

Abstract
The aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, contributes to carcinogenesis through its role in the regulation of cytochrome P450 1 (CYP1)-catalyzed metabolism of carcinogens. Here, we investigated genetic and epigenetic mechanisms that affect AhR expression. Analyses of the human AHR proximal promoter in MCF-7 human breast cancer cells using luciferase assays and electrophoretic mobility shift assays revealed multiple specificity protein (Sp) 1 binding sequences that are transcriptional activators in vitro. The regulation of AhR expression was evaluated in long-term estrogen exposed (LTEE) MCF-7 cells, which showed increased AhR expression, enhanced CYP1 inducibility, and increased capacity to form DNA adducts when exposed to the dietary carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. The increased AhR expression in LTEE cells was found not to result from increased mRNA stability, differential RNA processing, or decreased DNA methylation. Analysis of the AHR proximal promoter region using chromatin immunoprecipitation confirmed that enhanced expression of AhR in LTEE cells involves changes in histone modifications, notably decreased trimethylation of histone 3, lysine 27. Upon further examination of the GC-rich Sp1-binding region, we confirmed that it contains a polymorphic (GGGGC)n repeat. In a population of newborns from New York State, the allele frequency of (GGGGC)n was n = 4>5≫6, 2. Circular dichroism spectroscopy revealed the ability of sequences of this GC-rich region to form guanine-quadruplex structures in vitro. These studies revealed multiple levels at which AhR expression may be controlled, and offer additional insights into mechanisms regulating AhR expression that can ultimately impact carcinogenesis.
Keywords: aryl hydrocarbon receptor; long-term estrogen exposure; epigenetic; (GGGGC)n repeat polymorphism; guanine-quadruplex; 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
1. Introduction
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates the toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other environmental contaminants. The AhR regulates expression of enzymes that catalyze the metabolism of carcinogens including benzo[a]pyrene (BaP) and other polycyclic aromatic hydrocarbons (PAHs) [1–3], and heterocyclic aromatic amines, including 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Subsequent to ligand binding, the AhR shuttles from the cytoplasm into the nucleus, dimerizes with the AhR nuclear translocator, and regulates expression of target genes by binding to xenobiotic response elements in their enhancer regions. These include the genes encoding cytochromes P450 (CYP) of the CYP1 family: CYP1A1, CYP1A2, and CYP1B1. Induction of CYP1 enzymes through activation of the AhR leads to increased rates of carcinogen metabolism, which is generally a protective mechanism [4]. However, metabolic formation of PAH dihydrodiol epoxides, PAH ortho-quinones, heterocyclic amine N-hydroxy intermediates, and other reactive metabolites of environmental, dietary, and endogenous substrates can lead to DNA adduction, mutagenesis, and carcinogenesis [5].
Given the importance of AhR expression in the regulation of molecular responses to a multitude of chemical, physical, and biological signals [6], surprisingly few studies have been focused on the mechanisms that control expression of the human AHR gene. The promoters of both the human AHR and mouse Ahr genes do not contain TATA boxes; rather, they possess GC-rich regions that are general features of housekeeping genes [7]. The view that the AHR gene exhibits characteristics of a housekeeping gene is supported by the constitutive expression of the AhR in many cell types [6]. Previous work on the mouse Ahr promoter revealed functional Sp1 sites in the GC-rich region that is juxtaposed 5′ to the transcription start site [8]; a similar role of Sp1 transcription factors in transcriptional activation of the human AHR gene is likely [9]. The long-term regulation of AhR expression in MCF-7 human breast cancer cells was found to depend on medium serum and estrogen content, which suggests the involvement of epigenetic mechanisms in the regulation of AHR gene expression [10]. Recent studies indicate that changes in epigenetic regulation can contribute to diseases including breast cancer [11].
The hormonal and chemical mechanisms of carcinogenesis in the breast and other tissues are complex, and they are thought to involve interactions of estrogens and carcinogens of environmental and dietary origin. Life-long exposure to estrogens has been identified as a risk factor for the development of breast cancer [12–14], and a majority of human breast cancers are estrogen receptor (ER)-positive. Studies in this laboratory have focused on interactions of estrogens with the AhR, and have shown up-regulation of AhR mRNA and protein by long-term estrogen exposure (LTEE) of MCF-7 breast carcinoma cells to physiologic levels of 17β-estradiol (E2) in comparison with their expression in control cells cultured without added E2 [15, 16]. Cellular and molecular changes elicited by LTEE of MCF-7 cells include enhanced CYP1 inducibility, increased initial levels of BaP:DNA adducts after BaP exposure, enhanced estrogen-dependent tumor growth as xenografts, and marked changes in the expression of numerous genes, some of which showed antiestrogenic modulation when co-exposed to TCDD [15] or raloxifene [16], while others did not.
Because of the significant role of AhR expression in carcinogenesis, and particularly its role in the regulation of the metabolism of carcinogens, we undertook a comprehensive study of the regulation of AhR expression and activity in MCF-7 cells. Our genetic studies focused on the proximal promoter region, in which we characterized specificity protein (Sp) 1 binding. To evaluate epigenetic mechanisms that may be responsible for the differential expression of the AhR in control and LTEE cells, we investigated DNA methylation and the presence of histone methylation marks that are associated with transcriptional regulation. During the course of our studies we confirmed the presence of polymorphic (GGGGC)n repeats within this Sp1-binding region, and we evaluated the potential of the nucleotide sequences to form guanine-quadruplex structures.
2. Materials and Methods
2.1. Cell culture and media
The MCF-7 cells were those described in our previous studies [10, 15, 16] and were cultured in DC5 medium, consisting of Dulbecco’s Modified Eagle’s Medium without phenol red (Sigma, St. Louis, MO), supplemented with 5% (v/v) bovine calf serum (Cosmic calf serum, HyClone, Logan, UT), 100 µM non-essential amino acids (Sigma), 2 mM L-glutamine (Sigma), 10 µg/l human recombinant insulin (Gibco, Life Technologies, Grand Island, NY), and either 5 µg/ml plasmocin (Lonza, Allendale, NJ) or 0.5 µg/ml tiamulin fumarate (Sigma). Cell cultures were maintained for at least 12 passages in either DC5 containing 0.01% (v/v) DMSO (Sigma) vehicle (referred to as control cells), or 1 nM 17β-estradiol (E2; Steraloids, Newport, RI) (referred to as LTEE cells). Each passage was generally 10 days. LTEE cells that were cultured for an additional 35 days in the absence of added E2 are referred to as LTEE (-E2) cells. MDA-MB-231, 184-A1, MCF-10A, T47D, and ZR-75-1 cells were cultured as previously described [17]. All cultures were maintained in a 37 °C incubator with humidified air containing 5% CO2. When cells were exposed to AhR ligands for 24 h or less, the experiments were performed using conditioned medium, which consisted of DC5 medium recovered after exposure to control cells for at least 3 days.
2.2. Ethoxyresorufin-O-deethylase (EROD) assay
EROD assays were carried out using confluent MCF-7 cultures in 96-well plates as previously described [10]. In brief, control and LTEE cells that had been maintained in their respective media were seeded at 80% confluence in DC5 medium and treated the following day with medium containing 10 nM TCDD (Cambridge Isotope Laboratories, Andover, MA) or 0.1% DMSO vehicle for 72 h. For experiments involving PhIP (Toronto Research Chemicals, Toronto, ON, Canada), exposure of cells was performed as described in Section 2.4, and for inhibition experiments, PhIP was added during the EROD assay to cells pretreated with TCDD. EROD activities were normalized to protein content as measured using the BCA protein assay (Thermo Fisher, Pittsburg, PA).
2.3. Real-time polymerase chain reaction (PCR) analysis of mRNA levels after E2 withdrawal
Control, LTEE, and LTEE (-E2) cells were seeded at near confluence in DC5 medium in 6-well plates, and LTEE (-E2) cells were treated the following day with 1 nM E2 for 6 h, after which total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA). Reverse transcription and quantification of RNA levels were performed as previously described [10]. Real-time PCR analysis utilized primers specific for the AhR, CYP1B1, ERα, progesterone receptor (PGR), and AhR interacting protein (AIP) transcripts, which are listed in Supplementary Table 1. All oligodeoxynucleotides (ODNs) were synthesized by Integrated DNA Technologies (Coralville, IA).
2.4. Analysis of PhIP:DNA adducts in PhIP-exposed MCF-7 cells by liquid chromatography electrospray ionization triple-stage mass spectrometry (LC-ESI/MS3)
Control and LTEE MCF-7 cells were seeded in T-25 flasks in DC5 medium and, when confluent, were exposed for 24 h to 10 µM PhIP with or without 10 nM TCDD in 0.2% DMSO vehicle, or the vehicle alone, using conditioned medium. Genomic DNA was isolated using the PureGene DNA Purification System (Gentra Systems, Qiagen) with an additional protein digestion step using proteinase K (Sigma) at 200 µg/ml for 2 h at 37 °C prior to DNA precipitation. The quantity and the purity of DNA were determined by UV absorbance at 260/280 nm. The DNA adduct standard, N-(deoxyguanosin-8-yl)-PhIP (dG-C8-PhIP), and the [13C10]-dG labeled analog used as the internal standard were prepared by reaction of the N-acetoxy derivative of PhIP with dG (Sigma) or [13C10]-dG (Cambridge Isotope Laboratories), respectively, in 100 mM K2HPO4 buffer (pH 8.0). The internal standard was added prior to DNA digestion at a level of 1 adduct per 107 DNA bases. Digestion of the DNA and the solid-phase extraction of adducts with HyperSep filter SpinTips (Thermo Fisher) were performed as previously described [18].
The LC-ESI/MS3 analyses of dG-C8-PhIP adducts were conducted essentially as described [19]. Briefly, chromatographic separations were achieved using an Agilent 1100 Series capillary liquid chromatography system (Agilent Technologies, Santa Clara, CA) equipped with an Aquasil C18 column (0.32×250 mm) from Thermo Fisher. Samples (2 µl) were injected, and analytes were separated with a gradient program consisting of a hold at 100% A (solvent composition: 0.01% HCO2H and 10% CH3CN) for 2 min, followed by a linear gradient to 100% B (solvent composition: 95% CH3CN containing 0.01% HCO2H) over 30 min at a flow rate of 6 µl/min. The MS instrumentation used was a linear quadrupole ion trap mass spectrometer (LTQ, Thermo Fisher); Xcalibur version 2.07 software was used for data manipulations. Analyses were conducted in the positive-ionization mode and employed an Advance CaptiveSpray source from Michrom Bioresource (Auburn, CA). The source spray voltage was 1.5 kV, and no sheath gas, sweep gas, or auxiliary gas was employed. Other parameters were optimized as described [19]. The tandem MS/MS scan mode was employed to monitor the loss of deoxyribose from the protonated molecules of the adducts, ([M+H-116]+), followed by the consecutive-reaction-monitoring scan mode at the MS3 scan stage, to characterize the product ions of the aglycone adducts, [BH2]+. The total-ion counts produced at the MS3 scan stage were used for quantitative measurements. The MS3 transitions monitored were: m/z 490.1 → 374.1 → scan 160–600 for dG-C8-PhIP and m/z 500.1 → 379.1 → scan 160–600 for [13C10]-dG-C8-PhIP.
2.5. Site-directed mutagenesis, transient transfection, and luciferase assays of the AhR reporter construct
The AHR promoter-luciferase reporter plasmid, pGL3-hAhRP [20], which incorporates an AHR promoter insert that encompasses 5640 bp of the AHR 5′-flanking region, was used, and a deletion construct of this vector was prepared by restriction endonuclease digestion with KpnI (Gibco, Life Technologies) and ApaI (Fermentas, Thermo Fisher), yielding a construct encompassing 120 bp of the proximal AHR promoter region as described [20]. This deletion construct is referred to as AhRΔ(−120). The AHR promoter contains a GC-rich region directly upstream of the transcriptional start site (TSS). In the pGL3-hAhRP plasmid, this GC-rich region contained (GGGGC)4, which represents two additional Sp1 consensus binding sites in comparison with the Ensembl sequence for the AHR gene, which contains (GGGGC)2, found at -37 bp upstream of the TSS [21]. Since one report [22] indicated that the (GGGGC)2 was the most prominent allele, two repeats, or 10 bp, were removed from the AhRΔ(−120) deletion construct using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, Agilent Technologies) according to the manufacturer’s instructions, followed by introduction of point mutations in putative transcription factor binding sites using site-directed mutagenesis. Briefly, complementary forward and reverse primers were designed to introduce point mutations within the core sequence of the transcription factor binding sequence of interest in the AHR promoter, which were flanked by unaltered DNA sequences (Supplementary Table 1). These primers were included in a PCR reaction containing 10 ng of vector dsDNA and 2.5 U PfuUltra HF DNA polymerase (Stratagene, Agilent Technologies). PCR conditions are outlined in Supplementary Table 2. Template DNA was digested with 10 U DpnI, and cDNA plasmids containing the mutation were transformed into XL1-Blue supercompetent cells (Stratagene, Agilent Technologies). Colonies were selected with ampicillin, endonuclease-free DNA was prepared from each colony (PureYield Plasmid Midiprep System, Promega, Madison, WI), and regions of the AHR promoter were sequenced. AHR promoter sequences containing the target mutation were then subcloned into the original pGL3-basic vector (Promega).
For the determination of promoter activities of the constructs, control and LTEE MCF-7 cells were seeded in 24-well plates in DC5 medium. After 24 h, 50 ng of the phRG-TK Renilla control vector (Promega), 250 ng Bluescript-SK carrier plasmid (Stratagene, Agilent Technologies), and 200 ng of the AhRΔ(−120) plasmid or mutant subclone DNA were combined with 1.6 µl Lipofectamine 2000 for transfection according the manufacturer’s protocol (Life Technologies). This mixture was added to triplicate wells, which were then incubated for 48 h. Following transfection, cells were lysed, and luciferase activity was assayed using the Dual-Luciferase Reporter 1000 Assay System according to the manufacturer’s protocol (Promega). Luminescent signals were recorded using a Lumat LB9501 luminometer (Berthold, Germany), and firefly luciferase activities were normalized to Renilla luciferase activities [15].
2.6. Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were obtained using the CelLytic NuCLEAR Kit (Sigma). Sense and antisense ODNs corresponding to regions of the proximal promoter of the AHR gene were annealed in 10 mM Tris-HCl (pH 7.6), 50 mM NaCl, and 1 mM EDTA (sequences and locations are presented in Supplementary Table 1). ODN duplexes were labeled at the 5′ phosphate with [γ-32P]ATP (3000 Ci/mmol; PerkinElmer, Shelton, CT) using T4 polynucleotide kinase according to the manufacturer’s instructions (Fermentas, Thermo Fisher). For binding reactions, 0.5 µg of nuclear extract was first mixed with binding buffer (25 mM HEPES, 25 mM KCl, 10% glycerol, 1 mM EDTA, 2 mM MgCl2, 0.5 mM DTT, 1 µg poly(dI-dC), and 250 ng BSA; components from Sigma) for 20 min on ice; the radioactive probes (105 cpm per reaction) were then added for 15 min at room temperature. For supershift experiments, antibodies against Sp1 (2 µg) or Sp3 (1 µg) were added for an additional 15 min at room temperature. For competition experiments, unlabeled probe was incubated with the binding reaction for 10 min at room temperature after the 20-min incubation on ice and prior to the addition of radioactive probe for 20 min. Protein:DNA complexes were resolved by electrophoresis through 6% polyacrylamide gels. Radioactivity was imaged by exposing the gel to a phosphor screen for 20 h, which was then analyzed using a Typhoon 9400 scanner (GE Healthcare Life Sciences, Piscataway, NJ).
2.7. Determination of AhR mRNA stability
Control and LTEE cells were seeded in 6-well plates in DC5 medium containing 0.01% DMSO vehicle or 1 nM E2, respectively. There was a medium change on the following day using these same media, and after a 72-h culture period, cells were exposed to 1 µg/ml actinomycin D (Sigma) with DMSO as the vehicle, in duplicate, for 0, 1, 2, 6, and 10 h. RNA was recovered at each time point and purified using the RNeasy Mini Kit (Qiagen). Total RNA was reverse-transcribed using oligo-dT primers and Superscript III reverse transcriptase (Life Technologies), and the resulting cDNA was treated with RNase H (Fermentas, Thermo Fisher). The cDNA was amplified by real-time PCR using the LightCycler System (Roche Molecular Biochemicals, Indianapolis, IN) with AhR-specific primers corresponding to sequences in different exons (Supplementary Table 1) and quantified as previously described [10].
2.8. Real-time PCR of heterologous nuclear RNA (hnRNA) levels
RNA from control and LTEE MCF-7 cells was isolated as described [23], and hnRNA was reverse transcribed as above, but using reverse primers specific for a sequence of exon 11 of the AHR gene (Supplementary Table 1). Real-time PCR was carried out with AhR-specific forward and reverse primers complementary to sequences in intron 10 and exon 11, respectively (Supplementary Table 1). The amplification reaction was terminated in the linear range (32 cycles), and amplification efficiency was determined by using the equation, E = (RA/RB)(1/Ct,A-Ct,B)-1, in which E = amplification efficiency, RA and RB = arbitrary thresholds A and B in an individual curve, and Ct,A and Ct,B = threshold cycle numbers at the arbitrary thresholds A and B [24]. To determine the level of genomic DNA carryover in the isolation of RNA, identical reverse transcription reactions without reverse transcriptase were also amplified, and values were subtracted as background. Levels of AhR mRNA were determined by real-time PCR as described in Section 2.3.
2.9. Determination of DNA methylation in vitro
Genomic DNA from control and LTEE MCF-7 cells, and from 184-A1 and MCF-10A breast epithelial cells, was isolated using the Puregene DNA Isolation kit (Gentra Systems, Qiagen). Bisulfite modification was performed using the EZ DNA Methylation Kit as suggested by the manufacturer (Zymo Research, Irvine, CA). Briefly, 0.5 to 1 µg of DNA in 50 µl was denatured with 5 µl M-dilution Buffer for 15 min at 37 °C. DNA containing putative guanine-quadruplex-forming sequences was denatured with 7.5 µl M-dilution buffer for 30 min at 42 °C. After addition of the bisulfite-containing CT-Conversion Reagent (100 µl), samples were incubated at 50 °C for 18 h. Modified DNA was purified using Zymo-Spin I columns and was amplified immediately or stored at −20 °C.
The sense strand of bisulfite-modified genomic DNA was first amplified by PCR using a previously described method [25], but with primers specific for the AHR and CYP1B1 promoters (Supplementary Table 3). The PCR was performed with 1.5 mM MgCl2, 1 µM of each promoter-specific sense and anti-sense primer, 5 U of HotStarTaq polymerase (Qiagen), and 50 to 100 ng of bisulfite-modified genomic DNA. PCR amplification conditions are outlined in Supplementary Table 2. First-round PCR products were then used as template and were re-amplified by GC-tagged primers (Supplementary Table 4). PCR products were then purified with the Qiagen Gel Extraction Kit and subjected to direct-cycle sequencing by the Wadsworth Center Applied Genomic Technologies Core using primers described in Supplementary Table 4.
In order to amplify the GC-rich region of the AHR promoter (−104 bp to +193 bp), special PCR conditions were employed. PCR was performed in a volume of 50 µl with recombinant Taq DNA polymerase and (NH4)2SO4 buffer (Fermentas, Thermo Fisher) with addition of 6% propylene glycol (Sigma) [26], 3 mM MgCl2, and TaqStart Antibody (Clontech Laboratories, Mountain View, CA). Primer concentrations were 0.6 µM (for the first round) and 0.75 µM (for the second round). Thermal cycling was performed using a combination of slowdown [27] and touchdown methodologies (Supplementary Table 2).
2.10. Chromatin immunoprecipitation (ChIP) assay
ChIP was performed following the procedure of Svotelis et al. [28] with some modifications. Briefly, confluent control and LTEE MCF-7 cells were rinsed once with PBS, and then crosslinked with 1.1% p-formaldehyde (Sigma) in PBS for 10 min at room temperature. The reaction was quenched by the addition of 0.125 M glycine (Sigma) for 5 min. After collection of cells in the presence of protease inhibitors and lysis of cells with SDS, the lysates were diluted to the equivalent of 5×106 cells/ml. Aliquots (500 µl) were sonicated on ice using a Cole Palmer (Vernon Hills, IL) series 4710 Ultrasonic Homogenizer (power setting 60) with six 15-s pulses and rest periods of 45 s between pulses. An aliquot of lysate was reserved for analysis of input DNA. Each immunoprecipitate represented 1×106 cells. Lysates were pre-cleared with Protein A/G Plus agarose (Santa Cruz Biotechnology, Santa Cruz, CA), and DNA:protein complexes were immunoprecipitated using the following antibodies: anti-Sp1 (PEP2) and anti-Sp3 (D-20) from Santa Cruz Biotechnology; anti-histone H3 (#39163), anti-histone H3 trimethyl Lys27 (H3K27me3, #39157), and anti-histone H3 dimethyl Lys4 (H3K4me2, #39142) from Active Motif (Carlsbad, CA); and anti-RNA-polymerase II (RNA Pol II, #MMS-126R) from Covance (Princeton, NJ). Crosslinks were reversed for 4 h at 65 °C, and DNA was isolated using the Puregene DNA Purification Kit (Gentra Systems, Qiagen).
PCR was performed using recombinant Taq DNA polymerase, (NH4)2SO4 buffer (Fermentas, Thermo Fisher), and gene-specific primers (Supplementary Table 1) following the manufacturer’s protocol, but with addition of 6% propylene glycol [26], 1 to 1.6 mM MgCl2, and TaqStart Antibody (Clontech Laboratories). PCR conditions were similar to those used for the amplification of the GC-rich region for methylation analysis (Supplementary Table 2). Control regions of genomic DNA were amplified using primers specific for transcripts of the α-tubulin gene (TUBA1A) [29] and of the AIP gene, the expression of which varies minimally among cell lines and experimental conditions [30].
2.11. Determination of (GGGGC)n polymorphic repeats
To determine the allele frequency of polymorphic (GGGGC)n repeats in an anonymous population of New York State newborns, surplus DNA was obtained from the Newborn Screening Program of the New York State Department of Health (NYSDOH). This study was approved by the Institutional Review Board of the NYSDOH. The number of polymorphic (GGGGC)n repeats was also determined in a panel of 17 human-derived cell lines. PCR conditions were the same as those used for amplification of DNA after ChIP (Section 2.10). Protein was removed from the samples using StrataClean Resin (Stratagene, Agilent Technologies), and nucleotide sequencing was performed at the Wadsworth Center Applied Genomic Technologies Core.
2.12. Circular dichroism (CD) spectroscopy
Single-stranded ODNs were synthesized by Integrated DNA Technologies and represent sequences of the GC-rich region encompassed by the AHR proximal promoter, or are selected mutations thereof (Supplementary Table 1). ODNs were diluted to 5 µM in 10 mM Tris-HCl buffer (pH 7.65) with or without 150 mM KCl, and CD spectra over the range of 200 to 320 nm were recorded at 20 °C using a Jasco J-720 spectropolarimeter (Easton, MD) with a 0.5-cm path-length cuvette and spectra derived from10 replicates. Samples were subsequently melted at 85 to 90 °C for 10 min and slowly cooled overnight to establish the low-energy conformation (i.e., a guanine-quadruplex structure); CD spectra were then recorded as before. The CD spectra obtained were compared to reference spectra [31] generated by incubating DNA of known guanine quadruplex-forming sequences under the same conditions as those above.
2.13. Statistical analysis
Data were subjected to ANOVA followed by the Bonferroni multiple comparison test. Data are presented as the means ± standard error (SE) unless otherwise stated. For allele frequencies, chi-square analysis was performed using Sigma Stat (Systat Software, San Jose, CA), and 95% confidence intervals were calculated with a correction for continuity using the Confidence Interval of a Proportion program: http://faculty.vassar.edu/lowry/prop1.html, which is based on the method of Newcombe [32].
3. Results
3.1. Inducible EROD activity decreases over time in control but not in LTEE MCF-7 cells
In order to evaluate potential epigenetic effects of extended E2 exposure on responsiveness to AhR agonists in MCF-7 cells, we monitored TCDD-inducible EROD activity in control and LTEE cells between passages 12 and 59 (or approximately 4 to 20 months of continuous cell culture). We found that the CYP1-catalyzed EROD activity decreased over this period in control cells to the point that TCDD no longer induced EROD activity (Fig. 1A). In contrast, TCDD-inducible EROD activity in LTEE cells, which had increased over the first 12 passages [15], remained constant over the course of these experiments. Control cells exposed to the DMSO vehicle showed minimal EROD activity, while exposure to 10 nM TCDD for 72 h induced approximately the same level of EROD activity at passages 12 and 27. However, by passage 38, the TCDD-inducible EROD activity in control cells had decreased to levels comparable to those in LTEE cells without TCDD exposure. This near-total loss of EROD inducibility by TCDD remained evident throughout the course of the experiments.
Fig. 1.

Effects of LTEE on Ah-responsiveness and gene expression in MCF-7 cells. (A) Ah-responsiveness decreases in control cells with increasing time in culture. At the passages indicated, control and LTEE cultures were exposed to 0.1% DMSO vehicle or 10 nM TCDD for 48 h, and CYP1 activity was determined using the EROD assay with normalization to total protein content. Significant differences between TCDD-induced EROD activities in control and LTEE cells are shown. Data are the means ± SE; n = 5; ***, P < 0.001. (B) AhR expression remains up-regulated in LTEE cells after withdrawal of E2 and is unaffected by short-term re-exposure to 1 nM E2. RNA from control (white bars) and LTEE cells (black bars) cultured for 49 passages, and from LTEE cultures from which E2 had been withdrawn for 35 days (LTEE (-E2), gray bars) was quantified by real-time PCR. LTEE (-E2) cells were re-exposed to E2 for 6 h as indicated. Primers specific for AhR, CYP1B1, ERα, PGR, and AIP cDNA were used in PCR reactions. Significant differences between treatment groups are shown. Data are the means ± SE; n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
3.2. Withdrawal of E2 from LTEE cells alters gene expression
The differences between the levels of AhR expression and AhR-mediated CYP1 inducibility in control and LTEE cells, and the changes in these levels over time, suggested that the AHR gene is under epigenetic regulation. We therefore investigated whether LTEE cells cultured for 35 days in the absence of added E2, termed LTEE (-E2) cells, would display a persistence of the elevated AHR gene expression. LTEE (-E2) cells were also exposed to E2 for 6 h to investigate the response to short-term E2 re-exposure. As previously demonstrated, AhR and CYP1B1 mRNA levels were increased in LTEE cells relative to their levels in control cells [15, 16]. Subsequent withdrawal of E2 from LTEE cultures produced a variety of changes in gene expression (Fig. 1B). AhR mRNA levels in LTEE (-E2) cells remained significantly greater than the levels in control cells and did not change after withdrawal of E2, which suggests an epigenetic mechanism of regulation. Conversely, CYP1B1 mRNA levels, which were increased in LTEE cells relative to the levels in control cells, decreased in LTEE (-E2) cells, but remained significantly greater than mRNA levels in control cells; the level of reduction in LTEE (-E2) cells may correspond to the level of direct transcriptional regulation by ligand-bound ERα.
Levels of ERα mRNA were significantly reduced in LTEE cells in comparison with the levels in control cells, and were moderately reversed in LTEE (-E2) cells, an effect of estrogen exposure in MCF-7 cells that has been previously reported [33]. Interestingly, transcript levels from the well-characterized E2-regulated gene, PGR, were significantly increased in LTEE cells relative to those in control cells, but in LTEE (-E2) cells, the level of this transcript decreased to roughly 9% of its level in control cells. The mRNA transcribed from the AIP gene was used as a control transcript in these experiments; there were no significant differences in the levels of this transcript among the treatment groups. None of the transcripts we investigated responded significantly to a 6-h re-exposure to 1 nM E2 compared with their levels in LTEE (-E2) cells.
3.3. The exposure of LTEE cells to TCDD leads to marked increases in PhIP:DNA adduct formation
Having determined that the differences in AhR expression in control and LTEE cells correlated with the level of CYP1 inducibility in the EROD assay, we further investigated whether the AhR level could be a control point in a pathway leading to the formation DNA adducts. The consequences of increased AhR expression in LTEE MCF-7 cells on the susceptibility to DNA adduction by metabolites of PhIP, a dietary carcinogen, were examined using LC-ESI/MS3. Mass chromatograms (Supplementary Fig. 1A-D) from the analyses show that PhIP:DNA adducts were not detectable (<1 adduct in 108 bases) in DNA from control cells without TCDD exposure, whereas adducts in LTEE cells that had been exposed to 10 nM TCDD were readily detectable. Product-ion mass spectra at the MS3 scan stage for dG-C8-PhIP recovered from TCDD-treated LTEE cells (Supplementary Fig. 1E-F) showed ions that have been previously observed [18]. Quantitative LC-ESI/MS3 showed that when LTEE MCF-7 cells were exposed for 24 h to 10 µM PhIP and 10 nM TCDD, there was a marked enhancement of PhIP:DNA adduct formation in LTEE cells in comparison with that observed in control cells receiving PhIP and TCDD, and in LTEE cells without TCDD exposure (Fig. 2A). These data provide evidence for the role of AhR-regulated CYP1 family enzymes in the metabolic activation of PhIP in these cultures.
Fig. 2.

Effects of LTEE on PhIP:DNA adduct formation and CYP1 inducibility in MCF-7 cells. MCF-7 cells were cultured for 35 or more passages in DC5 (Control) or DC5 containing 1 nM E2 (LTEE). Cells were then seeded in DC5 medium and were exposed for 24 h to the DMSO vehicle or 10 µM PhIP with or without 10 nM TCDD, as indicated. (A) DNA was isolated and hydrolyzed, and PhIP adducts were analyzed by LC-ESI/MS3. Data shown are the means ± SE; n = 3. Significant differences between control and LTEE cells exposed to TCDD and PhIP (***, P < 0.001) and between LTEE cells treated with and without TCDD (###, P < 0.001) are indicated. (B) EROD activities were determined, and the data shown are the means ± SE; n = 5. Significant differences between control and LTEE cells with identical treatments are indicated (***, P < 0.001). (C) Dose-response curves for inhibition of TCDD-induced EROD activity of control and LTEE cells by PhIP, with the addition of PhIP coincident with TCDD exposure (upper) or during the EROD assay (lower). Data shown are the means ± SE; n = 5. Significant differences are indicated between LTEE cells treated with TCDD alone and TCDD plus the indicated concentration of PhIP (***, P < 0.001).
EROD assays were performed to determine the effects of exposure to PhIP and TCDD on the induction of CYP1 activity. As expected, TCDD markedly induced CYP1 activity in LTEE cells, while exposure to PhIP alone caused only a slight increase in EROD activity in LTEE cells. Co-treatment of LTEE cells with TCDD and PhIP showed a 53% inhibition of EROD activity compared with the activity observed when the cultures were exposed to TCDD alone (Fig. 2B). We then investigated whether this decrease was due to an inhibition of the CYP1 enzymatic activity in the EROD assay by PhIP, or whether PhIP antagonized the AhR-mediated CYP1 induction by TCDD. Co-treatment of cells with both PhIP and TCDD caused a significant decrease in EROD activity in LTEE cells compared with the activity caused by exposure to TCDD alone, which was evident at PhIP concentrations at or above 10−6 M (Fig. 2C). However, when PhIP was added during the EROD assay but after the cells had been pre-exposed to 10 nM TCDD for 24 h, 10−5 M PhIP did not inhibit metabolism of 7-ethoxyresorufin to resorufin (Fig. 2C); therefore, PhIP exposure antagonized the induction of CYP1 enzymes by TCDD, as opposed to directly inhibiting the CYP1 enzymatic activity. When TCDD was included together with PhIP, there was a marked increase in adduct formation in LTEE cells, despite the fact that PhIP lessened CYP1A induction in the EROD assay. These results indicate that the enhanced induction of CYP1 by TCDD in LTEE cells is more significant than the antagonism of the AhR by PhIP.
3.4. Analysis of regulatory elements within the AHR proximal promoter
Our previous studies showed that expression of AhR mRNA and protein was significantly up-regulated in LTEE cells in comparison with their levels in control cells [15, 16]. In order to investigate functional elements of the human AHR promoter that could be responsible for its elevated expression in LTEE cells, we utilized a luciferase-reporter plasmid and generated a deletion construct containing 120 bp of the AHR 5′-flanking region, termed AhRΔ(−120). AHR promoter activity was determined by luciferase assay in control and LTEE MCF-7 cells.
We identified five putative transcription factor binding sites within the AhRΔ(−120) deletion construct by searching DNA motif databases [34]. The five sites corresponded to transcription factors that were either up-regulated by LTEE in a cDNA array analysis performed previously in this laboratory [15], or are known to be involved in estrogen regulation. We altered each of these sequences through site-directed mutagenesis, and performed luciferase assays in which the mutant promoter activity was compared to the activity of the unaltered construct. The five sequences that were mutated were: three Sp1 binding motifs, a Krüppel-like Factor-4 (KLF4) recognition sequence, and a myogenin (Myo) recognition sequence. Each of the three Sp1 binding-sequence mutants showed about 2-fold lower activity than the native AhRΔ(−120) construct, indicating these elements are involved in transcriptional enhancement (Fig. 3). The KLF4 mutant had 1.7-fold increased activity over the native AhRΔ(−120) construct, indicating an inhibitory activity on transcription at this site. Alteration of the Myo site had no significant effect on luciferase activity of the resulting construct, suggesting that this element does not play a significant role in the regulation of AHR gene expression in MCF-7 cells.
Fig. 3.

Effect of site-directed mutagenesis of Sp1-binding sites on AHR promoter activity. Control (black bars) and LTEE (white bars) cells maintained for 16 passages were seeded in 24-well plates and the next day transfected with phRG-TK Renilla control vector and the AhRΔ(−120) deletion construct or AhRΔ(−120) mutant subclones (Sp1 -82, Sp1 -63, Sp1 -41, KLF4 -14, Myo -8). Cultures were incubated for 48 h, and luciferase assays were performed. Firefly luciferase activities were normalized to those of Renilla luciferase. Data are means ± SE; n = 3. Significant differences from the AhRΔ(−120) construct are shown; *, P < 0.05; ***, P < 0.001.
Mutational analyses of the AHR proximal promoter revealed sequence elements that are likely to be important in the regulation of AHR gene transcription. However, when the luciferase activities of the reporter constructs in control and LTEE cells were compared, there were no significant differences. Similarly, no difference in luciferase activity due to LTEE was observed when luciferase assays were performed with the full 5.6-kb pGL3-hAhRP plasmid (data not shown). These results indicate that other factors may contribute to the differences in AhR regulation in control and LTEE cells, such as regulatory DNA elements outside the region encompassed by these constructs, or contributions from epigenetic or post-transcriptional control mechanisms.
3.5. Sp transcription factors bind AHR proximal promoter sequences
Results from luciferase assays indicating the presence of functional Sp transcription-factor binding sites in the proximal AHR promoter led us to investigate whether Sp proteins are able to bind to these DNA elements. Nuclear extracts from control and LTEE MCF-7 cells were incubated with radiolabeled ODNs corresponding to three sites, referred to as Sp1 -82, Sp1 -63, and Sp1 -41, and the protein:DNA complexes were resolved by EMSA. The Sp1 and Sp3 protein:DNA complexes were then identified by supershift using antibodies against Sp1 and Sp3 (Fig. 4, lanes 3–5, 7–9). ODNs spanning the individual Sp1 binding sites showed similar electrophoretic mobility patterns with bound Sp1 protein in both control and LTEE nuclear extracts (Fig. 4, lanes 2, 6). This would suggest direct recruitment of Sp1 at the AHR proximal promoter. Sp3:DNA complexes were evident in control nuclear extracts (Fig. 4, lane 2), but in some lanes showed proportionally lower binding in LTEE than control nuclear extracts (lane 6 of Fig. 4). When longer incubation times of the binding reactions were used during competitive EMSAs, equivalent bands were observed for Sp3 protein:DNA complexes in control and LTEE nuclear extracts incubated with each of the probes (Supplementary Fig. 2).
Fig. 4.
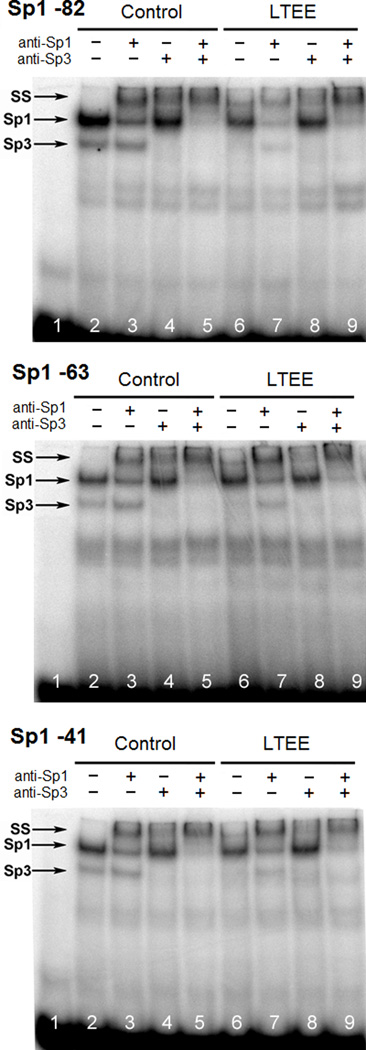
Sp proteins bind to ODNs representing sequences of the AHR proximal promoter. Nuclear extracts from control and LTEE cells at passage 26 were incubated with radiolabeled ODN probes encompassing each of the 3 Sp1 sites (Sp1 -82, Sp1 -63, Sp1 -41 relative to the TSS) in the AHR proximal promoter, and protein:DNA complexes were resolved by EMSA. Supershifting of protein:DNA complexes was achieved by incubating binding reactions with antibodies against Sp1 and/or Sp3, and complexes were resolved on 6% denaturing acrylamide gels. Lane 1 shows unbound probe. Protein:DNA complexes are indicated as containing either Sp1 or Sp3; SS = supershift; free probe is apparent at the bottom of each gel.
3.6. Stability of the AhR mRNA in control and LTEE MCF-7 cells
After determining that there was minimal difference in AHR promoter activity between control and LTEE cells using AHR-promoter-luciferase-reporter assays, we then investigated whether the increased level of AhR mRNA in LTEE cells relative to those in control cells was due to increased stability of the mRNA transcript. We found that the initial AhR transcript level in LTEE cells was 16-fold higher than that in control cells, but in the presence of actinomycin D the AhR transcript levels in LTEE cells decayed, with a half-life of 1.74 h, to levels below those in comparably treated control cells (Fig. 5A). AhR transcript levels in control cells remained constant over the course of the experiment. Therefore, a differential rate of AhR mRNA degradation was observed between control and LTEE cells, but increased AhR mRNA stability was not found to be responsible for the elevated levels of AhR mRNA in LTEE cells in comparison with control cells, because an increased rather than decreased rate of AhR mRNA decay was observed in LTEE cells.
Fig. 5.

Analyses of AhR transcript expression. (A) AhR mRNA is elevated in LTEE cells, but shows decreased stability. Control (black circles) and LTEE cells (white circles) were cultured for 23 passages and treated with actinomycin D for 0, 1, 2, 6, and 10 h in duplicate, and total RNA was isolated and reverse transcribed as indicated in Materials and Methods. Primers specific for AhR cDNA were used to determine mRNA levels by real-time PCR. Data are the means ± range of duplicates. (B) AhR hnRNA levels are enhanced in LTEE MCF-7 cells. Total RNA from control and LTEE cells maintained for 12 passages was isolated, and primers specific for sequences of intron 10/exon 11 of the AHR gene were used to amplify reverse-transcribed hnRNA by real-time PCR. Relative expression values were calculated using the ΔCt method as described in Materials and Methods. AhR mRNA levels were determined using real-time PCR. Data are the means ± SE; n = 3. Significant differences between control and LTEE are shown, *, P < 0.05.
3.7. AhR hnRNA is increased in LTEE cells
In order to determine whether the regulation of AhR expression is due to differences in rates of transcription or in processing of the RNA, levels of hnRNA isolated from control and LTEE cells were examined. Using primers that spanned the junction of intron 10 and exon 11 of the AhR cDNA, analysis by real-time PCR showed a 2.9-fold induction of hnRNA in LTEE cells compared with levels in control cells (Fig. 5B). This induction is similar to the 4.1-fold induction that was determined for the mRNA, and is consistent with the level of induction expected for cells at passage 12 (Fig. 1A). This result suggests that the rate of AHR gene transcription is higher in LTEE cells than in control cells.
3.8. AHR proximal promoter DNA is not methylated in control or LTEE MCF-7 cells
Since the expression of the AhR in control and LTEE cells appeared to be epigenetically regulated, we investigated the possibility that differential DNA methylation of the AHR promoter was responsible for the observed up-regulation of AhR expression in LTEE cells. Bisulfite-modified genomic DNA from control and LTEE cells, at passages 12 and 56, and from 184-A1 and MCF-10A breast epithelial cells was subjected to two rounds of PCR using methylation- and gene-specific primers. DNA sequencing results showed that, in a region of −265 to +193 bp surrounding the TSS, the 35 CpG sites of the AHR gene were not methylated in any of the cell lines examined (Table 1). Since CYP1B1 mRNA is also up-regulated in LTEE cells, we examined 119 CpG sites in the DNA of the CYP1B1 promoter in passage 12 control and LTEE MCF-7 cells, and in 184-A1 and MCF-10A cells. Eight sites were found to be methylated, but to a similar degree in each of the cell lines and between control and LTEE MCF-7 cells (Table 1); the region analyzed encompassed −1589 to +20 bp surrounding the CYP1B1 TSS.
Table 1.
Analysis of CpG methylation in the promoters of the CYP1B1 and AHR genes.
| Gene name |
Cell Lines | Number of CpG sites examined |
Number of CpG sites methylated |
Position of methylated CpG |
|---|---|---|---|---|
| CYP1B1 | 184-A1, MCF-10A, MCF-7 (control and LTEE for passage 12) | 119 | 8 (observed in all samples) | -579, -1452, -1454, -1458, -1465, -1518, -1535, -1537 |
| AHR | 184-A1, MCF-10A, MCF-7 (control and LTEE for passages 12 and 56) | 41 | 0 (not observed in any sample) | Not Applicable |
3.9. ChIP of the AHR proximal promoter reveals changes in histone methylation
Because persistent changes in AhR expression did not appear to be the result of DNA methylation, we employed ChIP to investigate whether differences in AhR expression in control and LTEE cells were the result of chromatin modifications. Primers were designed to amplify six overlapping regions both upstream and downstream of the TSS of the AHR promoter (Fig. 6). The results indicated similar recruitment of Sp1 and Sp3 transcription factors to region 3, which contains the highly GC-rich sequence and Sp transcription factor binding sites (Fig. 6). Pre-immunoprecipitated input DNA was included as a positive control for the PCR reactions; immunoprecipitations without antibodies were included as negative controls.
Fig. 6.

ChIP of the AHR promoter reveals changes in epigenetic marks of gene regulation. DNA from control and LTEE cells cultured for 36 passages was isolated after immunoprecipitation and subjected to PCR amplification for 28–32 cycles (for immunoprecipitation with Sp1, Sp3, H3K4me2 antibodies) or 30–34 cycles (for immunoprecipitation with Histone 3, H3K27me3, RNA Pol II antibodies) using 6 sets of primers covering ~400 bp surrounding the TSS; ChIP using RNA Pol II antibodies was performed in cells cultured for 48 passages. Amplifications of DNA from ChIPs containing no antibody (No Ab control) are included for each appropriate set of immunoprecipitated DNA. Primer regions 1–6, the TSS (arrow), and putative Sp1 binding sites (vertical black bars) are indicated in the AHR promoter (upper diagram). PCR product sizes are indicated by the input control, and the DNA ladders in the first and last lanes indicate 300, 200, and 100 bp.
We then examined the status of histones bound at the AHR proximal promoter in lysates from control and LTEE cells using antibodies against histone 3 and transcriptionally activating and repressive modifications: H3K4me2 and H3K27me3, respectively. PCR amplification of anti-histone 3-immunoprecipitated DNA showed an enrichment of histone 3 at promoter regions 3 and 4 in control cells as compared with LTEE cells (Fig. 6), with roughly equal histone 3 binding between control and LTEE cells at regions 1, 2, 5, and 6. We detected somewhat enhanced interaction of H3K4me2 and particularly strong interaction of H3K27me3 at all six regions of the AHR promoter in control cells compared with LTEE cells. These data indicate that in control cells, the AHR proximal promoter is more closely associated with markers of epigenetic silencing, concordant with the observed low AhR expression level in these cells compared with that in LTEE cells. RNA Pol II-precipitated DNA was enriched in regions 3 through 5 in LTEE cells, demonstrating a stronger association of the transcriptional machinery at the AHR TSS. In comparison with the AHR, exon 1 of the TUBA1A gene and exon 1 and promoter region of AIP showed similar recruitment of H3K4me2, H3K27me3, and RNA Pol II between control and LTEE cells (Supplementary Fig. 3).
3.10. Polymorphic repeats of (GGGGC)n in normal and cancer cell lines
When we employed standard PCR conditions for the amplification of the GC-rich region of the AHR gene promoter, we were unable to obtain satisfactory results. PCR products of varying sizes were obtained, and sequencing of the products revealed that the amplification reaction had in essence skipped over a portion of the sequence. We therefore investigated a variety of PCR conditions, primers, and additives, and we found that the inclusion of 6% propylene glycol [26] was the only procedural modification of those we tested that allowed for the correct amplification of the AHR proximal promoter sequence. During our investigation of the AHR promoter, we observed a polymorphic sequence that varied in the number of (GGGGC)n repeats among different human cell lines.
The Ensembl DNA sequence of the human AHR promoter contains (GGGGC)2 starting at -37 bp upstream of the TSS, and the initial report on this polymorphism indicated that (GGGGC)2 was the most prominent allele in the Japanese population that was studied [22]. However, sequence analysis of PCR products amplified from the AHR promoter in 17 cell lines using our PCR conditions revealed that the major polymorphic sequences at this site consist of (GGGGC)4 or (GGGGC)5 rather than (GGGGC)2. We found that ZR-75-1 and MDA-MB-231 breast cancer cells and GM04155 Leukemic T-cells were heterozygous for (GGGGC)4/5, while T47D breast cancer cells were heterozygous for (GGGGC)2/4. Results for all 17 cell lines are presented (Table 2). We next determined the allele frequency of this (GGGGC)n polymorphism in a population of New York State newborns. We found that the most prominent allele was (GGGGC)4, followed in incidence by (GGGGC)5 (Table 3). The (GGGGC)2 and (GGGGC)6 alleles were found to be rare. Differences in allele frequencies of the (GGGGC)n repeats among the four ethnic groups were not significant. We also observed what appear to be two previously unreported base changes in (GGGGC)4: C>A at the third repeat and G>T at the second repeat.
Table 2.
Polymorphic repeats in the AHR proximal promoter.
| Cell Line | Origin | Demographics | Homozygous/ Heterozygous |
(GGGGC)n Repeats |
|---|---|---|---|---|
| MCF-7 | Cancer (breast) | Caucasian F, 69 y | Homo | 5 |
| MCF-10A | Normal (breast) | Caucasian F, 36 y | Homo | 5 |
| MDA-MB-231 | Cancer (breast) | Caucasian F, 51 y | Het | 4/5 |
| 184-A1 | Normal (breast) | Unknown F | Homo | 4 |
| T47D | Cancer (breast) | Unknown F, 54 y | Het | 2/4 |
| ZR-75-1 | Cancer (breast) | Caucasian F, 63 y | Het | 4/5 |
| A2780 | Cancer (ovarian) | Unknown F | Homo | 4 |
| HOSE-1 | Normal (ovarian) | Unknown F | Homo | 4 |
| A549 | Cancer (lung) | Caucasian M, 58 y | Het | 4/5 |
| NCI-H23 | Cancer (lung) | African American M, 51 y | Homo | 4 |
| NCI-H292 | Cancer (bronchial) | African American F, 32 y | Homo | 4 |
| HEK293 | Normal (kidney) | Unknown | Homo | 4 |
| PANC-1 | Cancer (pancreas) | Caucasian M, 56 y | Homo | 4 |
| GM04155 | Cancer (T-cell) | Caucasian M, 11 y | Het | 4/5 |
| GM04679 | Cancer (B-cell) | African American M, 11 y | Homo | 4 |
| GM03638 | Cancer (B-cell) | Unknown F, 16 y | Homo | 5 |
| GM16726 | Cancer (B-cell) | African American M, 27 y | Homo | 5 |
Table 3.
AHR gene promoter polymorphisms in New York State newborns.
| Newborns | Alleles | (GGGGC)2 | (GGGGC)4 | (GGGGC)5 | (GGGGC)6 | *C/A | **G/T | |
|---|---|---|---|---|---|---|---|---|
| African-American | 32 | 64 | 1 | 53 | 9 | 1 | 1 | 0 |
| %*** | 1.6 (0.08–9.54) | 82.8 (70.9–90.7) | 14.1 (7.0–25.5) | 1.6 (0.08–9.54) | 1.6 (0.08–9.54) | 0 (0–7.05) | ||
| Asian | 32 | 64 | 0 | 42 | 22 | 0 | 0 | 0 |
| % | 0 (0–7.05) | 65.6 (52.6–76.8) | 34.4 (23.3–47.4) | 0 (0–7.05) | 0 (0–7.05) | 0 (0–7.05) | ||
| Caucasian | 32 | 64 | 0 | 48 | 15 | 1 | 0 | 1 |
| % | 0 (0–7.05) | 75 (62.4–84.6) | 23.4 (14.1–36.0) | 1.6 (0.08–9.54) | 0 (0–7.05) | 1.6 (0.08–9.54) | ||
| Hispanic | 32 | 64 | 1 | 39 | 23 | 1 | 3 | 0 |
| % | 1.6 (0.08–9.54) | 60.9 (47.9–72.6) | 35.9 (24.6–49) | 1.6 (0.08–9.54) | 4.7 (1.2–14) | 0 (0–7.05) | ||
| Total | 128 | 256 | 2 | 182 | 69 | 3 | 4 | 1 |
| % | 0.8 (0.13–3.1) | 71.1 (65.1–76.5) | 27.0 (21.7–32.9) | 1.2 (0.3–3.7) | 1.6 (0.5–4.2) | 0.4 (.02–2.5) |
(GGGGC)4: GGGGCGGGGCGGGGCGGGGC
C/A: GGGGCGGGGCGGGG(C/A)GGGGC
G/T: GGGGC(G/T)GGGCGGGGCGGGGC
Percent calculations are followed by the 95% confidence interval, in parentheses, with a correction for continuity (http://faculty.vassar.edu/lowry/prop1.html).
To determine the effect of the (GGGGC)n repeats on AHR gene transcription, we compared the promoter activity of two AhRΔ(−120) luciferase constructs that incorporated either (GGGGC)2 or (GGGGC)4. The construct containing the (GGGGC)4 elicited approximately twice the luciferase activity as did the construct containing (GGGGC)2 in vitro (Fig. 7). Additionally, mutation of one of the repeats in the construct that contained (GGGGC)2 (Fig. 3, Sp1 -41 site) reduced luciferase activity by approximately half. EMSAs were performed with ODNs containing (GGGGC)2 of the AHR proximal promoter (Supplementary Fig. 2, Sp1 -41); base substitutions specifically targeting the (GGGGC)n repeat sequence in the second putative Sp1 site (Mutant 2) markedly reduced Sp binding in both control and LTEE cells.
Fig. 7.

Effect of the number of (GGGGC)n repeat sequences on AHR promoter activity. LTEE MCF-7 cells were transiently transfected with polymorphic forms of the firefly luciferase AHR promoter construct, AhRΔ(−120), with (GGGGC)2 or (GGGGC)4 as indicated. After 44 h, the cells were lysed and assayed for luciferase activity. Data were normalized to the activity of the co-transfected phRG-TK Renilla luciferase construct, and are represented as the mean ± SE; n = 3; ***, P < 0.001.
3.11. AHR proximal promoter sequences have the capacity to form guanine-quadruplex structures
The sequence of the GC-rich region of the AHR proximal promoter suggests that it may be amenable to the formation of one or more of the guanine-quadruplex structures, which are secondary DNA structures consisting of a series of guanines that form stacked Hoogsteen-bonded tetrads [35]. We performed CD spectroscopy using ODNs (Supplementary Table 1) spanning the GC-rich region of the AHR promoter containing (GGGGC)2, the sequence of which had a high G-score when analyzed for the ability to form a guanine quadruplex [36]. CD spectra recorded for the AHR native forward sequence containing (GGGGC)2 produced positive peaks at ~260 nm and ~295 nm (Supplementary Fig. 4A); these spectra are consistent with either mixed parallel/antiparallel or propeller-type parallel structures [31]. CD spectra recorded for ODNs containing several point mutations targeting the guanine tetrads within the native sequence showed differences at ~260 and ~295 nm from spectra of the native sequence, suggesting a diminution in their abilities to form quadruplex structures compared with that of the native sequence. CD spectra were also recorded for ODNs containing AHR gene sequences of (GGGGC)n with n = 3, 4, or 5, and they were compared with spectra from the ODN that contained (GGGGC)2. In the presence of 150 mM KCl, spectra recorded for all of these ODNs showed peaks at ~260 nm, and these peaks increased in intensity as the number of (GGGGC)n repeats increased (Supplementary Fig. 4B). The spectra of the (GGGGC)3-, (GGGGC)4-, and(GGGGC)5-containing ODNs did not contain peaks at 295 nm as was observed in the spectrum of the (GGGGC)2-containing ODN; the spectra with only the peaks at 260 nm are indicative of four-stranded parallel structures [31].
4. Discussion
Most studies of the AhR have focused on its role in regulating gene expression, notably of the genes of the aryl hydrocarbon locus that encode phase I and II enzymes and phase III transporters. Recent studies have also identified additional roles of the AhR in regulation of the cell-cycle and immune function. In their treatise on AhR expression and regulating the regulator, Harper et al. [6] noted that there are a number of factors that have been shown to alter the short- and long-term expression of AhR in vivo and in vitro, including hormones, growth factors, developmental stage, and rates of cell proliferation. There are numerous examples in which alterations of the level of AhR expression determines the responsiveness of downstream pathways [6]. However, the underlying cellular and molecular mechanisms responsible for the observed changes in AhR expression remain for the most part elusive.
Our studies of both the short- and long-term regulation of AhR expression and function have utilized MCF-7 cells, the well characterized line of luminal A human breast cancer cells. In addition to rapid effects elicited by agents such as phorbol esters [37], we have observed that there are long-term regulatory effects on AhR expression, and exposure to E2 can be isolated as an independent variable that can affect the long-term expression of the AhR [15, 16]. The results reported here show that AhR expression is slowly diminished and is eventually lost in MCF-7 cells cultured long-term without E2 supplementation. As has been observed in other systems, we reported a close correlation between AhR levels and the inducibility of CYP1 enzymes [38]. The near-total loss of the AhR expression in control cells observed in the current study essentially eliminates CYP1 inducibility in MCF-7 cells. Our cellular and molecular studies show that alterations in the expression level of AhR markedly affect downstream CYP1 levels and inducibility of CYP1A1 and CYP1B1 gene expression, and support the contention that alterations in AhR expression in vivo will have physiological and toxicological consequences through effects on carcinogen metabolism.
The dietary carcinogen, PhIP, is primarily metabolized by CYP1A2 in the liver; however, extrahepatic CYP1A1 and CYP1B1 also catalyze the metabolism of PhIP to its mutagenic metabolite, N2-OH-PhIP. CYP1A1 has a higher catalytic efficiency than CYP1A2 and CYP1B1 for PhIP metabolism [39]. CYP1A1 and CYP1B1 are present or are inducible in extrahepatic tissues including cells derived from the mammary gland, in which bioactivation of PhIP may contribute to the formation of mutagenic DNA lesions [40]. Our current data show that both the increased level of AhR expression in LTEE MCF-7 cells and the activation of the AhR by TCDD are associated with increased levels of PhIP:DNA adducts formed in vitro. This synergism between estrogen and TCDD exposure in enhancing PhIP:DNA adducts suggest that the AhR is potentially a critical control point in the bioactivation of this carcinogen. Our results show that, with the addition of TCDD during exposure to PhIP, levels of adducts were increased in LTEE cells compared with control cells; the combination of the AhR ligand, TCDD, and PhIP exposure in LTEE cells was particularly deleterious. Although PhIP itself is a weak AhR agonist [40, 41], in the diet and in cooked meats, there may be co-exposure to PhIP and AhR ligands such as BaP, since both are formed by partial combustion and pyrolysis. While PhIP is a potent carcinogen in rodents, the potential role of PhIP in the etiology of human cancers including breast cancer remains unclear. Some epidemiologic investigations have shown that women who consume greater amounts of foods containing PhIP have a higher risk for breast cancer [42]; however, appreciable levels of PhIP:DNA adducts were not detected in DNA from breast cancer patients [43].
Our previous studies have shown that AhR mRNA and protein expression levels are up-regulated in LTEE MCF-7 cells, but the mechanism responsible for the effect was not identified [15, 16]. Given that AhR expression in mouse-derived cell lines is regulated by Sp1 [8], we investigated whether the transcriptional regulation of the AHR proximal promoter involved Sp1 and Sp3 transcription factor binding in human-derived cells. Luciferase-reporter assays of transcriptional activity showed that the human AHR proximal promoter contains functional Sp binding sites; however, the promoter activities were very similar when assayed in control and LTEE cells. These studies with naked DNA promoter constructs would indicate that other factors may be responsible for the increased and persistent expression of AhR in LTEE cells. We cannot discount the possibility that there are regions outside of those present in our constructs that are necessary for the regulation of the AHR gene by E2 or E2-mediated transcription factors, including the presence of distal EREs [44, 45]. While our results indicate the presence of Sp1 binding sites in the proximal promoter of the AHR gene that are likely to be important in the transcriptional regulation of AhR expression, analyses by AHR-promoter luciferase-reporter assay, competitive EMSA, and ChIP, when taken together, indicate that differential expression or binding of Sp1 and/or Sp3 do not appear to play a major role in the observed differences in AhR expression between control and LTEE cells.
The observations of increased levels of AhR hnRNA in LTEE in comparison with levels in control cells and enhanced rather than depressed rates of AhR mRNA decay in LTEE cells do not support a post-transcriptional mechanism as being responsible for the enhanced expression of AhR mRNA in LTEE relative to that in control cells. We therefore hypothesized that epigenetic mechanisms were responsible for both the diminishment of AhR inducibility by TCDD over time in control cells and the persistence of elevated AhR expression after E2 withdrawal for 35 days. Short-term exposure of MCF-7 cells to estrogen has not been reported to induce AhR expression in the early (3-or 4-h E2 exposure) or late (24-h E2 exposure) microarray expression metasignatures [46]. In comparison with those of the AhR, PGR transcript levels in LTEE (-E2) cells were decreased to below those in control cells, indicating that genes previously characterized as estrogen-responsive may not necessarily be subject to estrogen imprinting by LTEE.
It has been previously reported that expression of the AhR may be down-regulated by epigenetic mechanisms [47] including DNA hypermethylation [48]. The studies of Mulero-Navarro and coworkers on the 5′-flanking region of the AHR gene indicated that the CpG island that encompasses nucleotides −133 to +194, and spans the proximal promoter including the TSS, was subject to methylation [48]. Chronic myeloid leukemia K562 cells and acute lymphoblastic leukemia REH cells showed extensive methylation of CpG islands in this region of the AHR gene, and concomitantly reduced Sp1 binding and reduced AhR expression [48]. In a patient population, those subjects who showed AHR proximal promoter hypermethylation also showed reduced Sp1 binding in this region. In this same study, no methylation of this region was observed in DNA from MCF-7 cells. Given the greatly reduced level of AhR expression in control cells relative to LTEE MCF-7 cells, we investigated whether DNA methylation might be induced in this low-estrogen condition. However, we found this not to be the case; consistent with the results of Mulero-Navarro et al. [48], we did not detect DNA methylation of the AHR promoter region in either control or LTEE MCF-7 cells, nor did we observe methylation of the same region of DNA obtained from 184-A1 and MCF-10A cells, two non-tumor-derived cell lines of human breast origin. The observed loss of AhR expression in control MCF-7 cells that occurs without measurable methylation of the AHR proximal promoter and the neighboring CpG island suggests that the process of silencing in MCF-7 cells is mechanistically different than what has been observed in human leukemia cells.
While our data do not show evidence of DNA methylation of AHR proximal promoter, they do indicate that AhR expression is under the epigenetic regulation at the level of chromatin remodeling in MCF-7 cells. The loss of AHR gene expression over time suggests that a heterochromatin-like state develops in control cells. This is in contrast to LTEE cells, in which a euchromatin-like conformation likely develops from reduced association of AHR promoter DNA with H3K27me3. In a recent study, immunohistochemistry of cancerous breast tissue compared with normal breast tissue showed that lower levels of H3K27me3 in the tumors significantly correlated with shorter overall patient survival [49].
Methylation of H3K27 is catalyzed by the Polycomb group protein, enhancer of Zeste homologue 2, a histone methyltransferase which has been shown to be phosphorylated by estrogen- and xenoestrogen-induced PI3K signaling in MCF-7 cells [50]. Phosphorylation of enhancer of Zeste homologue 2 reduces its methyltransferase activity, which dissociates it from chromatin and facilitates gene expression [51]. There are numerous other characterized histone modifications relevant to cancer, such as the acetylation or methylation of histone 3 at lysine 9 [11, 52], that were not investigated in this study, but require evaluation to determine the full extent of histone modifications involved in the epigenetic regulation of the AHR gene in LTEE cells.
During the course of this investigation, we confirmed the presence of a genetic polymorphism in the proximal promoter region of the AHR promoter beginning at -37 bp relative to the TSS, which contains a cluster of Sp binding sequences. The (GGGGC)n polymorphism was previously reported in a study of a Japanese population [22]; however, data concerning the allele frequencies of the repeat were not presented, and there has been no further investigation of the polymorphism since its discovery in 2004. We suspect that the lack of characterization of this polymorphism has been due to the inherent difficulties in amplifying this GC-rich region using PCR. When we utilized the propylene glycol technique of Zhang et al. [26], we were able to determine the allelic frequencies of the (GGGGC)n repeats. We found that the majority of alleles in normal and cancerous cell lines we tested were found to contain (GGGGC)4. This was also found to be the case in a population of New York State newborns; the allele containing (GGGGC)5 was also common in the newborns, but alleles containing (GGGGC)2 or (GGGGC)6 were found to be rare. Our analyses of AHR promoter activity showing a significant increase in activity for the construct containing the (GGGGC)4 sequence compared with the construct containing the (GGGGC)2 sequence suggest that this polymorphism may be an important determinant of the expression of AhR.
Given that each additional repeat of GGGGC adds an additional consensus Sp1 binding site [53], greater numbers of repeats may allow for greater Sp binding and activation of gene transcription. We also show that, with a larger number of repeats, this region may be able to form more stable parallel guanine quadruplex structures as indicated by the CD spectra, which showed increasing peak amplitudes at 260 nm as the number of repeats increased. However, only the ODN containing (GGGGC)2 produced a CD spectrum compatible with either a mixed parallel/anti-parallel structure or the propeller-type parallel structure [31], either of which can be formed as a unimolecular quadruplex. In contrast, the spectra of ODNs containing (GGGGC)n with n = 3, 4, or 5 were consistent with four-stranded parallel structures, and these are more likely to be formed as tetramers in vitro [31].
The formation of guanine-quadruplex structures may be significant in this study for two reasons: (i) formation of these structures may be responsible for the difficulty in amplifying the region using PCR, and (ii) the structures may represent a physiologic mechanism whereby gene transcription is regulated. We noted the difficulty of amplifying sequences containing 5-repeats in genomic DNA from MCF-7 cells by PCR without a suitable denaturant, and suggest that a unimolecular guanine quadruplex may possibly form in the 5-repeat sequence in duplex DNA, causing polymerase arrest during PCR. Whether or not an AHR promoter guanine quadruplex forms in vivo is unknown; however, recent evidence suggests a prevalence of quadruplex-forming DNA motifs within 1-kb regions of several gene promoters [54], and extensive studies of promoter-based quadruplex formation in oncogenes such as c-MYC have shown the possibility of direct regulation of gene expression by guanine-quadruplex structures [55]. In some cases guanine-quadruplex formation appears to cause a reduction in transcriptional rates through polymerase arrest [56], while in other cases quadruplex formation appears to enhance rates of transcription [57]. The potential for regulation of AhR expression by quadruplex formation will be investigated in future studies. It is intriguing to speculate that the regulation of AhR expression in vivo involves guanine-quadruplex formation and is affected by the polymorphic (GGGGC)n repeat.
In summary, our studies of AhR expression in MCF-7 breast cancer cells established that there are functional Sp1 binding sites within the proximal promoter region of the AHR gene. Our studies with LTEE MCF-7 cells showed a marked difference between control and LTEE cells in AhR expression and enhanced capacity for bioactivation of PhIP to form PhIP:DNA adducts in LTEE cells. Investigations into the enhanced AhR expression in LTEE cells indicated that it was not a result of increased mRNA stability, differential RNA processing, or changes in proximal promoter DNA methylation. Analysis of the histones bound to the AHR proximal promoter confirmed that reduced expression of AhR in control cells is concomitant with changes in histone modifications, notably the increased interaction of H3K27me3, a histone mark associated with repression of gene transcription. We confirmed a genetic polymorphism within this region of Sp1 sites consisting of varying repeats, with sequences of (GGGGC)4 and (GGGGC)5 being most prominent in New York State newborns. Analyses of AHR promoter activity showed a significant difference between the (GGGGC)2 and (GGGGC)4 repeats, suggesting physiological consequences of the polymorphism on AhR expression. These studies reveal multiple levels at which AhR expression may be controlled, and offer additional insights into mechanisms regulating AhR expression that can ultimately impact carcinogenesis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 CA081243 (to DCS), R01 CA122320 (to RJT), R01 CA106186 (to SDS) and R21 CA104812 (to SDS). The authors gratefully acknowledge use of the Wadsworth Center’s Tissue Culture Facility and the Biochemistry and Applied Genomic Technologies Core Facilities. We thank our Wadsworth Center colleagues for the following DNA samples: from PANC-1 and HEK293 cells, Dr. Erasmus Schneider; from A549 cells, Dr. Xinxin Ding; and from NCI-H23 and NCI-H292 lung cancer cell lines, and four leukemia cell lines, Dr. Noel Espina. DNA from A2780 and HOSE-1 cells was a kind gift of Dr. Gareth Owen of the Pontifical Catholic University of Chile (Santiago, Chile). The AHR promoter-luciferase reporter plasmid pGL3-hAhRP was a kind gift from Drs. Sandra Wolff and Josef Abel of the Medical Institute of Environmental Hygiene at the Heinrich-Heine-University, Düsseldorf, Germany.
Abbreviations
- AIP
AhR interacting protein
- AhR
aryl hydrocarbon receptor
- BaP
benzo[a]pyrene
- CD
circular dichroism
- ChIP
chromatin immunoprecipitation
- CYP
cytochrome P450
- dG-C8-PhIP
N-(deoxyguanosin-8-yl)-PhIP
- E2
17β-estradiol
- EMSA
electrophoretic mobility shift assay
- ER
estrogen receptor
- EROD
ethoxyresorufin-O-deethylase
- H3K4me2
histone 3 dimethyl lysine 4
- H3K27me3
histone 3 trimethyl lysine 27
- hnRNA
heterologous nuclear RNA
- KLF4
Krüppel-like Factor-4
- LC-ESI/MS3
liquid chromatography-electrospray ionization triple-stage mass spectrometry
- LTEE
long-term estrogen exposure
- ODN
oligodeoxynucleotide
- PAH
polycylic aromatic hydrocarbon
- PCR
polymerase chain reaction
- PGR
progesterone receptor
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- RNA Pol II
RNA-polymerase II
- SE
standard error
- Sp
specificity protein
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TSS
transcriptional start site
Footnotes
Appendix A. Supplementary data
Supplementary Tables 1 through 4 and Supplementary Figures 1 through 4 are associated with this article.
Conflict of interest statement: The authors have nothing to disclose.
References
- 1.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 2.Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- 3.Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275:8432–8438. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 4.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 5.Xue W, Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol Appl Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol. 2006;72:267–279. doi: 10.1016/j.bcp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Garrison PM, Denison MS. Analysis of the murine AhR gene promoter. J Biochem Mol Toxicol. 2000;14:1–10. doi: 10.1002/(sici)1099-0461(2000)14:1<1::aid-jbt1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald CT, Nebert DW, Puga A. Regulation of mouse Ah receptor (Ahr) gene basal expression by members of the Sp family of transcription factors. DNA Cell Biol. 1998;17:811–822. doi: 10.1089/dna.1998.17.811. [DOI] [PubMed] [Google Scholar]
- 9.Racky J, Schmitz HJ, Kauffmann HM, Schrenk D. Single nucleotide polymorphism analysis and functional characterization of the human Ah receptor (AhR) gene promoter. Arch Biochem Biophys. 2004;421:91–98. doi: 10.1016/j.abb.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Spink DC, Katz BH, Hussain MM, Pentecost BT, Cao Z, Spink BC. Estrogen regulates Ah responsiveness in MCF-7 breast cancer cells. Carcinogenesis. 2003;24:1941–1950. doi: 10.1093/carcin/bgg162. [DOI] [PubMed] [Google Scholar]
- 11.Dalvai M, Bystricky K. The role of histone modifications and variants in regulating gene expression in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:19–33. doi: 10.1007/s10911-010-9167-z. [DOI] [PubMed] [Google Scholar]
- 12.Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001;344:276–285. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 13.Clamp A, Danson S, Clemons M. Hormonal risk factors for breast cancer: identification, chemoprevention, and other intervention strategies. Lancet Oncol. 2002;3:611–619. doi: 10.1016/s1470-2045(02)00875-6. [DOI] [PubMed] [Google Scholar]
- 14.Russo J, Hasan Lareef M, Balogh G, Guo S, Russo IH. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J Steroid Biochem Mol Biol. 2003;87:1–25. doi: 10.1016/s0960-0760(03)00390-x. [DOI] [PubMed] [Google Scholar]
- 15.Spink BC, Bennett JA, Pentecost BT, Lostritto N, Englert NA, Benn GK, et al. Long-term estrogen exposure promotes carcinogen bioactivation, induces persistent changes in gene expression, and enhances the tumorigenicity of MCF-7 human breast cancer cells. Toxicol Appl Pharmacol. 2009;240:355–366. doi: 10.1016/j.taap.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Englert NA, Spink BC, Spink DC. Persistent and non-persistent changes in gene expression result from long-term estrogen exposure of MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2011;123:140–150. doi: 10.1016/j.jsbmb.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Spink DC, Spink BC, Cao JQ, DePasquale JA, Pentecost BT, Fasco MJ, et al. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–298. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- 18.Bessette EE, Goodenough AK, Langouët S, Yasa I, Kozekov ID, Spivack SD, et al. Screening for DNA adducts by data-dependent constant neutral loss-triple stage mass spectrometry with a linear quadrupole ion trap mass spectrometer. Anal Chem. 2009;81:809–819. doi: 10.1021/ac802096p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nauwelaërs G, Bessette EE, Gu D, Tang Y, Rageul J, Fessard V, et al. DNA adduct formation of 4-aminobiphenyl and heterocyclic aromatic amines in human hepatocytes. Chem Res Toxicol. 2011;24:913–925. doi: 10.1021/tx200091y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolff S, Harper PA, Wong JM, Mostert V, Wang Y, Abel J. Cell-specific regulation of human aryl hydrocarbon receptor expression by transforming growth factor-beta(1) Mol Pharmacol. 2001;59:716–724. doi: 10.1124/mol.59.4.716. [DOI] [PubMed] [Google Scholar]
- 21.Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukushima-Uesaka H, Sai K, Maekawa K, Koyano S, Kaniwa N, Ozawa S, et al. Genetic variations of the AHR gene encoding aryl hydrocarbon receptor in a Japanese population. Drug Metab Pharmacokinet. 2004;19:320–326. doi: 10.2133/dmpk.19.320. [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun. 2002;294:347–353. doi: 10.1016/S0006-291X(02)00478-3. [DOI] [PubMed] [Google Scholar]
- 25.Han W, Cauchi S, Herman JG, Spivack SD. DNA methylation mapping by tag-modified bisulfite genomic sequencing. Anal Biochem. 2006;355:50–61. doi: 10.1016/j.ab.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Yang X, Meng L, Liu F, Shen C, Yang W. Enhanced amplification of GC-rich DNA with two organic reagents. Biotechniques. 2009;47:775–779. doi: 10.2144/000113203. [DOI] [PubMed] [Google Scholar]
- 27.Frey UH, Bachmann HS, Peters J, Siffert W. PCR-amplification of GC-rich regions: 'slowdown PCR'. Nat Protoc. 2008;3:1312–1317. doi: 10.1038/nprot.2008.112. [DOI] [PubMed] [Google Scholar]
- 28.Svotelis A, Gévry N, Gaudreau L. Chromatin immunoprecipitation in mammalian cells. Methods Mol Biol. 2009;543:243–251. doi: 10.1007/978-1-60327-015-1_16. [DOI] [PubMed] [Google Scholar]
- 29.Gregory RI, Randall TE, Johnson CA, Khosla S, Hatada I, O'Neill LP, et al. DNA methylation is linked to deacetylation of histone H3, but not H4, on the imprinted genes Snrpn and U2af1-rs1. Mol Cell Biol. 2001;21:5426–5436. doi: 10.1128/MCB.21.16.5426-5436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol Appl Pharmacol. 2007;220:320–332. doi: 10.1016/j.taap.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Paramasivan S, Rujan I, Bolton PH. Circular dichroism of quadruplex DNAs: applications to structure, cation effects and ligand binding. Methods. 2007;43:324–331. doi: 10.1016/j.ymeth.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. 1998;17:857–872. doi: 10.1002/(sici)1097-0258(19980430)17:8<857::aid-sim777>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 33.Saceda M, Lippman ME, Lindsey RK, Puente M, Martin MB. Role of an estrogen receptor-dependent mechanism in the regulation of estrogen receptor mRNA in MCF-7 cells. Mol Endocrinol. 1989;3:1782–1787. doi: 10.1210/mend-3-11-1782. [DOI] [PubMed] [Google Scholar]
- 34.Wingender E, Dietze P, Karas H, Knuppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun D, Liu WJ, Guo K, Rusche JJ, Ebbinghaus S, Gokhale V, et al. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol Cancer Ther. 2008;7:880–889. doi: 10.1158/1535-7163.MCT-07-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kikin O, D'Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spink BC, Fasco MJ, Gierthy JF, Spink DC. 12-O-tetradecanoylphorbol-13-acetate upregulates the Ah receptor and differentially alters CYP1B1 and CYP1A1 expression in MCF-7 breast cancer cells. J Cell Biochem. 1998;70:289–296. [PubMed] [Google Scholar]
- 38.Spink BC, Bennett JA, Lostritto N, Cole JR, Spink DC. Expression of the aryl hydrocarbon receptor is not required for the proliferation, migration, invasion, or estrogen-dependent tumorigenesis of MCF-7 breast cancer cells. Mol Carcinog. 2012 doi: 10.1002/mc.21889. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crofts FG, Sutter TR, Strickland PT. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by human cytochrome P4501A1, P4501A2 and P4501B1. Carcinogenesis. 1998;19:1969–1973. doi: 10.1093/carcin/19.11.1969. [DOI] [PubMed] [Google Scholar]
- 40.Thomas RD, Green MR, Wilson C, Weckle AL, Duanmu Z, Kocarek TA, et al. Cytochrome P450 expression and metabolic activation of cooked food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in MCF10A breast epithelial cells. Chem Biol Interact. 2006;160:204–216. doi: 10.1016/j.cbi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Kleman MI, Overvik E, Mason GG, Gustafsson JA. In vitro activation of the dioxin receptor to a DNA-binding form by food-borne heterocyclic amines. Carcinogenesis. 1992;13:1619–1624. doi: 10.1093/carcin/13.9.1619. [DOI] [PubMed] [Google Scholar]
- 42.Sinha R, Gustafson DR, Kulldorff M, Wen WQ, Cerhan JR, Zheng W. 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, a carcinogen in high-temperature-cooked meat, and breast cancer risk. J Natl Cancer Inst. 2000;92:1352–1354. doi: 10.1093/jnci/92.16.1352. [DOI] [PubMed] [Google Scholar]
- 43.Gu D, Turesky RJ, Tao Y, Langouët SA, Nauwelaërs GC, Yuan JM, et al. DNA adducts of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 4-aminobiphenyl are infrequently detected in human mammary tissue by liquid chromatography/tandem mass spectrometry. Carcinogenesis. 2012;33:124–130. doi: 10.1093/carcin/bgr252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 45.Badia E, Duchesne MJ, Semlali A, Fuentes M, Giamarchi C, Richard-Foy H, et al. Long-term hydroxytamoxifen treatment of an MCF-7-derived breast cancer cell line irreversibly inhibits the expression of estrogenic genes through chromatin remodeling. Cancer Res. 2000;60:4130–4138. [PubMed] [Google Scholar]
- 46.Ochsner SA, Steffen DL, Hilsenbeck SG, Chen ES, Watkins C, McKenna NJ. GEMS (Gene Expression MetaSignatures), a Web resource for querying meta-analysis of expression microarray datasets, 17β-estradiol in MCF-7 cells. Cancer Res. 2009;69:23–26. doi: 10.1158/0008-5472.CAN-08-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garrison PM, Rogers JM, Brackney WR, Denison MS. Effects of histone deacetylase inhibitors on the Ah receptor gene promoter. Arch Biochem Biophys. 2000;374:161–171. doi: 10.1006/abbi.1999.1620. [DOI] [PubMed] [Google Scholar]
- 48.Mulero-Navarro S, Carvajal-Gonzalez JM, Herranz M, Ballestar E, Fraga MF, Ropero S, et al. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis. 2006;27:1099–1104. doi: 10.1093/carcin/bgi344. [DOI] [PubMed] [Google Scholar]
- 49.Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay NV, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008;47:701–706. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol. 2010;24:993–1006. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 52.Lo PK, Sukumar S. Epigenomics and breast cancer. Pharmacogenomics. 2008;9:1879–1902. doi: 10.2217/14622416.9.12.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel DJ, Phan AT, Kuryavyi V. Human telomere, oncogenic promoter and 5'-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palumbo SL, Memmott RM, Uribe DJ, Krotova-Khan Y, Hurley LH, Ebbinghaus SW. A novel G-quadruplex-forming GGA repeat region in the c-myb promoter is a critical regulator of promoter activity. Nucleic Acids Res. 2008;36:1755–1769. doi: 10.1093/nar/gkm1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lew A, Rutter WJ, Kennedy GC. Unusual DNA structure of the diabetes susceptibility locus IDDM2 and its effect on transcription by the insulin promoter factor Pur-1/MAZ. Proc Natl Acad Sci U S A. 2000;97:12508–12512. doi: 10.1073/pnas.97.23.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.