] [PubMed] [Google Scholar]
- 96.Powderly WG. Long-term exposure to lifelong therapies. J Acquir Immune Defic Syndr. 2002;29:28–40. doi: 10.1097/00126334-200202011-00005. [DOI] [PubMed] [Google Scholar]
- 97.Carr A, Samaras K, Thorisdottir A, Kaufmann GR, Chisholm DJ, Cooper DA. Diagnosis, prediction and natural course of HIV-1 protease-inhibitor-associated lipodystrophy, hyperlipidaemia and diabetes mellitus: a cohort study. Lancet. 1999;353:2093–9. doi: 10.1016/S0140-6736(98)08468-2. [DOI] [PubMed] [Google Scholar]
- 98.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–51. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 99.Pore N, Gupta AK, Cerniglia GJ, Maity A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia. 2006;8:889–95. doi: 10.1593/neo.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Konings IR, Verweij J, Wiemer EA, Sleijfer S. The applicability of mTOR inhibition in solid tumors. Curr Cancer Drug Targets. 2009;9:439–50. doi: 10.2174/156800909788166556. [DOI] [PubMed] [Google Scholar]
- 101.Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008;17:1947–54. doi: 10.1517/13543780802556485. [DOI] [PubMed] [Google Scholar]
- 102.Murphy JD, Spalding AC, Somnay YR, Markwart S, Ray ME, Hamstra DA. Inhibition of mTOR radio-sensitizes soft tissue sarcoma and tumor vasculature. Clin Cancer Res. 2009;15:589–96. doi: 10.1158/1078-0432.CCR-08-1019. [DOI] [PubMed] [Google Scholar]
- 103.Shinohara ET, Cao C, Niermann K, Mu Y, Zeng F, Hallahan DE, et al. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene. 2005;24:5414–22. doi: 10.1038/sj.onc.1208715. [DOI] [PubMed] [Google Scholar]
- 104.Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291–7. [PubMed] [Google Scholar]